

Abstract
Synaptic plasticity in the adult brain is believed to represent the cellular mechanisms of learning and memory. Mitochondria are involved in the regulation of the complex processes of synaptic plasticity. This paper reviews the current knowledge on the regulatory roles of mitochondria in the function and plasticity of synapses and the implications of mitochondrial dysfunctions in synaptic transmission. First, the importance of mitochondrial distribution and motility for maintenance and strengthening of dendritic spines, but also for new spines/synapses formation is presented. Secondly, the major mitochondrial functions as energy supplier and calcium buffer organelles are considered as possible explanation for their essential and regulatory roles in neuronal plasticity processes. Thirdly, the effects of synaptic potentiation on mitochondrial gene expression are discussed. And finally, the relation between age-related alterations in synaptic plasticity and mitochondrial dysfunctions is considered. It appears that memory loss and neurodegeneration during aging are related to mitochondrial (dys)function. Although, it is clear that mitochondria are essential for synaptic plasticity, further studies are indicated to scrutinize the intracellular and molecular processes that regulate the functions of mitochondria in synaptic plasticity.
Keywords: Aging, ATP, calcium, learning and memory, long-term potentiation, mitochondria, synaptic plasticity
INTRODUCTION
Neuronal plasticity can be defined as structural and functional adaptations of neuronal circuits to changes due to learning and memory, environmental influences and brain damage [1, 2]. These dynamical processes in the neuronal system underlie the ability of the brain to change and to behaviourally adapt to a continuous changing environment. The two most important examples of neuroplasticity are long-term potentiation (LTP) and long-term depression (LTD) [3]. Both neurophysiological features can be found in neuronal synapses in response to a brief repeated stimulation. The cellular and molecular mechanisms behind this structural and functional changes of the neuronal network include pre- and postsynaptic neurotransmission alterations, cytoskeletal remodelling, membrane trafficking, gene transcription, protein synthesis and activation [4-7].
LTP is also assumed to be the cellular mechanism of learning and memory [8, 9]. Several studies have documented that changes in mitochondria, vital organelles presented in all eukaryotic cells, occur during synaptic activation and LTP [9]. For example, during LTP, mitochondrial energy production changes [10], mitochondrial calcium pump activity increases [11] and mitochondrial gene expression is enhanced [12].
The mitochondria are essential organelles found in all eukaryotic cells that are composed of compartments with specialized functions [13]. Mitochondria consist of outer membrane, intermembrane space, inner membrane and internal matrix. The outer and inner mitochondrial membrane have different properties. The outer mitochondrial membrane contains large numbers of integral proteins called porins. These porins form channels that allow small molecules (<500 Da) to freely diffuse from one side of the membrane to the other. In contrast, the inner membrane is not freely permeable, under physiological conditions. However, it contains specific transport proteins that regulate metabolite passage into and out of the matrix. Also, diverse proteins with different functions, including those involved in the electron transport chain and ATP synthesis [2].
The most prominent role of mitochondria is to produce cellular energy, in form of ATP through respiration, and to regulate cellular metabolism. Another important function of mitochondria in neurons is the regulation of calcium and redox signalling [14]. Mitochondria are able to remove calcium from the cytoplasm, in response to calcium influx into the cell and may also release calcium in response to certain stimuli [15, 16].
Given the oxidative, metabolic, and calcium buffering functions of the mitochondria, they are candidates to regulate and modify synaptic transmission and thus also related processes of functional and structural plasticity. For that matter, multiple lines of evidence have suggested that synaptic function and plasticity depend on mitochondria [2, 17-21].
A reciprocal influence between alteration of mitochondrial functionality and synaptic activity has already been reported for peripheral synapses [22]. Several pharmacological studies have revealed that the inhibition of mitochondrial activity results in impaired synaptic potentiation and neurotransmission [23, 24]. Acutely blocking mitochondrial function during intense stimulation also leads to depressed synaptic transmission [25, 26], whereas an increase in synaptic density is promoted in vitro through pharmacological enhancement of mitochondrial respiration [27]. These and other studies, which will be discussed here, suggest that mitochondria not only respond in various ways to synaptic activity but also regulate synaptic plasticity [16].
Here we review that mitochondria play important roles in regulating adult neuroplasticity. More specifically, we will discuss the role of mitochondria for dendritic spine growth and synaptogenesis, how mitochondrial ATP production and calcium buffering regulate neuroplasticity, and the relation between synaptic plasticity and mitochondrial gene expression. Finally, we review the role of mitochondrial dysfunctions on synaptic plasticity during aging.
DENDRITIC MITOCHONDRIA AND SYNAPTO- GENESIS
Synaptic plasticity in the mature nervous system involves structural and morphological modifications, such as dendritic spine growth and synaptogenesis [16, 28]. These modifications are the cellular response to changes in neuronal activity and are believed to be responsible for learning and memory [29, 30]. Mitochondria are present in axonal terminals and dendrites of neurons and are assumed to play a prominent role in synaptic plasticity [16].
Neuronal mitochondria are highly dynamic organelles that divide, fuse, and move purposefully within the different cell compartments of the neuron. The mitochondrial transport throughout the neuron and the recruitment of mitochondria to regions with high metabolic demands are essential for the proper functions of the neuronal network [14]. In dendrites, mitochondria are located mainly in the dendritic shafts and are also found to be associated with spines [31-33]. In response to synaptic stimulation, mitochondria redistribute toward dendritic protrusions and enhance their activity. It has been shown that the number of dendritic mitochondria increases simultaneously with synapse and spine morphogenesis, either following repetitive depolarization or in response of local electrical stimulation [27].
As reported by Li et al. [27], reducing the dendritic mitochondrial content led to loss of synapses and spines, whereas the number of spines and synapses significantly increased by accumulation of mitochondria in the dendrites. Furthermore, recent in vitro studies showed that the reduction of dendritic mitochondrial content through increased mitophagy leads to inhibition of dendrite growth during neuronal polarization [18] and to dendrite shortening in mature neuronal cultures [34]. Taken together, sufficient dendritic mitochondrial content is required for proper development and maintenance of dendrites, as well as synapse- and spine-formation.
Synaptic excitation affects the motility and subcellular distribution of mitochondria in dendrites. Electric stimulation in hippocampal organotypic slice culture leads to enlargement of the dendritic spines and recruitment of mitochondria to the active side [27]. On the other side, new spine and synapse formation is, in turn, enhanced by aggregation of mitochondria in dendrites [27]. Thus, there are mechanisms for mutual regulation of synaptic plasticity and mitochondrial distribution and activity.
