

Abstract
AIM
To identify and characterize the protective effect that L-carnitine exerted against an oxidative stress in C2C12 cells.
METHODS
Myoblastic C2C12 cells were treated with menadione, a vitamin K analog that engenders oxidative stress, and the protective effect of L-carnitine (a nutrient involved in fatty acid metabolism and the control of the oxidative process), was assessed by monitoring various parameters related to the oxidative stress, autophagy and cell death.
RESULTS
Associated with its physiological function, a muscle cell metabolism is highly dependent on oxygen and may produce reactive oxygen species (ROS), especially under pathological conditions. High levels of ROS are known to induce injuries in cell structure as they interact at many levels in cell function. In C2C12 cells, a treatment with menadione induced a loss of transmembrane mitochondrial potential, an increase in mitochondrial production of ROS; it also induces autophagy and was able to provoke cell death. Pre-treatment of the cells with L-carnitine reduced ROS production, diminished autophagy and protected C2C12 cells against menadione-induced deleterious effects.
CONCLUSION
In conclusion, L-carnitine limits the oxidative stress in these cells and prevents cell death.
Keywords: Superoxide anions, Mitochondria, Reactive Autophagy, Muscle, Carnitine, Oxygen species, Cell death
Core tip: The overall goal of this study was to identify and characterize the protective effect of L-carnitine on oxidative stress in muscle cells. We, first, induced an oxidative stress in cultured muscle cells and parameters associated with the stress were measured. In another set of experiments, cells were treated with L-carnitine and the same parameters measured. The comparison among the data allowed showing that L-carnitine was able to fight against oxidative stress and dramatically limit cell death. In conclusion, our results show that, at least in muscle cells, L-carnitine can be considered as a non-conventional antioxidant.
INTRODUCTION
During oxidative stress, a massive production of reactive oxygen species (ROS) induces alterations in cell structure. Oxidative stress may play a role in the onset of a wide variety of disorders[1] and participate at the ageing process[2]. ROS are able to interact with proteins, lipids and DNA leading to the formation of adducts accompanied with a loss of function[3]. These deleterious effects are normally kept under control by antioxidants. Most antioxidant systems are composed of an enzyme (e.g., superoxide dismutase, catalase, glutathione peroxidase[4]) usually under the control of a nutrient (vitamins or minerals); but besides these “classical” systems, several compounds have been shown to regulate metabolic activity inside the cells preventing the overproduction of ROS. These molecules are not strictly antioxidants but are able to reduce ROS production. Melatonin[5], creatine[6], coenzyme Q10[7] and carnitine[8] belong to this family.
An adult body contains around 25 g of L-carnitine[9]. L-Carnitine found in a human body is either derived from food stuff, especially from meat and dairy products[10], or derived from an endogenous synthesis. In mammals, L-carnitine is mainly synthesized in the liver, the testis and the kidney, this biosynthesis requires lysine and methionine as ultimate precursors and five enzymatic reactions[11,12].
Dietary or biosynthetic carnitine is excreted into the blood stream (at a concentration of 50-100 μmol/L) and distributed to organs and tissues depending on L-carnitine. Among these, muscle concentrates most of the L-carnitine: In a human body, around 98% of all the L-carnitine is found in skeletal and heart muscles and the ratio between plasma and muscle carnitine concentration is around 1:50[13]. None of these organs is able to synthesize L-carnitine and this molecule has to be imported from the blood stream using specific transporters[14].
The primary role of carnitine is to permit the transport of long chain fatty acids into mitochondria where they can enter the β-oxidation pathway. L-carnitine is also a cofactor for peroxisomal enzymes[15] and recognized as an inhibitor for HDAC[16]. L-carnitine has also been used as a protective agent against neurotoxic agents. The precise mechanisms involved are still to be clearly identified and several hypotheses remain. L-carnitine may exert its protective effects as a regulatory element in energy production and/or by interacting with the production of free radicals[17].
