

Abstract
Nicotinic acetylcholine receptors can be assembled from either homomeric or heteromeric pentameric subunit combinations. At the interface of the extracellular domains of adjacent subunits lies the acetylcholine binding site, composed of a principal component provided by one subunit and a complementary component of the adjacent subunit. Compared with neuronal nicotinic acetylcholine cholinergic receptors (nAChRs) assembled from α and β subunits, the α9α10 receptor is an atypical member of the family. It is a heteromeric receptor composed only of α subunits. Whereas mammalian α9 subunits can form functional homomeric α9 receptors, α10 subunits do not generate functional channels when expressed heterologously. Hence, it has been proposed that α10 might serve as a structural subunit, much like a β subunit of heteromeric nAChRs, providing only complementary components to the agonist binding site. Here, we have made use of site-directed mutagenesis to examine the contribution of subunit interface domains to α9α10 receptors by a combination of electrophysiological and radioligand binding studies. Characterization of receptors containing Y190T mutations revealed unexpectedly that both α9 and α10 subunits equally contribute to the principal components of the α9α10 nAChR. In addition, we have shown that the introduction of a W55T mutation impairs receptor binding and function in the rat α9 subunit but not in the α10 subunit, indicating that the contribution of α9 and α10 subunits to complementary components of the ligand-binding site is nonequivalent. We conclude that this asymmetry, which is supported by molecular docking studies, results from adaptive amino acid changes acquired only during the evolution of mammalian α10 subunits.
Introduction
Nicotinic acetylcholine (ACh) receptors (nAChRs) are members of the pentameric ligand-gated ion channel family (Nemecz et al., 2016). Seventeen nAChR subunits (α1–α10, β1–β4, δ, γ, and ε) have been identified in vertebrates (Nemecz et al., 2016), each of which has a large extracellular N-terminal region, four transmembrane helices (M1–M4), and an intracellular domain (Thompson et al., 2010). At the interface of the extracellular domains of adjacent subunits lies the ACh binding site, formed by six noncontiguous regions (loops A–F). Each binding site is composed of a principal component or (+) face provided by one subunit, which contributes three loops of highly conserved residues (loops A–C), and a complementary component (−) of the adjacent subunit, which contributes three loops (D–F) that have lower levels of sequence conservation between subunits (Brejc et al., 2001; Unwin, 2005; Dellisanti et al., 2007). Consequently, the components of the extracellular intersubunit binding sites are nonequivalent and their loops contribute differently to receptor function (Karlin, 2002).
nAChRs can be assembled from either homomeric or heteromeric subunit combinations (Millar and Gotti, 2009). Homomeric receptors, such as α7, have five equivalent ACh binding sites, each formed by the same principal and complementary components. ACh occupancy of one site is enough for activation of the homomeric human α7 nAChR, and also of a chimeric receptor containing the extracellular domain of α7 and the transmembrane domain of the serotonin type 3A (5-HT3A) receptor subunit (Rayes et al., 2009; Andersen et al., 2013). However, whereas occupancy of three nonconsecutive binding sites is required for maximal open-channel lifetime of the chimeric receptor, only one functional agonist binding site is required for maximal open-channel lifetime in α7.
In contrast to homomeric nAChRs, heteromeric receptors can have nonequivalent ACh binding sites provided by different subunit interfaces. For example, the Torpedo nAChR has two structurally different binding sites provided by the α(+)-δ(−) and α(+)-γ(−) subunit interfaces (Martinez et al., 2000), which bind agonists with different affinities (Blount and Merlie, 1989; Prince and Sine, 1999). According to the known stoichiometries of some neuronal nAChRs (Millar and Gotti, 2009), and by analogy to the muscle-type receptor, it was originally thought that heteromeric nAChRs have two agonist binding sites (Sine, 2002). In the case of neuronal nAChRs such as α4β2, α subunits were thought to only provide the (+) site to the binding interface, whereas β subunits were thought to provide the (−) site (Arias, 1997; Luetje and Patrick, 1991). However, it was subsequently shown that the composition of binding site interfaces is more complex. For example, the α4β2 receptor has two alternative stoichiometries, (α4)2(β2)3 and (α4)3(β2)2, leading to different binding site configurations and resulting in different functional and pharmacological properties (Carbone et al., 2009; Harpsøe et al., 2011; Mazzaferro et al., 2011); whereas (α4)2(β2)3 has two agonist binding sites provided by α(+)-β(−) interfaces, (α4)3(β2)2 has a third α(+)-α(−) binding interface (Hsiao et al., 2008; Mazzaferro et al., 2011).
The α9α10 receptor is an atypical member of the nAChR family. It is a heteromeric receptor composed only of α subunits (Elgoyhen et al., 1994, 2001; Sgard et al., 2002). Mammalian α9 subunits can form functional homomeric α9 receptors with an EC50 for ACh similar to that of the heteromeric α9α10 receptor (Elgoyhen et al., 1994, 2001). Hence, α9 subunits are capable of providing principal and complementary components to functional agonist binding sites. In contrast, rat and human α10 subunits do not lead to functional channels when expressed heterologously (Elgoyhen et al., 2001; Sgard et al., 2002). Consequently, it has been proposed that α10 might serve as a structural subunit, much like a β subunit of heteromeric receptors, providing only complementary components to the agonist binding site (Elgoyhen and Katz, 2012). A (α9)2(α10)3 stoichiometry has been determined for the rat recombinant receptor (Plazas et al., 2005), although expression of a 10-fold excess of α9 compared with α10 in Xenopus oocytes can lead to an additional receptor isoform with the stoichiometry (α9)3(α10)2 (Indurthi et al., 2014). However, the relative contribution of each subunit to the binding pockets of the heteromeric α9α10 receptor is unknown. By a combination of approaches (site-directed mutagenesis, expression studies, and molecular docking) we show that, contrary to previous assumptions, α10 subunits do contribute to the principal component of the binding site. Moreover, the contribution of α9 and α10 to the complementary component is nonequivalent. Our results demonstrate the versatility of nAChR subunits to generate diverse binding site interfaces with potentially different functional and/or pharmacological properties.
Materials and Methods
Expression of Recombinant Receptors in Xenopus laevis oocytes.
cDNAs encoding Gallus gallus (chick) or Rattus norvegicus (rat) α9 and α10 nAChR subunits were subcloned into a modified pGEMHE vector for expression studies in Xenopus laevis oocytes. Capped cRNAs were in vitro transcribed from linearized plasmid DNA templates using RiboMAX Large Scale RNA Production System (Promega, Madison, WI). Mutant subunits were produced using Quick Change XL II kit (Stratagene, La Jolla, CA). Amino acid sequences of rat and chicken α9, α10, and Torpedo α1 subunits were aligned using ClustalW (EMBL-EBI, Wellcome Genome Campus, Hinxton, Cambridgeshire). Residues were numbered according to the corresponding Torpedo α1 subunit mature protein (Karlin, 2002).
The maintenance of Xenopus laevis and the preparation and cRNA injection of stage V and VI oocytes have been described in detail elsewhere (Verbitsky et al., 2000). Typically, oocytes were injected with 50 nl of RNase-free water containing 0.01–1.0 ng of cRNA (at a 1:1 molar ratio when pairwise combined) and maintained in Barth’s solution at 18°C. Electrophysiological recordings were performed 2–6 days after cRNA injection under two-electrode voltage clamp with an Oocyte Clamp OC-725B or C amplifier (Warner Instruments Corp., Hamden, CT). Recordings were filtered at a corner frequency of 10 Hz using a 900BT Tunable Active Filter (Frequency Devices Inc., Ottawa, IL). Data acquisition was performed using a Patch Panel PP-50 LAB/1 interface (Warner Instruments Corp.) at a rate of 10 points per second. Both voltage and current electrodes were filled with 3 M KCl and had resistances of ∼1 MΩ. Data were analyzed using Clampfit from the pClamp 6.1 software (Molecular Devices, Sunnyvale, CA). During electrophysiological recordings, oocytes were continuously superfused (∼15 ml/min) with normal frog saline composed of: 115 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, and 10 mM HEPES buffer, pH 7.2. ACh was added to the perfusion solution for application. Unless otherwise indicated, the membrane potential was clamped to −70 mV. To minimize activation of the endogenous Ca2+ sensitive chloride current (Elgoyhen et al., 2001), all experiments were performed in oocytes incubated with the Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (acetoxymethyl ester) (100 µM) for 3 hours before electrophysiological recordings.