Although these various functions and features of mitochondria are described, the functional role of mitochondria in dendritic protrusions remains to be determined. As in axonal growth cones [35], it is possible that changes in ATP demand and required calcium buffering capacity underlie the number of mitochondria in growing spines.
MITOCHONDRIAL ATP AND SYNAPTIC PLASTICITY
In every biological system energy supply is fundamental in a wide range of cellular function. Neuronal functions supported by mitochondrial ATP production also include the assembly of the actin cytoskeleton for the development of pre- and postsynaptic compartments, membrane potential generation, synaptic vesicle recruitment and release, as well as protein phosphorylation reactions [33, 36-38]. All of these processes are critical for neuroplasticity and can be modified by changes in ATP production and release [9]. Mitochondria play important roles in controlling the complex processes of neuroplasticity, including neurotransmitter release and dendritic remodelling by generating energy in the form of ATP [2, 33].
As mentioned above, LTP (enduring increase in neuronal excitability) is an established experimental model of the cellular mechanisms of learning and memory [8]. Blocking the mitochondrial oxidative phosphorylation, the metabolic pathway in which mitochondria produce ATP, leads to significant impairment of LTP [39]. Uncoupling the respiratory chain from the oxidative phosphorylation by carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) but also inhibiting the mitochondrial complex I by rotenone in synaptosomes led to reduced amplitude of synaptic vesicle release as a result of dysfunctional mitochondria and impaired ATP production [20]. Furthermore, Verstreken et al. [37] demonstrated in Drosophila neuromuscular junctions that the recruitment of reserve synaptic-pool vesicles is sufficient for the maintenance of normal neurotransmission during intense stimulation and depends on mitochondrial ATP production. A lack of synaptic mitochondria as well as application of oligomycin, an inhibitor of mitochondrial ATP production, resulted in a disability to mobilize reserve pool vesicles, which was partially rescued by addition of ATP [37]. These findings suggest that mitochondrial energy production is critical not only for a proper transmitter release via vesicle exocytosis, but also for mobilization of reserve synaptic-pool vesicle and regulation of synaptic strength (see Fig. 1).
Fig. (1).
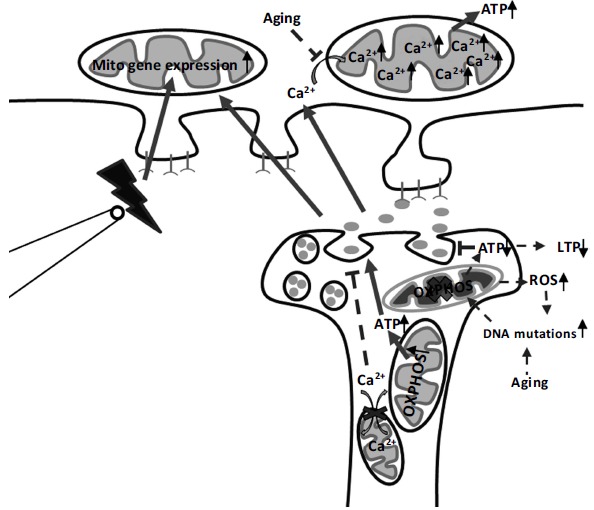
Mitochondrial function in adult synaptic plasticity. Long-term potentiation (LTP) (effects indicated by solid lines) leads to enhanced mitochondrial respiration and thereby ATP production. Cellular ATP concentration influences, on its side, neurotransmitter release. On the other hand, blocking the mitochondrial oxidative phosphorylation (OXPHOS) leads to decrease in ATP levels, drop in neurotransmitter release and significant impairment of LTP. Furthermore, inhibition of the mitochondrial calcium uptake and release machinery results also in impaired synaptic neurotransmission. On the postsynaptic side, mitochondrial calcium uptake as well as mitochondrial gene expression increase following high-frequency stimulation (indicated by and electrode) resulting in enhanced energy production. Therefore, mitochondria play important role in controlling synaptic activity by generating energy in the form of ATP and regulating the intracellular calcium homeostasis.
Signalling molecules such as glutamate and brain-derived neurotrophic factor BDNF regulate synaptic plasticity and also modify cellular energy metabolism [40, 41]. Glutamate plays critical roles in synaptic plasticity by activating receptors coupled to calcium influx [16]. It also regulates downstream signalling-pathways via activation of kinases (such as PKA, PKC and ERKs) and transcription factors that are important for long-term alterations of plasticity [42]. The protein activation processes are often ATP-dependent and glutamate also stimulates an increase in mitochondrial oxygen consumption and thereby ATP production [16, 43]. BDNF in turn modifies synaptic plasticity and has been shown to play a crucial role in hippocampus-dependent learning and memory [44]. BDNF could promote synaptic plasticity, in part, by enhancing mitochondrial energy production through increase of glucose utilization [45] and mitochondrial respiratory coupling at Complex I [46, 47]. Furthermore, a recent study showed that BNDF-signalling regulates mitochondrial transport and localization. In cultured hippocampal neurons, application of BDNF led to reduced mitochondrial motility in the axon and accumulation of mitochondria at presynaptic sites followed by an enhancement of the synaptic transmission [17].
Aging (effects indicated by dashed lines) results in increased incidence of DNA mutations. Certain DNA mutations lead to alteration in OXPHOS resulting in decreased ATP production and impaired LTP. In addition, DNA mutations increase reactive oxygen species (ROS) levels and oxidative stress. In parallel, high levels of ROS cause DNA mutations resulting in mitochondrial damage, which is a vicious circle. Furthermore, aging is associated with a loss of calcium uptake capacity of mitochondria. This leads to alterations in calcium homeostasis and a decrease in ATP production. Therefore, aged-related LTP deficits can be partially explained by mitochondrial dysfunctions associated with DNA mutations.
In summary, ATP is essential for several cellular mechanisms that are associated with different processes in synaptic plasticity. Experimental studies show that mitochondria play a role in neurotransmitter release via vesicle exocytosis, but also reserve pool vesicles mobilization and protein phosphorylation reactions. Mitochondrial energy production (ATP levels) is also influenced by signalling pathways that regulate synaptic plasticity (see Fig. 1). The available evidence indicates that the mitochondrial ATP production is homeostatically linked to synaptic plasticity. However, how mitochondrial energy metabolism and neuronal plasticity are inter-related requires further systematic studies.
MITOCHONDRIAL CALCIUM BUFFERING
Synaptic mechanisms of plasticity are besides ATP also calcium-dependent processes. Reduction of extracellular calcium as well as intracellular injections of the calcium binding ethylene glycol tetraacetic acid (EGTA) block the induction of LTP in hippocampal neurons [48, 49]. Furthermore, a persistent increase in the uptake and retention of calcium parallels hippocampal LTP [50]. Indeed, cellular, and specifically mitochondrial, calcium-buffering could be very important in LTP expression.