ROS production is associated with several muscle diseases[18] and it is clear that improving free radical metabolism could be beneficial to cells, animals or patients with muscle disorders[19]. The goal of this study was to evaluate the beneficial effect that L-carnitine treatment may exert on muscle cells undergoing an oxidative stress.
MATERIALS AND METHODS
Chemicals
Culture media were purchased from Lonza (Levallois-Perret, France). L-Carnitine (carnipure™) was a generous gift from Lonza. Probes used for cytometry analyses were purchased from Molecular probes (Cergy-Pontoise, France). All others chemicals were obtained from Sigma Aldrich (St Quentin Fallavier, France).
Cell culture
Murine myoblastic C2C12 cells were obtained from the European Collection of Cell Cultures (ECACC, Salisbury United Kingdom) and cultured according to their recommendations. Cells were grown in DMEM (Gibco, Cergy-Pontoise, France) with 10% heat-inactivated fetal calf serum (FCS; Gibco), 4 mmol/L L-glutamine (Gibco) at 37 °C in a humidified atmosphere with 5% CO2. Cells were trypsinized when they reached semi confluence. Dishes were seeded at a density of 2000 cells per square centimetre.
Treatments
Menadione was dissolved in 70% ethanol and added to culture medium (without phenol red) at the desired concentration (from 0 to 12 μmol/L); these varying concentrations were used as they may induce from moderate to severe oxidative stress[20]. Cells were treated for 1 to 24 h for all measured parameters. L-carnitine treated cells were cultured with L-carnitine at the final concentration of 500 μmol/L, at all the stages of the experiment. L-carnitine was dissolved in DPBS, filtrated on a 22 μm filter and added to the culture medium. This concentration was chosen as it remains in the physiological range (the extracellular concentration in carnitine is estimated to be in the 50-100 μmol/L range and in the muscle cells this concentration is at least 20 times higher) and as preliminary data showed that this concentration was efficient for limiting oxidative stress.
Flow cytometric analyses
All experiments were carried out with a Becton Dickinson Facscan equipped with a 488 nm Laser, and fluorescence signals were measured on three channels: FL1 (530/30 nm), FL2 (585/42 nm) and FL3 (670 nm LP). Data were acquired with the Cellquest software (BD Biosciences, Le Pont De Claix, France) and analyzed with the Cyflogic Software (http://www.cyflogic.com).
Propidium Iodide assay
After treatment with menadione and/or L-carnitine, the culture medium was removed and stored in a 15 mL tube; cells were rinsed with DPBS and trypsinized. The culture medium, the DPBS used for washing and the trypsinized cells were collected and centrifuged at 600 g for 3 min. The supernatant was discarded and the cells resuspended in DPBS. Propidium iodide was added to the cell suspension at a final concentration of 5 μg/mL. After a 5 min incubation, cells were analyzed with a flow cytometer[21]. FL3 fluorescence was recorded for 10000 cells. Lethal dose 50 was determined with the BioStat 2009 software (AnalystSoft) as described in[22].
Identification of LC3-II
LC3 has been used as a marker for autophagy. Cells treated with either L-carnitine, menadione (as before) and 3methyadenine (final concentration of 5 mmol/L) were harvested, washed twice in ice-cold PBS and resuspended in a RIPA solution (Tris-HCl, 20 mmol/L pH 8.0; 150 mmol/L NaCl, 2 mmol/L Na2EDTA; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS, 5 mmol/L sodium fluoride) containing protease inhibitors (0.1 mmol/L phenylmethylsulfonyl fluoride, 5 μg/mL aprotinin, 5 μg/mL pepstatin A and 1 μg/mL chymostatin). The lysate was spun at 13000 g for 20 min at 4 °C. Protein concentration was determined using the Bradford assay. Proteins (20 μg) were separated on a 15% SDS-PAGE and transferred onto a PVDF membrane. The membrane was first incubated with antibodies directed against LC3 (Sigma L8918; 1:2000) and then with secondary antibodies (Santa Cruz, France) coupled with HRP (1:5000). Immunoreactive proteins were visualized on a Biorad Imager and bands size and density were measured using the Image Lab Software. The data were normalized against actin as an internal control.