Concentration-response curves were normalized to the maximal agonist response in each oocyte. The mean and S.E.M. values of the responses are represented. Agonist concentration-response curves were iteratively fitted, using Prism 5 software (GraphPad Software Inc., La Jolla, CA), with the equation: I/Imax = AnH/(AnH + EC50nH), where I is the peak inward current evoked by the agonist at concentration A; Imax is the current evoked by the concentration of the agonist eliciting a maximal response; EC50 is the concentration of the agonist inducing a half-maximal current response; and nH is the Hill coefficient. Data were analyzed using Clampfit from the pClamp 6.1 software.
The effects of extracellular Ca2+ on the ionic currents through mutant α9α10 receptors were studied by measuring the amplitudes of the responses to an EC50 concentration of ACh upon varying the concentration of this cation from nominally 0 to 3 mM (Weisstaub et al., 2002). Amplitude values obtained at each Ca2+ concentration were normalized to that obtained in the same oocyte at a 1.8 mM. Values from different oocytes were averaged and expressed as the mean ± S.E.M.
Radioligand Binding.
Chimeric subunit cDNAs containing the extracellular N-terminal domain of the α9 or α10 subunit fused to the transmembrane and intracellular domain of the mouse 5-HT3A subunit have been described previously (Baker et al., 2004). The mammalian cell line tsA201 (derived from the human embryonic kidney 293 cell line) was obtained from Dr. William Green (University of Chicago, Chicago). Cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Paisley, United Kingdom) containing 2 mM L-GlutaMAX (Invitrogen) plus 10% heat-inactivated fetal calf serum (Sigma, Poole, United Kingdom) with penicillin (100 U/ml) and streptomycin (100 µg/ml) and were maintained in a humidified incubator containing 5% CO2 at 37°C. Cells were transiently transfected using Effectene transfection reagent (QIAGEN, Crawley, United Kingdom) according to the manufacturer’s instructions. In all cases, cells were transfected overnight and assayed for expression approximately 40–48 hours after transfection. To ensure that the levels of radioligand binding were not influenced by differences in the amount of subunit cDNA expressed, the amount of each subunit plasmid DNA and also the total amount of plasmid DNA were kept constant when subunits were expressed singly and in combination. This was achieved by the inclusion of empty plasmid expression vector when single subunits were transfected.
Binding studies with [3H]-α-bungarotoxin (α-BTX) in cell membrane preparations were performed essentially as described previously (Lansdell and Millar, 2000; Harkness and Millar, 2002). Membranes (typically, 10–100 µg of protein) were incubated with radioligand (final concentration 20 nM) for 150 minutes at 4°C in a total volume of 300 µl in the presence of protease inhibitors leupeptin (2 µg/ml) and pepstatin (1 µg/ml). Our standard protocol for determining nonspecific binding was the addition of 1 mM carbachol, 1 mM nicotine, and 10 µM methyllycaconitine to triplicate samples. Additional experiments were also performed in which nonspecific binding of [3H]-α-BTX was determined by displacement of the radioligand by ACh (1 mM). In all cases the levels of specific binding were determined by subtracting the level of nonspecific binding from the total binding (both of which were determined in triplicate). The data were determined as means of three independent experiments, each performed in triplicate. Radioligand binding was assayed by filtration onto 0.5% polyethylenimine-presoaked Whatman GF/B filters (Sigma-Aldrich, Dorset, England) followed by rapid washing (typically, five washes, each of 4 ml) with ice-cold 10 mM phosphate buffer using a Brandel cell harvester, and radioactivity was determined by scintillation counting. Care was taken to ensure that the number of receptor binding sites used for binding studies was low enough to avoid significant (>10%) ligand depletion at low concentrations of radioligand. Preliminary experiments were conducted to ensure that incubation times were long enough to enable radioligand binding to reach equilibrium. Protein concentrations were determined using bovine serum albumin standards (Bio-Rad, Hercules, CA).
Molecular Modeling and Docking.
Homology models of the extracellular domain of the chick and rat α9α10 nAChRs were created with SWISS MODEL (Schwede et al., 2003; Arnold et al., 2006; Bordoli et al., 2009) using the monomeric structure of the human α9 subunit as the template (Protein Data Bank ID 4UY2) (http://www.rcsb.org/pdb/explore/explore.do?structureId=4uy2) (Zouridakis et al., 2014). The monomeric models of these proteins were then structurally aligned to the pentameric structure of Lymnaea stagnalis AChBP bound to ACh (Protein Data Bank ID 3WIP) (http://www.rcsb.org/pdb/explore/explore.do?structureId=3wip) (Olsen et al., 2014) using the program STAMP (Russell and Barton, 1992) from visual molecular dynamics (Humphrey et al., 1996) to obtain pentameric models with a (α9)2(α10)3 stoichiometry bound to ACh. Four different types of possible binding site interfaces were included: α9α9, α9α10, α10α9, and α10α10. In each interface, the first subunit forms the principal face and the second forms the complementary face. The models were energy minimized to relax steric clashes using spdbviewer (Guex and Peitsch, 1997), and were used for docking studies after deletion of ACh from the models. Using AutoDock version 4.3 (Morris et al., 2009), ACh was docked into each of the four types of interfaces for rat and chick subunits. Two hundred genetic algorithm runs were performed for each condition. Residues R57, R111, and R117 were set as flexible to avoid steric and/or electrostatic effects that may impair ACh docking into the binding site.
Clustering of the results was done with AutoDock based on a root-mean-square deviation cutoff of 2.0 Å. Docking results were corroborated in three different procedures. The most representative docking result was plotted with Discovery Studio Visualizer 3.5 (Accelrys Software, San Diego, CA).
Double-Mutant Cycle Analysis.
The EC50 values were used to determine the coupling coefficient Ω based on the following equation:
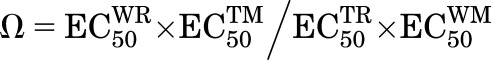 |
where WR corresponds to wild type; TM corresponds to the double mutant W55TR117M; WM corresponds to the single mutant R117M; and TR corresponds to the single mutant W55T. The coupling energy between residues was calculated by the following equation (Schreiber and Fersht, 1995):
Statistical Analysis.
Statistical significance was determined using analysis of variance (ANOVA) followed by the Bonferroni test. Some of our data sets did not fit to a standard Gaussian distribution when tested using Kolmogorov-Smirnov, D’Agostino-Pearson, or Shapiro-Wilk tests. In those cases, statistical significance was evaluated using nonparametric Mann-Whitney or Kruskal-Wallis tests followed by Dunn’s tests. A P < 0.05 was considered significant.
All drugs were obtained from Sigma-Aldrich (St. Louis, MO), except when otherwise indicated. ACh chloride was dissolved in distilled water as 100 mM stocks and stored aliquoted at −20°C. 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (acetoxymethyl ester) was stored at −20°C as aliquots of a 100 mM solution in dimethylsulfoxide, thawed, and diluted 1000-fold into Barth’s solution shortly before incubation of the oocytes. ACh solutions in Ringer’s saline were freshly prepared immediately before application.
Experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health (https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdf), and were approved by the Institution’s Animal Care and use Committee.
Results
The Principal Components of α9 and α10 Subunits Contribute Equally to Function of Rat α9α10 nAChRs.
To determine the contribution of the principal components of the α9 or α10 subunits to ligand binding and α9α10 nAChR function, we generated Y190T mutant subunits (Torpedo marmorata α1 numbering). Amino acid Y190 is a highly conserved key residue in loop C of α nAChR subunits (Karlin, 2002). It has been shown to interact with ACh in a crystal structure of a nAChR homolog from Lymnaea stagnalis (Olsen et al., 2014) and with α-BTX when crystallized with either the α1 (Dellisanti et al., 2007) and α9 receptor subunits (Zouridakis et al., 2014) or a α7/AChBP chimera (Huang et al., 2013). The substitution of Y190 by threonine profoundly reduces binding and gating of the muscle AChR (Chen et al., 1995) and prevents agonist-evoked responses in human α7 and α7/5-HT3A receptors (Andersen et al., 2013; Rayes et al., 2009). Additionally, as a consequence of loop C movement during ACh binding stabilization (Gao et al., 2006), Y190 has been reported to disrupt a salt bridge associated with the closed state of the receptor (Mukhtasimova et al., 2005).