The regulation of calcium concentration in neuronal cells is provided by plasma membrane pumps, cytoplasmic buffers, and two intracellular organelles: mitochondria and endoplasmic reticulum [51]. It has been shown that an increase in mitochondrial pump activity is associated with hippocampal LTP: mitochondrial calcium uptake was persistently increased after the induction of LTP in perforant path-dentate gyrus synapses [11]. Also, to increase the frequency of vesicular fusion events on the presynaptic side of hippocampal neurons, the regulator of adult synaptic plasticity BDNF requires inter alia full mitochondrial calcium stores [21]. In comparison, a pharmacological study of the neuromuscular junction on crayfish leg muscles suggests that synaptic alteration in response to intense stimulation is being produced by mechanism involving mitochondrial calcium uptake during stimulation and subsequent calcium release into cytoplasm [24]. The slow mitochondrial efflux of calcium results in a minutes-lasting plateau of calcium concentration, which in turn causes facilitation of the synaptic response [52]. Thus, mitochondria influence neurotransmitter release from axon terminals, which can be prevented by blocking the mitochondrial calcium release [16, 53].
As mentioned above, mitochondria consist of two membranes – inner and outer mitochondrial membrane. The permeability of the outer membrane is essential for the ability of mitochondria to regulate local calcium concentration levels [19] and is conferred by family of porin proteins that are substance of the permeability transition pore [54]. In order to study the synaptic modification due to impaired mitochondrial permeability during LTP, Levy et al. [55] blocked the mitochondrial permeability transition pore using low dose of cyclosporine A. Cyclosporine A treatment resulted in an increase in basal synaptic transmission and deficit in synaptic plasticity in hippocampal slices due to increase in the resting calcium concentration in presynaptic terminals. These data are consistent with earlier reports on the role of mitochondrial calcium buffering [24, 25] suggesting that mitochondria could regulate synaptic function and plasticity through their ability to take up and release calcium.
An electrophysiological study showed that synaptic plasticity is impaired in homozygote porin knockout mice [54]. In hippocampal slices both short-term and long-term synaptic plasticity were found to be impaired. Accordingly, the porin-deficient mice showed impaired fear conditioning and spatial learning abilities as compared to the wild-type mice. On the other hand, the reduced permeability of mitochondrial outer membrane in heterozygote porin knockout mice led to enhanced mitochondrial function and increased synaptic protein expression [19]. Mitochondrial membrane permeability regulates mitochondrial membrane potential and therefore mitochondrial functions such as oxidative phosphorylation and/or release of apoptotic factors. Thus, it is not surprising that fine-tuning the mitochondrial membrane permeability can affect the cell beneficially. Taken together, these results indicate that mitochondrial outer membrane permeability is essential for synaptic transmission due to regulation of local calcium concentration. Therefore the regulation of the mitochondrial permeability transition pore complex may play an essential role in synaptic plasticity.
In conclusion, mitochondrial calcium influx and release play important roles in regulating synaptic plasticity (see Fig. 1). Further studies of the mechanisms of the mitochondrial involvement in the local regulation of cellular calcium homeostasis and signalling would be necessary for the understanding basic cellular mechanism underlying neuronal plasticity [53].
THE EFFECT OF SYNAPTIC PLASTICITY ON MITOCHONDRIAL BIOGENESIS
Synaptic plasticity involves structural changes at the synapses as a result of molecular remodelling of synaptic proteins, which could lead to long-lasting alterations of the synaptic response to subsequent neurotransmitter release. Several studies have reported that mitochondrial alterations such as changes in mitochondrial transmembrane potential, calcium uptake and release as well as mitochondrial gene expression are associated with LTP [12, 56]. A study using differential screening of a cDNA libraries from high-frequency stimulation (50 trains of 10 pulses; 400 Hz, 25 ms, 250 ms duration pulses) and control stimulation (0.05 Hz baseline stimulation) conditions showed that, following high-frequency stimulation, the postsynaptic mitochondrial gene expression was elevated. The analysis indicated a general up-regulation in mitochondrial gene expression which persisted for up to two weeks [12].
Furthermore, modulators of hippocampal synaptic plasticity and learning and memory such as oestrogen, growth factors and nitric oxide [57-60] are also known to modify mitochondrial biogenesis [12, 61-63]. Thus mitochondrial biogenesis is promoted by several signalling pathways that also regulate neuroplasticity.
Taken together, these findings suggest that changes in mitochondrial gene expression may be required for the maintenance of synaptic potentiation. Further work will be required to establish specific roles for different mitochondrial genes in the regulation and maintenance of synaptic plasticity.
SYNAPTIC PLASTICITY, MITOCHONDRIA AND AGING
Ageing is associated with specific impairments of learning and memory. These impairments can be caused, at least partly, by altered synaptic plasticity mechanisms, including deficits in the induction and maintenance of LTP [64, 65]. Early studies of synaptic plasticity alterations reporting no difference in LTP induction in aged animals used high-frequency and high-amplitude stimulation protocols [66, 67]. However, when less robust protocols with lower stimulation intensity were used, aged animals show deficits in LTP induction, including an increased induction threshold [68, 69]. Examination of differences between young and old brains has also revealed that LTP maintenance is impaired in older animals [69, 70]. While in young animals LTP can last from hours to weeks [71], studies in the aged animals indicated a more rapid LTP decay in these preparations [67, 72]. In conclusion, aged-related LTP deficits are associated with increased threshold of LTP induction and decreased LTP maintenance duration.
Aged-related LTP deficits can arise from a number of different sources, including synaptic alterations, abnormalities in signal transduction pathways and different cellular functional alterations [64, 68, 70]. Here we concentrate on aged-related mitochondrial dysfunctions and their impact on synaptic plasticity in aging. As already mentioned, mitochondria play an important role in the maintenance of synaptic transduction through ATP generation and regulation of local intercellular calcium concentrations. Therefore, mitochondrial dysfunction and defects are predicted to affect various aspects of synaptic plasticity. Indeed, age-related mitochondrial dysfunctions are often associated with memory deficits and neurodegeneration diseases, such as Alzheimer’s disease and Parkinson’s disease [73, 74].