Tbars production
C2C12 cells (106 cells) were harvested and homogeneized in a Tris-HCl buffer (150 mmol/L, pH 7.1) and Tbars production determined as in[23]. The results were expressed as nmoles of MDA produced per mg of proteins. Protein amount was determined using the Bicinchoninic Acid Protein Assay (Pierce, France) with BSA as a standard.
Flow cytometric measurement of the superoxide production in the mitochondria with the MitoSox Red dye
MitoSoxRed labeling was carried out according as recommended by the manufacturer (Molecular Probes). Cells were harvested as above and resuspended in fresh medium. The reaction was carried out for 15 min at 37 °C in the dark with a final concentration of 5 μmol/L of MitoSox Red[24]. The cells were then centrifuged at 600 × g for 3 min and resuspended in DPBS. Analysis was performed using a Facscan cytometer (BD - Bioscience), using 10000 cells and fluorescence was recorded with the FL3 channel.
Flow cytometric measurement of the mitochondrial transmembrane potential (Δψm) using the 3,3’-dihexyloxacarbocyanine iodide dye
After treatment, the cells were harvested, the culture medium was removed and kept in a 15 mL tube; the cells were washed with DPBS and trypsinized. The culture medium, washing DPBS and trypsinized cells were pooled and centrifuged at 600 g during 3 min. The cell pellet was resuspended in fresh medium. dihexyloxacarbocyanine iodide [DiOC6(3)] labeling was carried out according to Molecular Probes recommendations; briefly cells were incubated 20 min at 37 °C with 20 nmol/L of DiOC6(3), cells were then centrifuged at 600 × g for 3 min then resuspended in DPBS. The analysis was performed using a Facscan cytometer, counting 10000 cells and recording fluorescence with the FL1 channel.
Cellular organization
Cells were grown on Labtek slides and treated as described above. After treatment, cells were labeled with MitoSox Red[25] and Hoechst 33342 (final concentration of 10 μg/mL). After staining, cells were fixed with 4% formaldehyde in DPBS during 1 h. Cells were mounted in Dakocytomation fluorescence medium (Dako, Copenhagen, Denmark) and after solidification of the mounting medium, cells were observed with a LSM confocal microscope (Zeiss) in plane mode with an EC Plan-Neofluar objective (40 ×/1.30 Oil DIC M27).
Statistical analysis
Results are expressed as the mean ± SD. The statistical significance of differences between treatments was determined with KaleidaGraph (Synergy software) using the ANOVA test with Dunnett Post Hoc test.
RESULTS
Menadione alters mitochondrial membrane potential in C2C12 cells, L-carnitine restores mitochondrial membrane potential
The effect of menadione on mitochondrial integrity was estimated by monitoring potential variations using the potential sensitive fluorochrome DiOC6(3). In the absence of menadione, no effect of L-carnitine was observed. In the presence of menadione, a significant decrease in the mitochondrial transmembrane potential was observed (Figure 1). This decrease appeared to be dose-dependent: While 77% of the cells were able to accumulate the fluorochrome in the absence of menadione, only 51% were concentrating the Dioc in the presence of 6 μmol/L of menadione and the percentage decreased to respectively 8% and 6% at concentrations of menadione of 9 and 12 μmol/L. L-carnitine treatment appeared to prevent menadione-induced loss of mitochondrial transmembrane potential. As shown on Figure 1, the number of cells able to preserve their mitochondrial transmembrane potential was much higher among the L-carnitine treated cells than among untreated cells. At 9 μmol/L of menadione and in the presence of L-carnitine, 70% of the cells exhibited fluorescence. At 12 μmol/L of menadione and in the presence of carnitine, the percentage of mitochondrial integrity was much lower than in control cells, but was still higher than in menadione-only treated cells.