We first evaluated specific total binding of [3H]-α-BTX in nAChRs carrying the Y190T mutation. As previously described (Baker et al., 2004), due to undetectable expression levels in cell lines when expressing wild-type α9 or α10 subunits (Baker et al., 2004), binding studies were performed with chimeric subunits containing the extracellular domain of rat α9 or α10 subunits fused to the C-terminal domain of the 5-HT3A subunit (referred to as α9χ and α10χ, respectively). Specific binding of [3H]-α-BTX was observed in cells transiently transfected with either α9χ or α10χ, indicating membrane targeting of homomeric receptors (Fig. 1). The coexpression of α9χ and α10χ resulted in significantly higher levels of [3H]-α-BTX specific binding, which is likely to be a consequence of more efficient assembly of the chimeric subunits into heteromeric complexes as previously described (Baker et al., 2004). Specific binding of [3H]-α-BTX to α9χα10χ was 6-fold higher than observed with α9χ expressed alone (n = 3, P < 0.0001, Kruskal-Wallis test followed by Dunn’s test).
Fig. 1.

Effect of the Y190T mutation on [3H]-α-BTX binding. Specific binding levels of [3H]-α-BTX (final concentration 20 nM) to wild-type and mutated (Y190T) subunit combinations expressed in mammalian tsA201 cells. Data are mean and S.E.M. of three independent experiments, each of which was performed in triplicate.
The introduction of the Y190T substitution into either α9χ or α10χ (α9χY190T or α10χY190T) resulted in a complete loss of specific binding of [3H]-α-BTX when expressed as either homomeric or heteromeric (double-mutant) receptors (Fig. 1). However, when either α9χY190T or α10χY190T was coexpressed with their nonmutated counterpart subunit (α9χ or α10χ), specific [3H]-α-BTX binding was observed, indicating that both α9 and α10 subunits can contribute to the principal component of the extracellular ligand binding site. Specific binding was 6-fold (n = 3) and 4-fold (n = 3) lower for α9χY190Tα10χ and α9χα10χY190T, respectively, compared with wild-type α9χα10χ (P < 0.0001, Kruskal-Wallis test followed by Dunn’s test). However, specific binding of α9χY190Tα10χ was 4-fold higher than that observed for homomeric α10χ receptors, suggesting that mutant (Y190T) subunits efficiently assemble into heteromeric receptors (P = 0.0472, Mann-Whitney test).
To examine whether Y190T mutants are capable of forming functional channels, receptors were heterogously expressed in Xenopus laevis oocytes. Figure 2A shows representative responses to increasing concentrations of ACh for wild-type and Y190T mutant receptors. Both α9Y190Tα10 and α9α10Y190T complexes formed functional channels. Maximal ACh-evoked currents were similar for wild-type α9α10 and α9α10Y190T mutants (Table 1) and were an order of magnitude larger than those previously reported for α9 homomeric receptors (Elgoyhen et al., 2001), indicating that the resultant responses are not due to the expression of α9 homomeric wild-type receptors. Moreover, responses of α9Y190Tα10 receptors derive from the incorporation of α9Y190T mutant subunits to the heteromeric receptor since α9Y190T homomeric receptors lack functional ligand binding sites (Fig. 1) and rat and human α10 homomers are nonfunctional (Elgoyhen et al., 2001; Sgard et al., 2002). Double-mutant α9Y190Tα10Y190T receptors failed to respond to either 1 or 30 mM ACh (n = 8), a result consistent with the lack of binding sites (Fig. 1). As displayed in Fig. 2B, the Y190T substitution in either α9 or α10 produced a shift of the ACh concentration-response curve to the right and an increase in the ACh EC50 of two orders of magnitude (Table 1). The increase in the EC50 of α9α10Y190T mutant receptors compared with wild-type receptors once again indicates that Y190T mutants are assembly competent, and that responses do not derive from homomeric α9 wild-type receptors. Taken together, these results suggest that both α9 and α10 can contribute with their principal components to the binding site and that the integrity of both is necessary for wild-type receptor function.
Fig. 2.

Effect of the Y190T mutation on the response to ACh of rat α9α10 receptors. (A) Representative traces of responses evoked by increasing ACh concentrations in oocytes expressing wild-type (upper panel), α9Y190Tα10 (middle panel), and α9α10Y190T (lower panel) receptors. (B) Concentration-response curves to ACh performed in oocytes expressing wild-type (○), α9Y190Tα10 (□), and α9α10Y190T (◊) receptors. Peak current values were normalized and refer to the maximal peak response to ACh in each case. The mean and S.E.M. of 5–8 experiments per group are shown.
TABLE 1.
Maximal evoked currents and concentration-response curve parameters
The number of experiments (n) represents independent oocytes from 3 to 6 different frogs. Asterisks (*) indicate the results are significantly different from the control wild-type α9α10. Comparisons of EC50 values for wild-type, mutant α9, mutant α10, or double-mutant receptors for each mutated residue were performed with one-way ANOVA followed by the Bonferroni test.
| Species |
Receptor |
Imax |
EC50 |
p |
n |
|---|---|---|---|---|---|
| nA | µM | ||||
| Rat | α9α10 | 298 ± 48 | 18 ± 3 | 8 | |
| α9Y190Tα10 | 112 ± 6 | 2254 ± 155 | <0.0001* | 5 | |
| α9α10Y190T | 336 ± 91 | 850 ± 170 | <0.0001* | 6 | |
| α9CC/SSα10 | 402 ± 103 | 148 ± 9 | <0.0001* | 8 | |
| α9α10CC/SS | 571 ± 113 | 147 ± 17 | <0.0001* | 17 | |
| α9CC/SSα10CC/SS | 360 ± 119 | 405 ± 13 | <0.0001* | 6 | |
| α9W55Tα10 | 42 ± 4 | 1022 ± 35 | <0.0001* | 5 | |
| α9α10W55T | 177 ± 81 | 36 ± 1 | 0.0665 | 6 | |
| α9α10R117M | 107 ± 38 | 31 ± 5 | 0.0655 | 5 | |
| α9α10 W55T/R117M | 245 ± 83 | 768 ± 135 | 0.0011* | 11 | |
| Chicken | α9α10 | 100 ± 12 | 16 ± 2 | 6 | |
| α9W55Tα10 | 59 ± 8 | 357 ± 75 | <0.0001* | 6 | |
| α9α10W55T | 159 ± 32 | 334 ± 13 | <0.0001* | 6 |
To further analyze the participation of the principal components of both α9 and α10 to receptor function, we mutated to serine the double cysteines of loop C, C192S/C193S [i.e., double cysteine to serine (CC/SS)], a hallmark of nAChR α subunits (Karlin, 2002). Figure 3A shows representative responses to increasing concentrations of ACh evoked in Xenopus laevis oocytes expressing mutant receptors bearing the CC/SS substitution in either α9 or α10 subunits, or both. Surprisingly, the CC/SS double-mutant receptors were functional. The CC/SS substitution in either α9 or α10 produced a similar shift of the ACh concentration-response curve to the right and an increase in the ACh EC50 of one order of magnitude (EC50: wild type = 18 ± 3 µM; α9CC/SSα10 = 148 ± 9 µM, n = 8, P < 0.0001; α9α10CC/SS = 147 ± 17 µM, n = 17, P < 0.0001, one-way ANOVA followed by the Bonferroni test) (Fig. 3B; Table 1). Further shift of the concentration-response curve and an increase of the ACh EC50 were observed in double-mutant CC/SS receptors (405 ± 13 µM, n = 6, P < 0.0001 compared with wild type, one-way ANOVA followed by the Bonferroni test).
Fig. 3.

Effect of the CC192/193SS (CC/SS) mutations on the response to ACh of rat α9α10 receptors. (A) Representative traces of responses evoked by increasing ACh concentrations in oocytes expressing α9CC/SSα10 (upper panel), α9α10CC/SS (middle panel), and α9CC/SSα10CC/SS (lower panel) receptors. (B) Concentration-response curves to ACh performed in oocytes expressing wild-type (○), α9CC/SSα10 (□), α9α10CC/SS (◊), and α9CC/SSα10CC/SS (△) receptors. Peak current values were normalized and refer to the maximal peak response to ACh in each case. The mean and S.E.M. of 6–17 experiments per group are shown.
Nonequivalent Contribution of α9 and α10 Complementary Components to Rat α9α10 nAChR Receptor Function.