Ultrastructural analyses of mitochondrial morphology have demonstrated the appearance of age-related changes in these organelles, such as: microvacuolization, broken cristae, and accumulation of paracrystalline inclusions [70, 75]. Together with a decrease in the membrane potential of the inner mitochondrial membrane and decrease levels of some complexes of the electron transport chain (ETC) associated with aging [76], these changes could explain the impaired mitochondrial energy metabolism in the old brain. The age-related alteration in mitochondrial oxidative phosphorylation processes resulting in decreased ATP production and energy deficit not only may cause an impaired LTP induction and maintenance, moreover changes in cellular bioenergetics may as well induce neuronal apoptosis and neurodegeneration [69, 70].
The changes in mitochondrial bioenergetics could be due to a complex interplay between the aged-related accumulation of DNA mutations and abnormalities in redox-sensitive signalling pathways [70, 77]. Aging results in genetic instability and increased incidence of mutations both in nuclear and in mitochondrial DNA [78]. Certain DNA mutations may impair the ETC in the mitochondrial membrane and thereby the ATP synthesis. On the other hand, an increase in reactive oxygen species (ROS) levels causing oxidative stress is also associated with aging and age-related synaptic impairments [79]. Interestingly, deficiency in the ETC may result in increased production of ROS and, in parallel, high levels of ROS may cause DNA mutations resulting in mitochondrial damage and ECT deficits [80, 81]. However, whether DNA mutations and/or increased ROS levels are the cause or the consequence of aging is not a subject of this review.
Calcium plays a very important role for the increasing mitochondrial ATP production during neuronal stimulation [71]. Neuronal stimulation enhances mitochondrial calcium uptake resulting in an increased mitochondrial calcium concentration and mitochondrial depolarization [82, 83]. The ETC is thereby activated and the proton extrusion rate elevates inducing a recovery of the resting mitochondrial membrane potential and an increase in the ATP production. Additionally, calcium activates also some of the key enzymes of the Krebs cycle [84]. In vitro studies demonstrated that in aged neurons the mitochondrial repolarisation after stimulation was delayed which could be a sign of limited mitochondrial calcium uptake capacity [85, 86]. Moreover, quantitative analysis showed that the delay of mitochondria repolarisation correlates strongly with the recovery of the resting intracellular calcium concentration. Thus, age-related loss of calcium uptake capacity of mitochondria appears to impair the recovering of resting calcium values after stimulation and lead to alterations in calcium homeostasis (see Fig. 1).
CONCLUSION
Synaptic plasticity in the mature nervous system involves structural modifications, such as dendritic spine growth or retraction, strengthening of synaptic transmission and even new synapses formation [16]. The plasticity processes occur in response to environmental stimuli and associated with changes in synaptic activity and are believed to be responsible for learning and memory [29, 30, 33]. In this review it is shown that, because of their molecular and functional complexity, mitochondria are not just involved in synaptic plasticity in the adult brain, but could also be essential regulators of these complex processes.
Li et al. [27] demonstrated that synapse formation was enhanced by mitochondrial aggregation in the dendrites. The dynamic mitochondrial distribution into the different neuron compartments is regulated by synaptic activity. Importantly, new synapse formation caused by potentiating stimuli depend critically on the proper distribution and function of mitochondria and is impaired by their absence [87]. Mitochondria are essential components of synaptic activity, mainly because of their functions as energy producer and calcium buffering organelles. ATP is required for maintenance and restoration of ion gradients, which enable synaptic transmission [33], but also for many different protein phosphorylation reactions involved in the regulation of LTP and other forms of synaptic plasticity [9, 38]. Verstreken et al. [37] demonstrated that during intense stimulation neurons with impaired mitochondrial functions could not maintain normal neurotransmitter release like controls. The evidence suggests that this effect was due to absence of mitochondrial ATP, which is required for the mobilization of reserve pool synaptic vesicles, since the synaptic transmission was rescued by ATP addition. The ability of mitochondria to rapidly buffer calcium during intense stimulation is also required for synaptic plasticity [55]. Blocking the mitochondrial calcium uptake resulted in a transient increase in presynaptic calcium levels and impaired neurotransmission during intense stimulation [24].
The studies reviewed herein demonstrate that mitochondria are essential for both synaptic activity and plasticity, since they are required for the energy supply and metabolic control of the synapse but also for the regulation of calcium concentration and signalling pathways (see Fig. 1). Mitochondria respond to synaptic plasticity in several ways i.e. aggregation of mitochondria in the pre- and post-synapses, elevation of ATP production, increase in mitochondrial calcium pump activity, and up-regulation of mitochondrial biogenesis. Moreover, accumulating evidence place mitochondrial alterations during aging in the focus of neurodegeneration studies [70, 73, 74]. The relation between mitochondrial dysfunctions and impaired synaptic plasticity in the old brain suggests a key role for mitochondria in the process of memory decline with aging. However, the precise involvement of mitochondria in the regulation of the cellular processes of learning and memory remains poorly understood. Advances in understanding the molecular and cell biology of mitochondria could lead not only to encoding the secret of memory but also to new approaches into the research of neurodegenerative diseases like different types of dementia.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Malenka R.C., Nicoll R.A. Long-term potentiation--a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [http://dx.doi.org/ 10.1126/science.285.5435.1870]. [PMID: 10489359]. [DOI] [PubMed] [Google Scholar]