Figure 1.
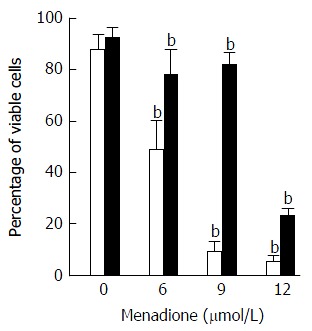
Protection against menadione-induced mitochondrial depolarization with L-carnitine. C2C12 cells were either untreated (white histogram) or pretreated with 500 μmol/L of L-carnitine (dark histogram) and incubated for 24 h with desired concentration of menadione (from 0 to 12 μmol/L). Mitochondria integrity was evaluated using DiOC6(3) probe staining; fluorescent cells were counted using a cytometer. Results are presented as the mean value ± SD. Two different values (P < 0.005) are indicated by a star above the two histograms.
Mitochondrial morphology is altered by menadione
To evaluate intracellular organization of the nucleus and the mitochondria, cells were stained with Hoechst 33342 and MitosoxRed. Cells mounted in fluorescence medium were observed with a LSM confocal microscope in plane mode (Figure 2). Control cells exhibited well identified mitochondria, with a homogeneous repartition in the cytoplasm; the same organization was observed in cells treated with L-carnitine. After treatment with 9 μmol/L of menadione during 24 h, the mitochondrial network appeared damaged with most of the mitochondria located around the nucleus. Such modifications were not observed on cells simultaneously treated with 500 μmol/L L-carnitine and with 9 μmol/L menadione for 24 h. In these conditions, mitochondria and cell structure were similar to those of control cells.
Figure 2.
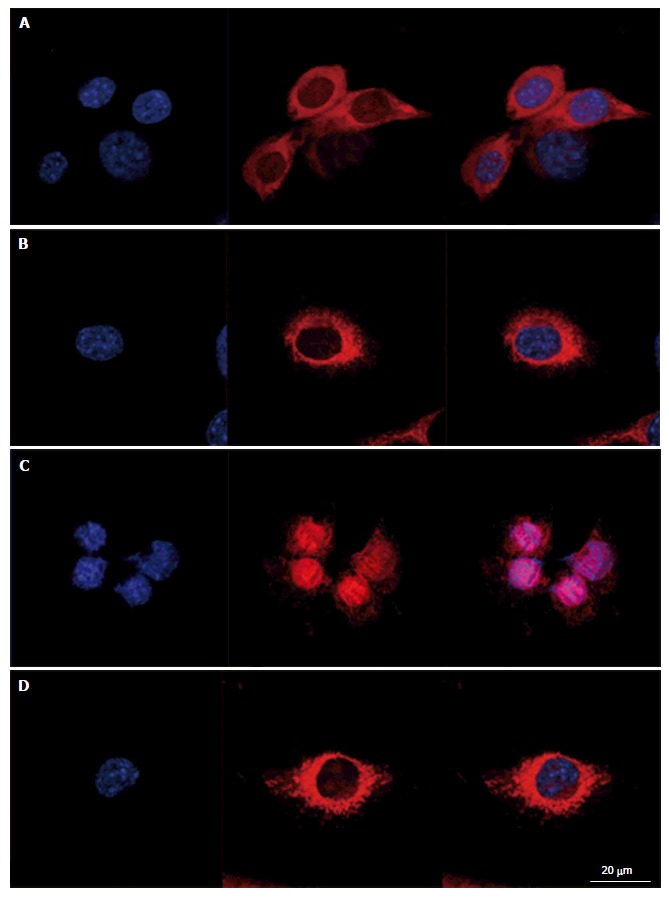
Prevention of menadione-induced mitochondrial distribution with L-carnitine. C2C12 cells were either untreated or pretreated with 500 μmol/L of L-carnitine and incubated for 24 h with menadione (0 and 9 μmol/L). Nucleus and mitochondria morphology was evaluated after staining with Hoechst 33342 and MitoSoxRed, respectively. From left to right, staining with nuclei, mitochondria and both. Cells mounted in fluorescence medium were observed with a LSM confocal microscope. A: C2C12 untreated with menadione and untreated with L-carnitine; B: C2C12 pretreated with L-carnitine and untreated with menadione; C: C2C12 untreated with L-carnitine and treated with 9 μmol/L of menadione; D: C2C12 pretreated with L-carnitine and treated with 9 μmol/L of menadione.