To determine the contribution of the complementary faces of either α9 or α10 to rat α9α10 nAChR function, we generated W55T mutant subunits. Amino acid W55 is highly conserved within loop D of nAChR subunits, which contributes to the complementary face of the ligand binding site (Karlin, 2002). The crystal structure of the ACh binding protein from Lymnaea stagnalis bound to ACh shows a cation-π interaction of W55 with this agonist (Olsen et al., 2014). Moreover, the substitution of W55 by threonine in an α7/5-HT3A chimera renders a receptor that binds α-BTX but impairs competition of [3H]-α-BTX by ACh, leading to nonfunctional receptors (Rayes et al., 2009). In addition, mutagenesis analysis in the Torpedo electric organ nAChR has demonstrated that W55 is part of the ACh binding pocket of nAChRs (Xie and Cohen, 2001).
Figure 4 shows binding experiments performed with [3H]-α-BTX in wild-type and W55T mutant α9α10 receptors. In contrast to previous findings reported for the α7/5-HT3A subunit chimera (Rayes et al., 2009), no detectable specific binding was observed with homomeric α9χW55T receptors. In contrast, homomeric α10χW55T receptors showed significant levels of specific binding, similar to levels observed with homomeric α10χ (2.5 ± 0.6 and 1.4 ± 0.5 fmol/mg, respectively, P = 0.229, Mann-Whitney test). Consistent with these results, heteromeric receptors containing a mutant α9χW55T subunit (α9χW55Tα10χ and α9χW55Tα10χW55T) showed binding levels similar to those observed with either α10χ or α10χW55T when expressed alone (P = 0.1–0.7, Mann-Whitney test). Moreover, receptors composed of wild-type α9χ subunits and mutated 10χ (α9χα10χW55T) displayed specific binding levels similar to those observed with wild-type heteromeric α9χα10χ receptors (P = 0.114, Mann-Whitney test). Taken together, these results indicate that the conserved amino acid W55 in loop D is involved in the binding site of the α9α10 receptor only when provided by the α9 subunit. This appears to suggest that the α9 subunit contributes to the complementary component of the binding site of α9α10 nAChRs and that the (−) faces of α9 and α10 are nonequivalent.
Fig. 4.

Effect of the W55T mutation on [3H]-α-BTX binding. Specific binding of [3H]-α-BTX (final concentration 20 nM) to wild-type and mutated (W55T) subunit combinations expressed in mammalian tsA201 cells. Data are mean and S.E.M. of three independent experiments, each of which was performed in triplicate.
An important question is whether ACh binds to α9χα10χW55T receptors. To discriminate between total and specific binding of [3H]-α-BTX we used a standard protocol in which a mixture of cold ligands were used to determine nonspecific binding. To confirm whether ACh itself is able to displace binding of [3H]-α-BTX we repeated these binding experiments and used only ACh to displace bound [3H]-α-BTX. For both wild-type (α9χα10χ) and mutated (α9χα10χW55T) nAChRs, bound [3H]-α-BTX was displaced as efficiently with ACh alone as with our standard mixture of nonradioactive competing ligands, confirming that the ACh binding site is retained in α9χα10χW55T. This indicates that the W55 mutation has a different effect in α10 to that observed with the α9 subunit and its previously reported effect in α7 (Rayes et al., 2009), and suggests that W55 contributes differently to the ACh binding site of the α9α10 receptor when provided by the α9 or α10 subunit. Since W55 is a highly conserved key residue present in loop D of nicotinic subunits that contributes to complementary components of binding sites (Karlin, 2002), the present results are consistent with the conclusion that α10 either does not contribute to the (−) face of the binding site of the α9α10 receptor or that W55 of α10 is not readily accessible within the binding pocket. If the latter is the case, then the contributions of the (−) faces of α9 and α10 to the binding interface are nonequivalent. To further examine these possibilities, the functional responses of W55T mutated receptors were studied in Xenopus laevis oocytes.
Figure 5A shows representative responses to increasing concentrations of ACh in Xenopus laevis oocytes expressing wild-type rat α9α10 receptors or W55T mutant receptors. Double-mutant α9α10 receptors failed to evoke currents at 1 or 30 mM ACh (n = 15). The W55T substitution in α9 produced a displacement of the concentration-response curve to ACh to the right with a 60-fold increase in the EC50 (EC50: wild type = 18 ± 3 µM, α9W55Tα10 = 1022 ± 35 µM, P < 0.0001, one-way ANOVA followed by the Bonferroni test, n = 5–8) (Table 1). On the other hand, the W55T substitution in α10 produced only a slight (although nonsignificant) increase in the receptor EC50 (EC50: wild type = 18 ± 3 µM, α9α10W55T = 36 ± 1 µM, P = 0.0665 one-way ANOVA followed by the Bonferroni test, n = 6) (Table 1). Maximal evoked currents of α9α10W55T receptors were not significantly different from those of wild-type α9α10 receptors (Imax: wild type = 298 ± 48 nA, α9α10W55T = 177 ± 81 nA, P = 0.1826, Mann-Whitney test, n = 6) (Table 1) and one order of magnitude larger than those reported for α9 homomeric receptors (Rothlin et al., 1999; Katz et al., 2000), indicating that α10W55T is incorporated into a α9α10W55T heteromeric receptor.
Fig. 5.

Effect of the W55T mutation on the response to ACh of rat α9α10 receptors. (A) Representative traces of responses evoked by increasing ACh concentrations in oocytes expressing α9W55Tα10 (upper panel) and α9α10W55T (lower panel) receptors. (B) Concentration-response curves to ACh performed in oocytes expressing wild-type (○), α9W55Tα10 (□), and α9α10W55T (◊) receptors. Peak current values were normalized and refer to the maximal peak response to ACh. The mean and S.E.M. of 5–8 experiments per group are shown. (C) Bar diagram illustrating the modulation of the α9α10W55T receptor by extracellular Ca2+ exerts. Current amplitudes obtained at different Ca2+ concentrations in each oocyte were normalized with respect to that obtained at 1.8 mM in the same oocyte. The mean and S.E.M. of three experiments per group are shown.
To further rule out the possibility that the modest effect observed in responses to ACh of α9α10W55T receptors is due to the lack of incorporation of the α10W55T subunit into a heteromeric assembly, we analyzed the Ca2+ sensitivity of the resultant receptors. Homomeric α9 receptors are only blocked by extracellular Ca2+, whereas heteromeric α9α10 receptors are potentiated in the submillimolar range and blocked at higher concentrations of this divalent cation (Katz et al., 2000; Weisstaub et al., 2002). Figure 5C shows the modulation profile obtained at a concentration of ACh close to the EC50 (30 µM) value and the application of increasing concentrations of extracellular Ca2+. Peak current amplitudes at each Ca2+ concentration in each oocyte were normalized to those obtained at 1.8 mM. Similar to that reported for wild-type receptors (Elgoyhen et al., 2001; Weisstaub et al., 2002), a biphasic Ca2+ modulation profile was observed with maximal responses at 0.5 mM. A one-way ANOVA followed by multiple comparisons indicated that the difference in normalized mean current amplitude between nominal 0 and 0.5 mM Ca2+ is significant (P = 0.019, Kruskal-Wallis test followed by Dunn’s test). This result demonstrates the occurrence of Ca2+ potentiation and thus confirms the incorporation of α10W55T subunits into pentameric receptors.
The functional results indicate that both α9 and α10 contribute to the (−) face of the intersubunit interface, but that their contribution is nonequivalent. Thus, if α10 did not contribute at all to the (−) face, the shift in the ACh concentration-response curve of double-mutated W55T receptors should resemble that of α9W55T receptors instead of rendering nonfunctional receptors (Fig. 5B).
The α9 and α10 Subunits Contribute Equally to the Complementary Component of the ACh Binding Site in the Chicken α9α10 nAChR.
The asymmetric contribution of α9 and α10 subunits to the (−) face of the ACh binding site might result from the adaptive evolution that occurred only in mammalian CHRNA10 genes. This resulted in important nonsynonymous amino acid substitutions in the coding region of the α10 nAChR subunits, including that of loop D (Franchini and Elgoyhen, 2006; Elgoyhen and Franchini, 2011; Lipovsek et al., 2012). If this were the case, then both α9 and α10 should equally contribute to the (−) face of the intersubunit interface in a nonmammalian vertebrate species. Figure 6A shows representative responses to increasing concentrations of ACh evoked in Xenopus laevis oocytes expressing chicken α9α10 wild-type and W55T mutant receptors. Double-mutant receptors failed to evoke currents at 1 or 30 mM ACh (n = 10). The W55T substitution in either α9 or α10 produced similar shifts in the ACh concentration-response curves to the right (Fig. 6) and a one order of magnitude increase in the receptor EC50 (EC50: wild type = 16 ± 2 µM, α9W55Tα10 = 357 ± 75 µM, α9α10W55T = 334 ± 13 µM, P < 0.0001, one-way ANOVA followed by the Bonferroni test, n = 6) (Table 1). This result suggests that, in contrast to the situation with rat α9α10 receptors, in chicken the (−) face of both α9 and α10 subunits equally contribute to receptor function.