- 2.Cheng A., Hou Y., Mattson M.P. Mitochondria and neuro- plasticity. ASN Neuro. 2010;2(5):e00045. doi: 10.1042/AN20100019. [http://dx.doi.org/ 10.1042/AN20100019]. [PMID: 20957078]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malenka R.C., Bear M.F. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [http://dx.doi.org/10.1016/ j.neuron.2004.09.012]. [PMID: 15450156]. [DOI] [PubMed] [Google Scholar]
- 4.Shepherd J.D., Huganir R.L. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [http://dx.doi.org/10.1146/annurev.cellbio. 23.090506.123516]. [PMID: 17506699]. [DOI] [PubMed] [Google Scholar]
- 5.Bramham C.R. Local protein synthesis, actin dynamics, and LTP consolidation. Curr. Opin. Neurobiol. 2008;18(5):524–531. doi: 10.1016/j.conb.2008.09.013. [http://dx.doi.org/10.1016/j.conb.2008.09.013]. [PMID: 18834940]. [DOI] [PubMed] [Google Scholar]
- 6.Greer P.L., Greenberg M.E. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59(6):846–860. doi: 10.1016/j.neuron.2008.09.002. [http://dx.doi.org/ 10.1016/j.neuron.2008.09.002]. [PMID: 18817726]. [DOI] [PubMed] [Google Scholar]
- 7.Shah M.M., Hammond R.S., Hoffman D.A. Dendritic ion channel trafficking and plasticity. Trends Neurosci. 2010;33(7):307–316. doi: 10.1016/j.tins.2010.03.002. [http://dx.doi.org/10.1016/j.tins.2010.03.002]. [PMID: 20363038]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris R.G. NMDA receptors and memory encoding. Neuropharmacology. 2013;74:32–40. doi: 10.1016/j.neuropharm.2013.04.014. [http://dx.doi.org/10.1016/ j.neuropharm.2013.04.014]. [PMID: 23628345]. [DOI] [PubMed] [Google Scholar]
- 9.Mattson M.P., Liu D. Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem. Biophys. Res. Commun. 2003;304(3):539–549. doi: 10.1016/s0006-291x(03)00627-2. [http:// dx.doi.org/10.1016/S0006-291X(03)00627-2]. [PMID: 12729589]. [DOI] [PubMed] [Google Scholar]
- 10.Wieraszko A. Changes in the hippocampal slices energy metabolism following stimulation and long-term potentiation of Schaffer collaterals-pyramidal cell synapses tested with the 2-deoxyglucose technique. Brain Res. 1982;237(2):449–457. doi: 10.1016/0006-8993(82)90456-5. [http:// dx.doi.org/10.1016/0006-8993(82)90456-5]. [PMID: 7083005]. [DOI] [PubMed] [Google Scholar]
- 11.Stanton P.K., Schanne F.A. Hippocampal long-term potentiation increases mitochondrial calcium pump activity in rat. Brain Res. 1986;382(1):185–188. doi: 10.1016/0006-8993(86)90130-7. [http://dx.doi.org/10.1016/0006-8993(86) 90130-7]. [PMID: 2945618]. [DOI] [PubMed] [Google Scholar]
- 12.Williams J.M., Thompson V.L., Mason-Parker S.E., Abraham W.C., Tate W.P. Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Brain Res. Mol. Brain Res. 1998;60(1):50–56. doi: 10.1016/s0169-328x(98)00165-x. [http://dx.doi.org/10.1016/ S0169-328X(98)00165-X]. [PMID: 9748499]. [DOI] [PubMed] [Google Scholar]
- 13.Bruce A., Johnson A., Lewis J., Raff M., Roberts K. 2007. [Google Scholar]
- 14.Mattson M.P., Gleichmann M., Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60(5):748–766. doi: 10.1016/j.neuron.2008.10.010. [http://dx.doi.org/10.1016/j.neuron.2008.10.010]. [PMID: 19081372]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simpson P.B. The local control of cytosolic Ca2+ as a propagator of CNS communication--integration of mitochondrial transport mechanisms and cellular responses. J. Bioenerg. Biomembr. 2000;32(1):5–13. doi: 10.1023/a:1005552126516. [http://dx.doi.org/10.1023/A:1005552126516]. [PMID: 11768762]. [DOI] [PubMed] [Google Scholar]
- 16.Mattson M.P. Mitochondrial regulation of neuronal plasticity. Neurochem. Res. 2007;32(4-5):707–715. doi: 10.1007/s11064-006-9170-3. [http://dx.doi.org/10. 1007/s11064-006-9170-3]. [PMID: 17024568]. [DOI] [PubMed] [Google Scholar]
- 17.Su B., Ji Y.S., Sun X.L., Liu X.H., Chen Z.Y. Brain-derived neurotrophic factor (BDNF)-induced mitochondrial motility arrest and presynaptic docking contribute to BDNF-enhanced synaptic transmission. J. Biol. Chem. 2014;289(3):1213–1226. doi: 10.1074/jbc.M113.526129. [http:// dx.doi.org/10.1074/jbc.M113.526129]. [PMID: 24302729]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brot S., Auger C., Bentata R., Rogemond V., Ménigoz S., Chounlamountri N., Girard-Egrot A., Honnorat J., Moradi-Améli M. Collapsin response mediator protein 5 (CRMP5) induces mitophagy, thereby regulating mitochondrion numbers in dendrites. J. Biol. Chem. 2014;289(4):2261–2276. doi: 10.1074/jbc.M113.490862. [http://dx.doi.org/ 10.1074/jbc.M113.490862]. [PMID: 24324268]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manczak M., Sheiko T., Craigen W.J., Reddy P.H. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J. Alzheimers Dis. 2013;37(4):679–690. doi: 10.3233/JAD-130761. [PMID: 23948905]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivannikov M.V., Sugimori M., Llinás R.R. Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J. Mol. Neurosci. 2013;49(1):223–230. doi: 10.1007/s12031-012-9848-8. [http://dx.doi.org/10.1007/s12031-012-9848-8]. [PMID: 22772899]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amaral M.D., Pozzo-Miller L. 2012.
- 22.Nguyen P.V., Atwood H.L. Altered impulse activity modifies synaptic physiology and mitochondria in crayfish phasic motor neurons. J. Neurophysiol. 1994;72(6):2944–2955. doi: 10.1152/jn.1994.72.6.2944. [PMID: 7897501]. [DOI] [PubMed] [Google Scholar]
- 23.Alnaes E., Rahamimoff R. On the role of mitochondria in transmitter release from motor nerve terminals. J. Physiol. 1975;248(2):285–306. doi: 10.1113/jphysiol.1975.sp010974. [http://dx.doi.org/10.1113/jphysiol.1975.sp010974]. [PMID: 50439]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang Y., Zucker R.S. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18(3):483–491. doi: 10.1016/s0896-6273(00)81248-9. [http://dx.doi.org/10.1016/S0896-6273(00)81248-9]. [PMID: 9115741]. [DOI] [PubMed] [Google Scholar]