L-carnitine prevents menadione-induced free radical generation in the mitochondria
The global ROS production was evaluated by the measure of thiobarbituric reactive species. Tbars production in C2C12 cells was determined at intervals from 0 to 24 h after treatment with four different concentrations of menadione (0 μmol/L: Figure 3A; 6 μmol/L: Figure 3B; 9 μmol/L: Figure 3C and 12 μmol/L: Figure 3D). In the absence of menadione, no effect on Tbars production was observed and L-carnitine supplementation remained without effect. In the presence of 6 μmol/L of menadione, Tbars production increased after 6 h of treatment, and was found to be maximal after 8 h of treatment. L-carnitine supplementation fully inhibited this increase and no differences were found among L-carnitine treated cells. With a treatment of 9 μmol/L of menadione, Tbars production was increased earlier than before and a significant difference was observed after 2 h of treatment. The effect of menadione was maximal after 4 h of treatment. Again, L-carnitine addition fully abolished the effect of menadione and in the presence of L-carnitine, no increase in Tbars production was observed. In the presence of 12 μmol/L of menadione, the increase in Tbars production was rapid and appeared to be maximal after 2 h of treatment. L-carnitine supplementation was able to prevent this increase, even if one can observe a slight increase after 24 h of treatment (Figure 3A).
Figure 3.
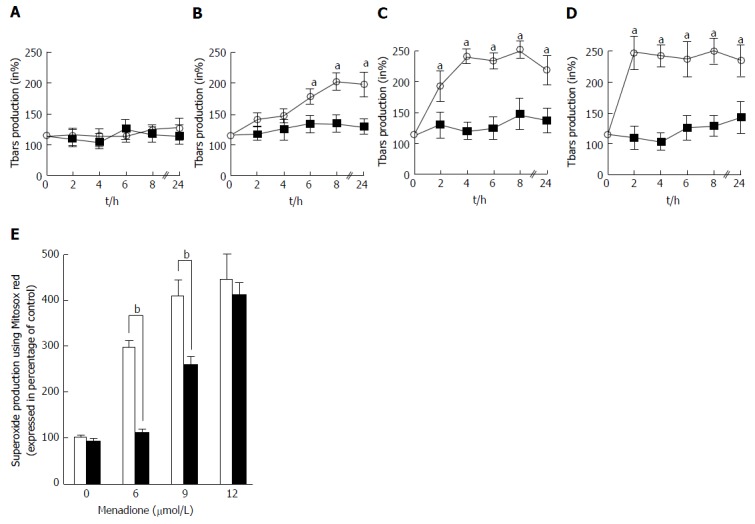
Characterization of reactive oxygen species production. A-D: Tbars production was determined in C2C12 cells in the presence of various amounts of menadione from 1 to 24 h. Results were expressed in percentage of the control cell Tbars production. Tbars production was analyzed in the presence of 0 (A), 6 (B), 9 (C) and 12 μmol/L (D) of menadione in control cells (empty circles and dashed line) and in cells pre-treated with 500 μmol/L of carnitine (black squares and full line). An asterisk on top of a symbol indicates a significant difference (P < 0.05); E: Superoxide anion production at the mitochondrial level on menadione-treated C2C12 cells with MitoSoxRed. C2C12 cells were either untreated (white histogram) or pretreated with 500 μmol/L of L-carnitine (black histogram) and incubated for 24 h with desired concentration of menadione (from 0 to 12 μmol/L). MitoSoxRed (MSR) is a cell permeable dye that is targeted to the mitochondria, it is oxidized by superoxide and exhibit red fluorescence. Superoxide production was measured by cytometry after staining with MSR. Results are presented as the mean value ± SD. A star on the top of two histograms indicates statistical difference (P < 0.005) between these two values (Untreated vs L-carnitine treated cells).