Fig. 6.

Effect of the W55T mutation on the response to ACh of chicken α9α10 receptors. (A) Representative traces of responses evoked by increasing ACh concentrations in oocytes expressing wild-type (upper panel), α9W55Tα10 (middle panel), and α9α10W55T (lower panel) chick receptors. (B) Concentration-response curves to ACh performed in oocytes expressing wild-type (○), α9W55Tα10 (□), and α9α10W55T (◊) chick receptors. Peak current values were normalized and refer to the maximal peak response to ACh. The mean and S.E.M. of six experiments per group are shown.
Molecular Docking of ACh in α9α10 Receptors.
To gain further insight into the contribution of the subunit components to ACh binding, we modeled different subunit arrangements to take into account the four possible subunit interfaces [α9(+)α9(−), α9(+)α10(−), α10(+)α10(−), and α10(+)α9(−)] in rat and chicken receptors, and performed molecular docking studies. To evaluate the capability of each interface to bind ACh, we compared the best binding energy (BBE) (Fig. 7A) and the frequency of conformations that bind the agonist in the correct orientation in the binding pocket (Fig. 7B). For all interfaces, the conformations considered as favorable were those showing the previously described cation-π interactions between the amino group of ACh and aromatic residues of the binding pocket (W55, Y93, W149, and Y190) (Dougherty, 2007; Hernando et al., 2012) (Fig. 7C). In these conformations, and for all interfaces, ACh shows the capability to form hydrogen bonds with D119 and Y197, which are equivalent to conserved H bonds of different nAChRs (Tomaselli et al., 1991; Lester et al., 2004; Hernando et al., 2012) (Fig. 7C). The BBE did not show important differences among the different models, except for the homomeric rat α10α10 interface. At this interface, the BBE was about −3.5 kcal/mol compared with −5 to −6 kcal/mol for all of the others (Fig. 7A).
Fig. 7.

Docking of ACh into homology-modeled α9/α10 binding-site interfaces. ACh was docked in the correct orientation into the two possible models for heteromeric interfaces of rat and chicken receptors. The BBE (A) and the percentage of favorable conformations (B) for bound ACh were averaged from three different runs for each interface. (C) Representative models of ACh docked into the different interfaces. The main π-cation interactions are shown with straight lines and the H-bonds are shown with dashed lines.
The main difference in the docking results among interfaces was detected in the frequency of favorable conformations (Fig. 7B). In rat, the most frequent conformations with ACh in the correct orientation at the binding site was observed at the interface in which α10 contributes to the principal and α9 to the complementary face [α10(+)α9(−) interface], with a BBE of −4.8 kcal/mol (Fig. 7). Models with rat the α10 subunit placed in the complementary face [α9(+)α10(−) or α10(+)α10(−)] showed a significant reduction of the frequency of conformations with ACh docked in the correct orientation (Fig. 7B). In the case of α10(+)α10(−), ACh only showed a favorable orientation at the binding site in less than 2% of the conformations in most of the docking conformations (Fig. 7B).
In chicken heteromeric interfaces, no significant differences were observed in the frequency of favorable conformations between the α9(+)α10(−) and α10(+)α9(−) interfaces. Thus, in contrast to the rat nAChR, this suggests that α10 contributes similarly to both the principal and complementary faces of the chicken receptor (Fig. 7). When comparing homomeric interfaces, rat α10(+)α10(−) appears to be very unfavorable for ACh binding (i.e., the lowest frequency of conformations with ACh in the correct orientation and the highest BBE). In chicken, both homomeric interfaces appear to be similarly favorable for ACh binding, but less favorable than the heteromeric ones (Fig. 7).
Taken together, the in silico studies support the experimental data indicating that in rat the contribution of α9 and α10 to complementary components is nonequivalent. In contrast, α9 can form relatively appropriate interfaces for ACh binding when placed at either the principal or complementary faces. Moreover, the modeling supports the functional data for chicken receptors, where α10 equally contributes to principal and complementary components.
α10 Residue 117 in Loop E of the (−) Face Is a Major Determinant of Functional Differences.
Given that the main key interactions at the binding site with aromatic residues are conserved in all models in conformations where ACh is bound in the correct orientation (Fig. 7), we analyzed in more detail other residues that might account for the fact that W55 is not a major determinant of rat α10 subunit complementary components, compared with rat α9 and chicken α9 and α10. Analysis of the model of ACh bound to the four different types of interfaces [α9(+)α9(−), α9(+)α10(−), α10(+)α10(−), and α10(+)α9(−)] shows that the residues on a radial distribution of 5 Å are the same for the principal components (Y93, S148, W149, Y190, C192, and Y197) and for most of the complementary components (W55, R57, R79, N107, V109, T/M/R117, and D119). They only differ at position 117, where the rat α10 positively charged arginine (R117) which is highly conserved in mammalian α10 subunits, is substituted by a noncharged methionine in chicken α10 and a threonine or methionine in nonmammalian α10 subunits (Figs. 7A and 8A); for an extended number of species see Lipovsek et al. (2012, 2014). Interestingly, all α9 subunits carry a threonine at this position. Moreover, the appearance of the R117 nonsynonymous amino acid substitution in mammalian species has been under positive selection pressure (Franchini and Elgoyhen, 2006). In many docking conformations R117 was placed toward the cavity (Fig. 7C). Moreover, R117 had to be set as flexible to avoid steric and/or electrostatic effects that impair ACh docking into the correct binding site (see Materials and Methods). In addition, rat α10 subunits have a negatively charged glutamic acid residue E59 in loop D, which is highly conserved and has been also positively selected in mammalian species (Franchini and Elgoyhen, 2006), compared with noncharged residues in nonmammalian α10 and α9 subunits (Fig. 8A).
Fig. 8.

The α9 and α10 subunit sequence alignments and distribution of charged residues. (A) Sequence alignments of part of the (−) face of α9 and α10 from different vertebrate species. Conserved W55 and mammalian positively selected E59 and R117 are shaded. (B) Distance (Å) of protein charged groups from the nitrogen atom of ACh in chicken and rat receptors. The analysis was made using the theoretical models constructed by homology modeling described in Materials and Methods. The results are shown for the four types of interfaces: α9(+)α9(−), α9(+)α10(−), α10(+)α10(−), and α10(+)α9(−). Positively charged groups are represented by black circles, whereas the negatively charged groups are represented by white circles. The identity of each residue is shown.
Because R117 and E59 are charged residues, due to the long-range nature of electrostatic interactions, we analyzed the distance distribution of protein-charged groups from the positively charged N atom of ACh (Fig. 8B). In all interfaces, the conserved residues observed on a radial distribution of 10 Å from this N atom were D119(−), R57(−), R79(−), D169(−), and D199(+) in order of increasing distance. Here, the plus and minus signs correspond to the presence of residues in either the principal (+) or complementary (−) face, respectively, and not to the charge of each residue. The most significant difference was the positively charged R117 at a distance of ∼8 to 9 Å from the ACh amino group, which was only present in the complementary site of rat α10. This relative excess in positively charged residues in rat α10 could result in an unfavorable interaction with the ligand through electrostatic repulsion and thus may perturb the binding site. Interestingly, the negatively charged E59 is close to R117. Although this residue could partially compensate for the positive charge of R117, it is located more than 10 Å from ACh, and thus its effect on the ligand is lower than that of R117. Moreover, the analysis of positively and negatively charged residues in the entire N-terminal domain of rat and chick subunits indicates that the global balance is neutral in rat α10, whereas it is strongly negative in rat α9 and chicken α9 and α10 subunits. The difference is due to an excess of basic residues (R and K) in rat α10 compared with the other subunits (Table 2). Overall, these observations further confirm that the complementary faces of rat α9 and α10 subunits are nonequivalent and that R117 in the complementary component of α10 might account for functional differences.
TABLE 2.