- 25.Billups B., Forsythe I.D. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 2002;22(14):5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [PMID: 12122046]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medler K., Gleason E.L. Mitochondrial Ca(2+) buffering regulates synaptic transmission between retinal amacrine cells. J. Neurophysiol. 2002;87(3):1426–1439. doi: 10.1152/jn.00627.2001. [PMID: 11877517]. [DOI] [PubMed] [Google Scholar]
- 27.Li Z., Okamoto K., Hayashi Y., Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–887. doi: 10.1016/j.cell.2004.11.003. [http://dx.doi.org/ 10.1016/j.cell.2004.11.003]. [PMID: 15607982]. [DOI] [PubMed] [Google Scholar]
- 28.Wong R.O., Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci. 2002;3(10):803–812. doi: 10.1038/nrn941. [http://dx.doi.org/10.1038/nrn941]. [PMID: 12360324]. [DOI] [PubMed] [Google Scholar]
- 29.Yuste R., Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu. Rev. Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [http://dx.doi.org/10.1146/ annurev.neuro.24.1.1071]. [PMID: 11520928]. [DOI] [PubMed] [Google Scholar]
- 30.Carlisle H.J., Kennedy M.B. Spine architecture and synaptic plasticity. Trends Neurosci. 2005;28(4):182–187. doi: 10.1016/j.tins.2005.01.008. [http://dx.doi.org/10.1016/j.tins.2005.01.008]. [PMID: 15808352]. [DOI] [PubMed] [Google Scholar]
- 31.Cameron H.A., Kaliszewski C.K., Greer C.A. Organization of mitochondria in olfactory bulb granule cell dendritic spines. Synapse. 1991;8(2):107–118. doi: 10.1002/syn.890080205. [http://dx.doi.org/10.1002/syn. 890080205]. [PMID: 1715612]. [DOI] [PubMed] [Google Scholar]
- 32.Popov V., Medvedev N.I., Davies H.A., Stewart M.G. Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: a three-dimensional ultrastructural study. J. Comp. Neurol. 2005;492(1):50–65. doi: 10.1002/cne.20682. [http://dx.doi.org/10.1002/cne.20682]. [PMID: 16175555]. [DOI] [PubMed] [Google Scholar]
- 33.Valenti D., de Bari L., De Filippis B., Henrion-Caude A., Vacca R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: an overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014;46(Pt 2):202–217. doi: 10.1016/j.neubiorev.2014.01.012. [http://dx.doi.org/10.1016/j.neubiorev.2014. 01.012]. [PMID: 24548784]. [DOI] [PubMed] [Google Scholar]
- 34.Cherra S.J., III, Steer E., Gusdon A.M., Kiselyov K., Chu C.T. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 2013;182(2):474–484. doi: 10.1016/j.ajpath.2012.10.027. [http://dx.doi.org/10.1016/j.ajpath.2012.10.027]. [PMID: 23231918]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris R.L., Hollenbeck P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993;104(Pt 3):917–927. doi: 10.1242/jcs.104.3.917. [PMID: 8314882]. [DOI] [PubMed] [Google Scholar]
- 36.Attwell D., Laughlin S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001;21(10):1133–1145. doi: 10.1097/00004647-200110000-00001. [http://dx.doi.org/10.1097/00004647-200110000-00001]. [PMID: 11598490]. [DOI] [PubMed] [Google Scholar]
- 37.Verstreken P., Ly C.V., Venken K.J., Koh T.W., Zhou Y., Bellen H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47(3):365–378. doi: 10.1016/j.neuron.2005.06.018. [http://dx.doi.org/10.1016/j.neuron. 2005.06.018]. [PMID: 16055061]. [DOI] [PubMed] [Google Scholar]
- 38.Stefani G., Onofri F., Valtorta F., Vaccaro P., Greengard P., Benfenati F. Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic vesicles. J. Physiol. 1997;504(Pt 3):501–515. doi: 10.1111/j.1469-7793.1997.501bd.x. [http://dx.doi.org/10.1111/ j.1469-7793.1997.501bd.x]. [PMID: 9401959]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunha R.A., Vizi E.S., Ribeiro J.A., Sebastião A.M. Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J. Neurochem. 1996;67(5):2180–2187. doi: 10.1046/j.1471-4159.1996.67052180.x. [http://dx.doi.org/10.1046/j.1471-4159.1996.67052180.x]. [PMID: 8863529]. [DOI] [PubMed] [Google Scholar]
- 40.Trenkner E., el Idrissi A., Harris C. Balanced interaction of growth factors and taurine regulate energy metabolism, neuronal survival, and function of cultured mouse cerebellar cells under depolarizing conditions. Adv. Exp. Med. Biol. 1996;403:507–517. doi: 10.1007/978-1-4899-0182-8_55. [http://dx.doi.org/10.1007/978-1-4899-0182-8_55]. [PMID: 8915389]. [DOI] [PubMed] [Google Scholar]
- 41.Mattson M.P., Lovell M.A., Furukawa K., Markesbery W.R. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J. Neurochem. 1995;65(4):1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [http://dx.doi.org/10.1046/j.1471-4159.1995.65041740.x]. [PMID: 7561872]. [DOI] [PubMed] [Google Scholar]
- 42.Roberson E.D., English J.D., Adams J.P., Selcher J.C., Kondratick C., Sweatt J.D. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J. Neurosci. 1999;19(11):4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [PMID: 10341237]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuchmann S., Buchheim K., Heinemann U., Hosten N., Buttgereit F. Oxygen consumption and mitochondrial membrane potential indicate developmental adaptation in energy metabolism of rat cortical neurons. Eur. J. Neurosci. 2005;21(10):2721–2732. doi: 10.1111/j.1460-9568.2005.04109.x. [http://dx.doi.org/10.1111/j.1460-9568.2005.04109.x]. [PMID: 15926920]. [DOI] [PubMed] [Google Scholar]
- 44.Lu B., Wang K.H., Nose A. Molecular mechanisms underlying neural circuit formation. Curr. Opin. Neurobiol. 2009;19(2):162–167. doi: 10.1016/j.conb.2009.04.004. [http://dx.doi.org/10.1016/j.conb.2009.04.004]. [PMID: 19457653]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burkhalter J., Fiumelli H., Allaman I., Chatton J.Y., Martin J.L. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J. Neurosci. 2003;23(23):8212–8220. doi: 10.1523/JNEUROSCI.23-23-08212.2003. [PMID: 12967982]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markham A., Cameron I., Franklin P., Spedding M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. Eur. J. Neurosci. 2004;20(5):1189–1196. doi: 10.1111/j.1460-9568.2004.03578.x. [http://dx.doi.org/10.1111/j.1460-9568.2004.03578.x]. [PMID: 15341590]. [DOI] [PubMed] [Google Scholar]
- 47.Markham A., Cameron I., Bains R., Franklin P., Kiss J.P., Schwendimann L., Gressens P., Spedding M. Brain-derived neurotrophic factor-mediated effects on mitochondrial respiratory coupling and neuroprotection share the same molecular signalling pathways. Eur. J. Neurosci. 2012;35(3):366–374. doi: 10.1111/j.1460-9568.2011.07965.x. [http://dx.doi. org/10.1111/j.1460-9568.2011.07965.x]. [PMID: 22288477]. [DOI] [PubMed] [Google Scholar]
- 48.Dunwiddie T., Madison D., Lynch G. Synaptic transmission is required for initiation of long-term potentiation. Brain Res. 1978;150(2):413–417. doi: 10.1016/0006-8993(78)90293-7. [http://dx.doi.org/10.1016/0006-8993(78)90293-7]. [PMID: 209851]. [DOI] [PubMed] [Google Scholar]
- 49.Lynch G., Larson J., Kelso S., Barrionuevo G., Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305(5936):719–721. doi: 10.1038/305719a0. [http://dx.doi.org/10.1038/305719a0]. [PMID: 6415483]. [DOI] [PubMed] [Google Scholar]
- 50.Baimbridge K.G., Miller J.J. Calcium uptake and retention during long-term potentiation of neuronal activity in the rat hippocampal slice preparation. Brain Res. 1981;221(2):299–305. doi: 10.1016/0006-8993(81)90779-4. [http://dx. doi.org/10.1016/0006-8993(81)90779-4]. [PMID: 7284771]. [DOI] [PubMed] [Google Scholar]
- 51.Thayer S.A., Miller R.J. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J. Physiol. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [http://dx.doi.org/10.1113/ jphysiol.1990.sp018094]. [PMID: 2213592]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delaney K.R., Tank D.W. A quantitative measurement of the dependence of short-term synaptic enhancement on presynaptic residual calcium. J. Neurosci. 1994;14(10):5885–5902. doi: 10.1523/JNEUROSCI.14-10-05885.1994. [PMID: 7931551]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang F., He X.P., Russell J., Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J. Cell Biol. 2003;163(3):511–523. doi: 10.1083/jcb.200307027. [http://dx.doi.org/10.1083/jcb.200307027]. [PMID: 14610054]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weeber E.J., Levy M., Sampson M.J., Anflous K., Armstrong D.L., Brown S.E., Sweatt J.D., Craigen W.J. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J. Biol. Chem. 2002;277(21):18891–18897. doi: 10.1074/jbc.M201649200. [http://dx.doi.org/10.1074/jbc.M201649200]. [PMID: 11907043]. [DOI] [PubMed] [Google Scholar]
- 55.Levy M., Faas G.C., Saggau P., Craigen W.J., Sweatt J.D. Mitochondrial regulation of synaptic plasticity in the hippocampus. J. Biol. Chem. 2003;278(20):17727–17734. doi: 10.1074/jbc.M212878200. [http://dx.doi.org/10. 1074/jbc.M212878200]. [PMID: 12604600]. [DOI] [PubMed] [Google Scholar]
- 56.Bindokas V.P., Lee C.C., Colmers W.F., Miller R.J. Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J. Neurosci. 1998;18(12):4570–4587. doi: 10.1523/JNEUROSCI.18-12-04570.1998. [PMID: 9614233]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foy M.R., Baudry M., Diaz Brinton R., Thompson R.F. Estrogen and hippocampal plasticity in rodent models. J. Alzheimers Dis. 2008;15(4):589–603. doi: 10.3233/jad-2008-15406. [PMID: 19096158]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harooni H.E., Naghdi N., Sepehri H., Rohani A.H. The role of hippocampal nitric oxide (NO) on learning and immediate, short- and long-term memory retrieval in inhibitory avoidance task in male adult rats. Behav. Brain Res. 2009;201(1):166–172. doi: 10.1016/j.bbr.2009.02.011. [http://dx.doi.org/10.1016/j.bbr.2009.02.011]. [PMID: 19428630]. [DOI] [PubMed] [Google Scholar]
- 59.Minichiello L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009;10(12):850–860. doi: 10.1038/nrn2738. [http://dx.doi.org/10.1038/ nrn2738]. [PMID: 19927149]. [DOI] [PubMed] [Google Scholar]
- 60.Brinton R.D. Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci. 2009;30(4):212–222. doi: 10.1016/j.tips.2008.12.006. [http://dx.doi.org/10.1016/j.tips.2008.12.006]. [PMID: 19299024]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gutsaeva D.R., Carraway M.S., Suliman H.B., Demchenko I.T., Shitara H., Yonekawa H., Piantadosi C.A. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J. Neurosci. 2008;28(9):2015–2024. doi: 10.1523/JNEUROSCI.5654-07.2008. [http://dx.doi.org/10.1523/JNEUROSCI. 5654-07.2008]. [PMID: 18305236]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klinge C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008;105(6):1342–1351. doi: 10.1002/jcb.21936. [http://dx. doi.org/10.1002/jcb.21936]. [PMID: 18846505]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Renton J.P., Xu N., Clark J.J., Hansen M.R. Interaction of neurotrophin signaling with Bcl-2 localized to the mitochondria and endoplasmic reticulum on spiral ganglion neuron survival and neurite growth. J. Neurosci. Res. 2010;88(10):2239–2251. doi: 10.1002/jnr.22381. [http://dx.doi.org/10.1002/jnr.22381]. [PMID: 20209634]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burke S.N., Barnes C.A. Senescent synapses and hippocampal circuit dynamics. Trends Neurosci. 2010;33(3):153–161. doi: 10.1016/j.tins.2009.12.003. [http://dx.doi.org/10.1016/j.tins.2009.12.003]. [PMID: 20071039]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Foster T.C. Challenges and opportunities in characterizing cognitive aging across species. Front. Aging Neurosci. 2012;4:33. doi: 10.3389/fnagi.2012.00033. [http://dx.doi.org/10.3389/fnagi.2012.00033]. [PMID: 23189053]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barnes C.A. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 1979;93(1):74–104. doi: 10.1037/h0077579. [http://dx.doi.org/10.1037/ h0077579]. [PMID: 221551]. [DOI] [PubMed] [Google Scholar]
- 67.Landfield P.W., Lynch G. Impaired monosynaptic potentiation in in vitro hippocampal slices from aged, memory-deficient rats. J. Gerontol. 1977;32(5):523–533. doi: 10.1093/geronj/32.5.523. [http://dx.doi.org/10.1093/ geronj/32.5.523]. [PMID: 196001]. [DOI] [PubMed] [Google Scholar]