Number of charged residues in rat and chicken α9 and α10 subunits
The basic-acidic balance was calculated as the difference in the number of basic (R and K) compared with acidic (D and E) amino acid residues.
| Species | Subunit | Acidic (D and E) | Basic (R and K) | Basic-Acidic Balance |
|---|---|---|---|---|
| Rat | α9 | 34 | 16 | −18 |
| α10 | 24 | 24 | 0 | |
| Chick | α9 | 33 | 18 | −15 |
| α10 | 28 | 18 | −10 |
We introduced the R117M substitution in the rat α10 subunit and expressed it in Xenopus oocytes with rat α9 (Fig. 9A). The α9α10R117M receptors were functional and their ACh EC50 values, although slightly higher, did not significantly differ from that of wild-type receptors (Table 1). However, when W55 of α10R117M subunits was mutated to threonine, a 43-fold shift in the ACh concentration-response curve to the right was observed (EC50: wild type = 18 ± 3 µM, α9α10 W55T/R117M = 768 ± 135 µM, P = 0.0011, one-way ANOVA followed by the Bonferroni test, n = 5–11) (Fig. 9; Table 1). Thus, it appears that when the R117 is removed, W55 contributes to the (−) face of rat α10 subunits.
Fig. 9.

Effect of the R117M mutation on rat α9α10 receptors. (A) Concentration-response curves to ACh performed in oocytes expressing wild-type (○), α9α10R117M (□), and α9α10W55T/R117M (◊) double-mutant rat receptors. Peak current values were normalized and refer to the maximal peak response to ACh. The mean and S.E.M. of 5–11 experiments per group are shown. (B) Scheme for double-mutant cycle analysis. ΔΔG values corresponding to each mutant are shown. These values were calculated as −RTln(EC50 mutant/EC50 wild type). The coupling parameter, Ω, was calculated as indicated in Materials and Methods.
The typical way to analyze a system in which two mutations are evaluated individually and in tandem is by mutant cycle analysis (Schreiber and Fersht, 1995; Corradi et al., 2007). Such analysis reveals whether the contributions from a pair of residues are additive or if the effects of mutations are coupled. We calculated the changes due to R117M and W55T mutations in the free energy of the responses using the EC50 values (Fig. 9B). Single-mutants α10W55T and α10R117M decreased the free energy (−0.40 and −0.32 kcal/mol, respectively); the change in the free energy of the double mutant was significantly different from the sum of the changes occurring in the two single mutants (−2.19 kcal/mol). To quantify energetic coupling between α10W55 and α10R117 we analyzed the changes in the free energy of coupling by double-mutant thermodynamic cycles. When the EC50 values are cast as a mutant cycle, the coupling coefficient is 12.4, which corresponds to free energy coupling of −1.47 kcal/mol. Taken together these results indicate that the effects of the mutations are not independent and that the residues are coupled in their contribution to function (Schreiber and Fersht, 1995; Corradi et al., 2007).
Discussion
The present study shows that, contrary to previous assumptions, the α10 subunit contributes to the principal face of the ligand binding site in the heteromeric α9α10 nAChR. Moreover, we show that the contribution of rat α9 and α10 subunits to the complementary face is nonequivalent. It is worth noting that conotoxin RgIA, which potently blocks α9α10 nAChRs (Ellison et al., 2006), was initially reported to bind to the α9(+)α10(−) interface based on molecular modeling, docking, and molecular dynamics simulations (Pérez et al., 2009). However, mutagenesis experiments have shown that conotoxins RgIA (Azam and McIntosh, 2012; Azam et al., 2015) and Vc1.1 (Yu et al., 2013) bind to the α10(+)α9(−) interface, further indicating that α10 contributes to the principal component of the binding site for antagonist as well as agonist binding.
The lack of [3H]-α-BTX binding to homomeric (α9χY190T and α10χY190T) and heteromeric (α9χY190Tα10χY190T) nAChRs is in agreement with the observation that Y190 in loop C of the principal component interacts with α-BTX when crystallized with either the α1 (Dellisanti et al., 2007), α9 (Zouridakis et al., 2014), or an α7/AChBP chimera (Huang et al., 2013). Moreover, Y190 has been shown to interact with ACh in a crystal structure of a nAChR homolog from Lymnaea stagnalis (Olsen et al., 2014). Therefore, the lack of binding of [3H]-α-BTX to Y190T mutant receptors most likely also indicates disrupted ACh binding sites. These binding experiments with Y190T mutated receptors, together with the expression studies, indicate that both α9 and α10 can contribute to the principal component of the agonist binding site.
The fact that the mutation of the CC/SS mutant, a hallmark of nAChR α subunits, in either α9 or α10 produced similar rightward shifts in the concentration-response curves to ACh further indicates that both subunits can equally contribute to the principal components of the binding site. The observation that α9CC/SSα10CC/SS double-mutant receptors were functional, albeit with a further increase in the ACh EC50 value, indicates that the ACh binding pocket is not completely disrupted in the absence of the continuous double cysteines of the principal component. This is in line with the observation that in the crystal structure of the Lymnaea stagnalis nAChR bound to ACh this agonist is wedged in between the disulfide bridge of the double cysteine, but that interactions occur with aromatic residues (Olsen et al., 2014). Likewise, mutation of the CC in the Aplysia californica AChBP produces a 10-fold decrease in affinity but does not abolish ACh binding (Hansen and Taylor, 2007). Thus, it has been shown that loop C contributes to the molecular recognition of the agonist by moving into a capped position and locking the agonist in place (Celie et al., 2004; Gao et al., 2005, 2006; Olsen et al., 2014). Movement of loop C is also involved in the initial steps that lead from binding to gating of the receptor (Sine and Engel, 2006).
The observation that the W55T mutation in loop D of the complementary component of the α9 (but not the α10) receptor subunit impaired [3H]-α-BTX binding most likely suggests a disrupted agonist binding site, and therefore that α9 contributes to the complementary component of the ligand binding site. In a crystal structure of α-BTX bound to a pentameric α7/AChBP chimera, while Y190 in loop C is the main contributor to the high-affinity toxin interaction through π-cation and hydrogen bond interactions (Huang et al., 2013; Sine et al., 2013) W55 contacts F32 of the toxin and its mutation produces mild but significant reduction of α-BTX binding affinity (Sine et al., 2013). The notion that α9 contributes to the complementary face of the binding site is further supported by the docking analysis, where in rat receptors the most frequent conformations with ACh in the correct orientation at the binding site were observed at the interface in which α10 contributes to the principal (+) and α9 to the complementary face (−) interface [α10(+)α9(−)]. Expression studies of mutant W55T receptors also indicate that α9 complementary components contribute to receptor function. The increase in ACh apparent affinity of α9W55Tα10 might also result from reduced gating kinetics. In this regard, mutations in this residue in the muscle receptor affect channel gating due to a reduction in the channel opening rate constant (Akk, 2002).
The fact that the α9χα10χW55T mutation bound [3H]α-BTX (and this was displaced by ACh), together with the finding that the α9α10W55T mutant receptors had similar ACh apparent affinity and macroscopic currents to wild-type receptors, indicates that either α10 does not contribute to the complementary face of the binding pocket or that α10 might inefficiently provide the (−) face since W55 in loop D cannot make the proper cation-π interactions with ACh. The latter is rather unexpected, since W55 is a key contributor of the (−) face to ACh binding in all nAChRs (Karlin, 2002; Olsen et al., 2014). However, it can explain the observation that α10 contributes to the complementary face in the presence of disrupted α9(−) faces, as observed in functional studies with α9W55Tα10 receptors. Therefore, one could conclude that in rat heteromeric α9α10 receptors the contribution of α10 to the complementary component is nonequivalent to that of α9 since it does not involve equally W55, a key residue for ACh binding and gating. This resembles what has been described for the Torpedo and muscle embryonic nAChRs, where the contribution of the γ and δ subunits to the (−) face is nonequivalent (Sine and Claudio, 1991; Martin et al., 1996; Xie and Cohen, 2001). Overall the functional results are in line with the in silico modeling, which showed a significant reduction in the frequency of conformations with ACh docked in the correct orientation with the rat α10 subunit placed in the complementary face, α9(+)α10(−) or α10(+)α10(−).
The observation that in chicken receptors the introduction of the W55T mutation in either α9 or α10 produced similar shifts in the ACh apparent affinity of resultant heteromeric receptors indicates that both α9 and α10 can equally contribute to the (−) face of the binding pocket. This is supported by the observation that, contrary to that observed for rat receptors, in chicken molecular docking studies indicate that the frequency of ACh bound in the correct orientation is similar for either α9(+)α10(−) or α10(+)α9(−) interfaces. This might explain that, in contrast to that observed for rat subunits (Elgoyhen et al., 2001; Sgard et al., 2002), chicken homomeric α10 receptors are functional when expressed in Xenopus laevis oocytes (Lipovsek et al., 2014).
The asymmetry between rat and chicken receptors most likely derives from the acquisition of nonsynonymous substitutions in the complementary face of mammalian α10 subunits (Franchini and Elgoyhen, 2006). R117 present in mammalian α10 subunits, but replaced by a noncharged methionine or threonine in nonmammalian α10 subunits and threonine in vertebrate α9 subunits (Fig. 8), might account for the fact that W55 does not equivalently contribute to receptor function when comparing rat α10 to rat α9, chicken α9, and chicken α10 subunits. Its presence might result in a positively charged environment that would perturb the access of the quaternary ammonium of ACh to the binding pocket. This resembles what has been recently described in the crystal structure of the α4β2 nAChR, where three hydrophobic groups on the (−) side of the β2 subunit are replaced by polar side chains on the (−) side of the α4 subunit. It has been suggested that this difference in chemical environment may affect agonist binding to α4–α4 interfaces in the (α4)3(β2)2 stoichiometry, being a polar environment less favorable for agonist binding (Morales-Perez et al., 2016). Understanding the underlying mechanisms accounting for the perturbation produced by R117 in the (−) face of the rat α10 subunit would require further experiments, including determination of the crystal structure of the α9α10 receptor bound to ACh. However, by double-mutant cycle analysis we have been able to show that W55 and R117 are coupled to each other in their contribution to nAChR function. Thus, the mutation at one site has structural or energetic impact at a second site. Typically, a value of Ω that deviates significantly from 1 is interpreted as a direct interaction between residues, such as that provided by a hydrogen bond or a salt bridge. However, the molecular structure of the α9α10 nAChR (Fig. 7) shows that W55 and R117 are not in close apposition and appear separated by about 10 Å, thus suggesting that the coupling does not arise from a direct interaction. The occurrence of long-range functional coupling between residues in which a direct interaction is precluded has been described in the mouse muscle nAChR (Gleitsman et al., 2009).
In conclusion, we have demonstrated that whereas both α9 and α10 contribute to the principal component of α9α10 nAChRs their contribution to the complementary face of the binding pocket in rat α9α10 nAChRs is nonequivalent. This results from the adaptive evolutionary amino acid changes acquired by mammalian α10, which rendered a divergent branch within the clade of vertebrate α10 subunits (Lipovsek et al., 2012).
Abbreviations
- 5-HT3A
serotonin type 3A
- α-BTX
α-bungarotoxin
- ACh
acetylcholine
- ANOVA
analysis of variance
- BBE
best binding energy
- CC/SS
double cysteine to serine
- nAChR
nicotinic acetylcholine receptor
Authorship Contributions
Participated in research design: Boffi, Gill-Thind, Corradi, Collins, Lipovsek, Moglie, Plazas, Craig, Millar, Bouzat, Elgoyhen.
Conducted experiments: Boffi, Marcovich, Gill-Thind, Corradi, Collins, Craig.
Performed data analysis: Boffi, Gill-Thind, Corradi, Moglie, Plazas, Craig, Millar, Bouzat, Elgoyhen.
Wrote or contributed to the writing of the manuscript: Boffi, Millar, Bouzat, Elgoyhen.
Footnotes
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica, Argentina; Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina; Universidad Nacional del Sur, Argentina; and the National Institutes of Health National Institute on Deafness and Other Communication Disorders [Grant RO1DC001508].
T.C. was supported by a Biotechnology and Biological Sciences Research Council (BBSRC) doctoral training account Ph.D. studentship [BB/D526961/1]. J.K.G.-T. was supported by a BBSRC Collaborative Award in Science and Engineering Ph.D. studentship [BB/F017146/1] with additional financial support from Eli Lilly & Co., Ltd.
Current affiliation: Department of Functional Neuroanatomy, Institute for Anatomy and Cell Biology, University of Heidelberg, Heidelberg, Germany.
Current affiliation: Centre for Developmental Neurobiology, King’s College, London, United Kingdom.
References
- Akk G. (2002) Contributions of the non-α subunit residues (loop D) to agonist binding and channel gating in the muscle nicotinic acetylcholine receptor. J Physiol 544:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen N, Corradi J, Sine SM, Bouzat C. (2013) Stoichiometry for activation of neuronal α7 nicotinic receptors. Proc Natl Acad Sci USA 110:20819–20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias HR. (1997) Topology of ligand binding sites on the nicotinic acetylcholine receptor. Brain Res Brain Res Rev 25:133–191. [DOI] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. [DOI] [PubMed] [Google Scholar]
- Azam L, McIntosh JM. (2012) Molecular basis for the differential sensitivity of rat and human α9α10 nAChRs to α-conotoxin RgIA. J Neurochem 122:1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, Papakyriakou A, Zouridakis M, Giastas P, Tzartos SJ, McIntosh JM. (2015) Molecular interaction of α-conotoxin RgIA with the rat α9α10 nicotinic acetylcholine receptor. Mol Pharmacol 87:855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker ER, Zwart R, Sher E, Millar NS. (2004) Pharmacological properties of α9α10 nicotinic acetylcholine receptors revealed by heterologous expression of subunit chimeras. Mol Pharmacol 65:453–460. [DOI] [PubMed] [Google Scholar]
- Blount P, Merlie JP. (1989) Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron 3:349–357. [DOI] [PubMed] [Google Scholar]
- Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 4:1–13. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411:269–276. [DOI] [PubMed] [Google Scholar]
- Carbone AL, Moroni M, Groot-Kormelink PJ, Bermudez I. (2009) Pentameric concatenated (α4)2(β2)3 and (α4)3(β2)2 nicotinic acetylcholine receptors: subunit arrangement determines functional expression. Br J Pharmacol 156:970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. (2004) Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41:907–914. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang Y, Akk G, Sine S, Auerbach A. (1995) Activation kinetics of recombinant mouse nicotinic acetylcholine receptors: mutations of alpha-subunit tyrosine 190 affect both binding and gating. Biophys J 69:849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi J, Spitzmaul G, De Rosa MJ, Costabel M, Bouzat C. (2007) Role of pairwise interactions between M1 and M2 domains of the nicotinic receptor in channel gating. Biophys J 92:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L. (2007) Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 Å resolution. Nat Neurosci 10:953–962. [DOI] [PubMed] [Google Scholar]
- Dougherty DA. (2007) Cation-π interactions involving aromatic amino acids. J Nutr 137:1504S–1508S; discussion 1516S–1517S. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Franchini LF. (2011) Prestin and the cholinergic receptor of hair cells: positively-selected proteins in mammals. Hear Res 273:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. (1994) α9: An acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79:705–715. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Katz E. (2012) The efferent medial olivocochlear-hair cell synapse. J Physiol Paris 106:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J. (2001) α10: A determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci USA 98:3501–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison M, Haberlandt C, Gomez-Casati ME, Watkins M, Elgoyhen AB, McIntosh JM, Olivera BM. (2006) α-RgIA: A novel conotoxin that specifically and potently blocks the α9α10 nAChR. Biochemistry 45:1511–1517. [DOI] [PubMed] [Google Scholar]
- Franchini LF, Elgoyhen AB. (2006) Adaptive evolution in mammalian proteins involved in cochlear outer hair cell electromotility. Mol Phylogenet Evol 41:622–635. [DOI] [PubMed] [Google Scholar]
- Gao F, Bren N, Burghardt TP, Hansen S, Henchman RH, Taylor P, McCammon JA, Sine SM. (2005) Agonist-mediated conformational changes in acetylcholine-binding protein revealed by simulation and intrinsic tryptophan fluorescence. J Biol Chem 280:8443–8451. [DOI] [PubMed] [Google Scholar]
- Gao F, Mer G, Tonelli M, Hansen SB, Burghardt TP, Taylor P, Sine SM. (2006) Solution NMR of acetylcholine binding protein reveals agonist-mediated conformational change of the C-loop. Mol Pharmacol 70:1230–1235. [DOI] [PubMed] [Google Scholar]