- 68.Burgdorf J., Zhang X.L., Weiss C., Matthews E., Disterhoft J.F., Stanton P.K., Moskal J.R. The N-methyl-D-aspartate receptor modulator GLYX-13 enhances learning and memory, in young adult and learning impaired aging rats. Neurobiol. Aging. 2011;32(4):698–706. doi: 10.1016/j.neurobiolaging.2009.04.012. [http://dx.doi.org/10.1016/j.neurobiolaging. 2009.04.012]. [PMID: 19446371]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lister J.P., Barnes C.A. Neurobiological changes in the hippocampus during normative aging. Arch. Neurol. 2009;66(7):829–833. doi: 10.1001/archneurol.2009.125. [http://dx.doi.org/10.1001/archneurol.2009.125]. [PMID: 19597084]. [DOI] [PubMed] [Google Scholar]
- 70.Dorszewska J. Cell biology of normal brain aging: synaptic plasticity-cell death. Aging Clin. Exp. Res. 2013;25(1):25–34. doi: 10.1007/s40520-013-0004-2. [http://dx.doi.org/10.1007/s40520-013-0004-2]. [PMID: 23740630]. [DOI] [PubMed] [Google Scholar]
- 71.Toescu E.C., Verkhratsky A. Ca2+ and mitochondria as substrates for deficits in synaptic plasticity in normal brain ageing. J. Cell. Mol. Med. 2004;8(2):181–190. doi: 10.1111/j.1582-4934.2004.tb00273.x. [http://dx.doi.org/10.1111/j.1582-4934.2004.tb00273.x]. [PMID: 15256066]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barnes C.A., McNaughton B.L. An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses. Behav. Neurosci. 1985;99(6):1040–1048. doi: 10.1037//0735-7044.99.6.1040. [http://dx.doi.org/10.1037/0735-7044.99.6. 1040]. [PMID: 3843538]. [DOI] [PubMed] [Google Scholar]
- 73.Troulinaki K., Bano D. Mitochondrial deficiency: a double-edged sword for aging and neurodegeneration. Front. Genet. 2012;3:244. doi: 10.3389/fgene.2012.00244. [http://dx.doi.org/10.3389/fgene.2012.00244]. [PMID: 23248639]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schon E.A., Przedborski S. Mitochondria: the next (neurode) generation. Neuron. 2011;70(6):1033–1053. doi: 10.1016/j.neuron.2011.06.003. [http://dx.doi.org/ 10.1016/j.neuron.2011.06.003]. [PMID: 21689593]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toescu E.C., Myronova N., Verkhratsky A. Age-related structural and functional changes of brain mitochondria. Cell Calcium. 2000;28(5-6):329–338. doi: 10.1054/ceca.2000.0167. [http://dx.doi.org/10.1054/ceca. 2000.0167]. [PMID: 11115372]. [DOI] [PubMed] [Google Scholar]
- 76.Gilmer L.K., Ansari M.A., Roberts K.N., Scheff S.W. Age-related changes in mitochondrial respiration and oxidative damage in the cerebral cortex of the Fischer 344 rat. Mech. Ageing Dev. 2010;131(2):133–143. doi: 10.1016/j.mad.2009.12.011. [http://dx.doi.org/10.1016/j.mad.2009. 12.011]. [PMID: 20080122]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yin F., Jiang T., Cadenas E. Metabolic triad in brain aging: mitochondria, insulin/IGF-1 signalling and JNK signalling. Biochem. Soc. Trans. 2013;41(1):101–105. doi: 10.1042/BST20120260. [http://dx.doi.org/ 10.1042/BST20120260]. [PMID: 23356266]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sedelnikova O.A., Horikawa I., Zimonjic D.B., Popescu N.C., Bonner W.M., Barrett J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004;6(2):168–170. doi: 10.1038/ncb1095. [http://dx.doi.org/ 10.1038/ncb1095]. [PMID: 14755273]. [DOI] [PubMed] [Google Scholar]
- 79.Brawek B., Löffler M., Wagner K., Huppertz H.J., Wendling A.S., Weyerbrock A., Jackisch R., Feuerstein T.J. Reactive oxygen species (ROS) in the human neocortex: role of aging and cognition. Brain Res. Bull. 2010;81(4-5):484–490. doi: 10.1016/j.brainresbull.2009.10.011. [http://dx.doi. org/10.1016/j.brainresbull.2009.10.011]. [PMID: 19854245]. [DOI] [PubMed] [Google Scholar]
- 80.Sastre J., Pallardó F.V., Viña J. Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life. 2000;49(5):427–435. doi: 10.1080/152165400410281. [http://dx.doi.org/10.1080/152165400410281]. [PMID: 10902575]. [DOI] [PubMed] [Google Scholar]
- 81.Indo H.P., Davidson M., Yen H.C., Suenaga S., Tomita K., Nishii T., Higuchi M., Koga Y., Ozawa T., Majima H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7(1-2):106–118. doi: 10.1016/j.mito.2006.11.026. [http://dx.doi.org/10.1016/ j.mito.2006.11.026]. [PMID: 17307400]. [DOI] [PubMed] [Google Scholar]
- 82.Nicholls D.G., Vesce S., Kirk L., Chalmers S. Interactions between mitochondrial bioenergetics and cytoplasmic calcium in cultured cerebellar granule cells. Cell Calcium. 2003;34(4-5):407–424. doi: 10.1016/s0143-4160(03)00144-1. [http://dx.doi.org/10.1016/S0143-4160(03)00144-1]. [PMID: 12909085]. [DOI] [PubMed] [Google Scholar]
- 83.Toescu E.C., Verkhratsky A. Neuronal ageing from an intraneuronal perspective: roles of endoplasmic reticulum and mitochondria. Cell Calcium. 2003;34(4-5):311–323. doi: 10.1016/s0143-4160(03)00142-8. [http://dx. doi.org/10.1016/S0143-4160(03)00142-8]. [PMID: 12909078]. [DOI] [PubMed] [Google Scholar]
- 84.Gunter T.E., Buntinas L., Sparagna G., Eliseev R., Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 2000;28(5-6):285–296. doi: 10.1054/ceca.2000.0168. [http://dx.doi.org/10.1054/ceca. 2000.0168]. [PMID: 11115368]. [DOI] [PubMed] [Google Scholar]
- 85.Toescu E.C., Verkhratsky A. Assessment of mitochondrial polarization status in living cells based on analysis of the spatial heterogeneity of rhodamine 123 fluorescence staining. Pflugers Arch. 2000;440(6):941–947. doi: 10.1007/s004240000390. [http://dx.doi.org/10.1007/ s004240000390]. [PMID: 11041562]. [DOI] [PubMed] [Google Scholar]
- 86.Xiong J., Verkhratsky A., Toescu E.C. Changes in mitochondrial status associated with altered Ca2+ homeostasis in aged cerebellar granule neurons in brain slices. J. Neurosci. 2002;22(24):10761–10771. doi: 10.1523/JNEUROSCI.22-24-10761.2002. [PMID: 12486169]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y.C., Zhai X.Y., Ohsato K., Futamata H., Shimada O., Atsumi S. Mitochondrial accumulation in the distal part of the initial segment of chicken spinal motoneurons. Brain Res. 2004;1026(2):235–243. doi: 10.1016/j.brainres.2004.08.016. [http://dx.doi.org/10.1016/j.brainres.2004. 08.016]. [PMID: 15488485]. [DOI] [PubMed] [Google Scholar]