- Gleitsman KR, Shanata JA, Frazier SJ, Lester HA, Dougherty DA. (2009) Long-range coupling in an allosteric receptor revealed by mutant cycle analysis. Biophys J 96:3168–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Taylor P. (2007) Galanthamine and non-competitive inhibitor binding to ACh-binding protein: evidence for a binding site on non-α-subunit interfaces of heteromeric neuronal nicotinic receptors. J Mol Biol 369:895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkness PC, Millar NS. (2002) Changes in conformation and subcellular distribution of α4β2 nicotinic acetylcholine receptors revealed by chronic nicotine treatment and expression of subunit chimeras. J Neurosci 22:10172–10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpsøe K, Ahring PK, Christensen JK, Jensen ML, Peters D, Balle T. (2011) Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors. J Neurosci 31:10759–10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando G, Bergé I, Rayes D, Bouzat C. (2012) Contribution of subunits to Caenorhabditis elegans levamisole-sensitive nicotinic receptor function. Mol Pharmacol 82:550–560. [DOI] [PubMed] [Google Scholar]
- Hsiao B, Mihalak KB, Magleby KL, Luetje CW. (2008) Zinc potentiates neuronal nicotinic receptors by increasing burst duration. J Neurophysiol 99:999–1007. [DOI] [PubMed] [Google Scholar]
- Huang S, Li SX, Bren N, Cheng K, Gomoto R, Chen L, Sine SM. (2013) Complex between α-bungarotoxin and an α7 nicotinic receptor ligand-binding domain chimaera. Biochem J 454:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. [DOI] [PubMed] [Google Scholar]
- Indurthi DC, Pera E, Kim HL, Chu C, McLeod MD, McIntosh JM, Absalom NL, Chebib M. (2014) Presence of multiple binding sites on α9α10 nAChR receptors alludes to stoichiometric-dependent action of the α-conotoxin, Vc1.1. Biochem Pharmacol 89:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A. (2002) Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci 3:102–114. [DOI] [PubMed] [Google Scholar]
- Katz E, Verbitsky M, Rothlin CV, Vetter DE, Heinemann SF, Elgoyhen AB. (2000) High calcium permeability and calcium block of the α9 nicotinic acetylcholine receptor. Hear Res 141:117–128. [DOI] [PubMed] [Google Scholar]
- Lansdell SJ, Millar NS. (2000) The influence of nicotinic receptor subunit composition upon agonist, α-bungarotoxin and insecticide (imidacloprid) binding affinity. Neuropharmacology 39:671–679. [DOI] [PubMed] [Google Scholar]
- Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. (2004) Cys-loop receptors: new twists and turns. Trends Neurosci 27:329–336. [DOI] [PubMed] [Google Scholar]
- Lipovsek M, Fierro A, Pérez EG, Boffi JC, Millar NS, Fuchs PA, Katz E, Elgoyhen AB. (2014) Tracking the molecular evolution of calcium permeability in a nicotinic acetylcholine receptor. Mol Biol Evol 31:3250–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovsek M, Im GJ, Franchini LF, Pisciottano F, Katz E, Fuchs PA, Elgoyhen AB. (2012) Phylogenetic differences in calcium permeability of the auditory hair cell cholinergic nicotinic receptor. Proc Natl Acad Sci USA 109:4308–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetje CW, Patrick J. (1991) Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci 11:837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Czajkowski C, Karlin A. (1996) The contributions of aspartyl residues in the acetylcholine receptor γ and δ subunits to the binding of agonists and competitive antagonists. J Biol Chem 271:13497–13503. [DOI] [PubMed] [Google Scholar]
- Martinez KL, Corringer PJ, Edelstein SJ, Changeux JP, Mérola F. (2000) Structural differences in the two agonist binding sites of the Torpedo nicotinic acetylcholine receptor revealed by time-resolved fluorescence spectroscopy. Biochemistry 39:6979–6990. [DOI] [PubMed] [Google Scholar]
- Mazzaferro S, Benallegue N, Carbone A, Gasparri F, Vijayan R, Biggin PC, Moroni M, Bermudez I. (2011) Additional acetylcholine (ACh) binding site at α4/α4 interface of (α4β2)2α4 nicotinic receptor influences agonist sensitivity. J Biol Chem 286:31043–31054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar NS, Gotti C. (2009) Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 56:237–246. [DOI] [PubMed] [Google Scholar]
- Morales-Perez CL, Noviello CM, Hibbs RE. (2016) X-ray structure of the human α4β2 nicotinic receptor. Nature 538:411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Free C, Sine SM. (2005) Initial coupling of binding to gating mediated by conserved residues in the muscle nicotinic receptor. J Gen Physiol 126:23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemecz Á, Prevost MS, Menny A, Corringer PJ. (2016) Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron 90:452–470. [DOI] [PubMed] [Google Scholar]
- Olsen JA, Balle T, Gajhede M, Ahring PK, Kastrup JS. (2014) Molecular recognition of the neurotransmitter acetylcholine by an acetylcholine binding protein reveals determinants of binding to nicotinic acetylcholine receptors. PLoS One 9:e91232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez EG, Cassels BK, Zapata-Torres G. (2009) Molecular modeling of the α9α10 nicotinic acetylcholine receptor subtype. Bioorg Med Chem Lett 19:251–254. [DOI] [PubMed] [Google Scholar]
- Plazas PV, Katz E, Gomez-Casati ME, Bouzat C, Elgoyhen AB. (2005) Stoichiometry of the α9α10 nicotinic cholinergic receptor. J Neurosci 25:10905–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince RJ, Sine SM. (1999) Acetylcholine and epibatidine binding to muscle acetylcholine receptors distinguish between concerted and uncoupled models. J Biol Chem 274:19623–19629. [DOI] [PubMed] [Google Scholar]
- Rayes D, De Rosa MJ, Sine SM, Bouzat C. (2009) Number and locations of agonist binding sites required to activate homomeric Cys-loop receptors. J Neurosci 29:6022–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin CV, Katz E, Verbitsky M, Elgoyhen AB. (1999) The α9 nicotinic acetylcholine receptor shares pharmacological properties with type A γ-aminobutyric acid, glycine, and type 3 serotonin receptors. Mol Pharmacol 55:248–254. [DOI] [PubMed] [Google Scholar]
- Russell RB, Barton GJ. (1992) Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins 14:309–323. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Fersht AR. (1995) Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J Mol Biol 248:478–486. [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D, Bertrand D, Besnard F. (2002) A novel human nicotinic receptor subunit, α10, that confers functionality to the α9-subunit. Mol Pharmacol 61:150–159. [DOI] [PubMed] [Google Scholar]
- Sine SM. (2002) The nicotinic receptor ligand binding domain. J Neurobiol 53:431–446. [DOI] [PubMed] [Google Scholar]
- Sine SM, Claudio T. (1991) γ- and δ-subunits regulate the affinity and the cooperativity of ligand binding to the acetylcholine receptor. J Biol Chem 266:19369–19377. [PubMed] [Google Scholar]
- Sine SM, Engel AG. (2006) Recent advances in Cys-loop receptor structure and function. Nature 440:448–455. [DOI] [PubMed] [Google Scholar]
- Sine SM, Huang S, Li SX, daCosta CJ, Chen L. (2013) Inter-residue coupling contributes to high-affinity subtype-selective binding of α-bungarotoxin to nicotinic receptors. Biochem J 454:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lester HA, Lummis SC. (2010) The structural basis of function in Cys-loop receptors. Q Rev Biophys 43:449–499. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, McLaughlin JT, Jurman ME, Hawrot E, Yellen G. (1991) Mutations affecting agonist sensitivity of the nicotinic acetylcholine receptor. Biophys J 60:721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 346:967–989. [DOI] [PubMed] [Google Scholar]
- Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB. (2000) Mixed nicotinic–muscarinic properties of the α9 nicotinic cholinergic receptor. Neuropharmacology 39:2515–2524. [DOI] [PubMed] [Google Scholar]
- Weisstaub N, Vetter DE, Elgoyhen AB, Katz E. (2002) The α9α10 nicotinic acetylcholine receptor is permeable to and is modulated by divalent cations. Hear Res 167:122–135. [DOI] [PubMed] [Google Scholar]
- Xie Y, Cohen JB. (2001) Contributions of Torpedo nicotinic acetylcholine receptor γTrp-55 and δTrp-57 to agonist and competitive antagonist function. J Biol Chem 276:2417–2426. [DOI] [PubMed] [Google Scholar]
- Yu R, Kompella SN, Adams DJ, Craik DJ, Kaas Q. (2013) Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J Med Chem 56:3557–3567. [DOI] [PubMed] [Google Scholar]
- Zouridakis M, Giastas P, Zarkadas E, Chroni-Tzartou D, Bregestovski P, Tzartos SJ. (2014) Crystal structures of free and antagonist-bound states of human α9 nicotinic receptor extracellular domain. Nat Struct Mol Biol 21:976–980. [DOI] [PubMed] [Google Scholar]