The mitochondrial generation of ROS was evaluated by analyzing C2C12 cells stained with MitosoxRed by flow cytometry. Carnitine treatment did not alter the basal production of mitochondrial ROS. Menadione treatment was found to induce an increase in the production of ROS in the mitochondria in a dose dependent manner. At a concentration of 6 μmol/L of menadione, a significant increase in the number of cells producing ROS was observed (Figure 3B) and when the concentration of menadione reached 9 μmol/L, more than 90% of the cells produced ROS. L-carnitine treatment was able to decrease the mitochondrial production of ROS for concentrations of menadione less than 12 μmol/L.
L-carnitine limits the autophagy process induced by menadione in C2C12 cells
Microtubule-associated protein light chain 3 (LC3) is considered as one of the more accurate markers for autophagy[26]. During the initiation of autophagy, a cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine leading to the formation of LC3-II[27]. Due to its hydrophobic property, the PE group increases the migration of the LC3 protein in a SDS-PAGE gel. We examined the changes in endogenous LC3 after menadione and carnitine treatments in C2C12 cells.
Menadione significantly increased the level of LC3-II proteins in a dose-dependent manner (Figure 4). L-carnitine treatment did not modify autophagy in control cells (i.e., untreated with menadione). At the concentrations of 6 and 9 μmol/L of menadione, adding carnitine decreases the level of LC3-II significantly suggesting a protective effect of carnitine on autophagy. At the concentration of 12 μmol/L of menadione, L-carnitine supplementation remained ineffective in reducing autophagy. At this concentration, the level of LC3-II was found to be the same between L-carnitine treated and untreated cells.
Figure 4.
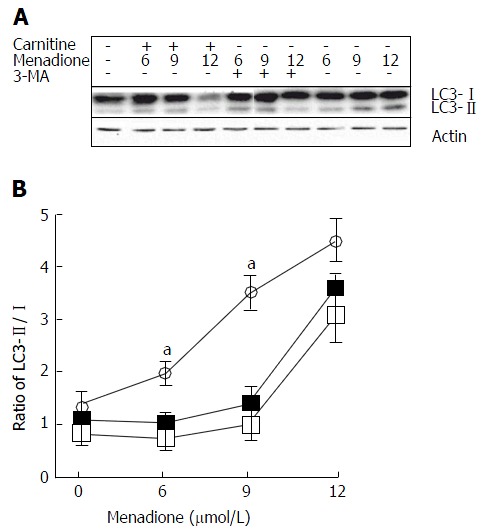
Menadione induces autophagy in C2C12 cells. A: Western blot for the determination of LC3 I and II forms. Initial cells were either treated with L-carnitine, menadione or 3-MA; B: LC3-I and II isoforms were detected by western blotting and quantified. The ratio between I and II forms was calculated and plotted in this figure. The black squares represent L-carnitine treated cells whereas empty circles represent C2C12 cells that were not pretreated with L-carnitine. Empty squares represent cells treated with 5 mmol/L of 3-MA. Each point is the mean of 4 independent experiments (± standard values). For each concentration of menadione, a star indicates a significant difference between untreated and L-carnitine treated cells (P < 0.05).
Menadione induces cell death in C2C12 cells, L-carnitine has a protective effect
Cell death was evaluated by flow cytometry by measuring membrane permeability to propidium iodide (PI) (Figure 5). Treatment with menadione induced a significant decrease in cell survival. At a concentration of 6 μmol/L of menadione, only 51% of the cells remain unstained by PI. The cell survival decreased as the menadione concentration increased.
Figure 5.
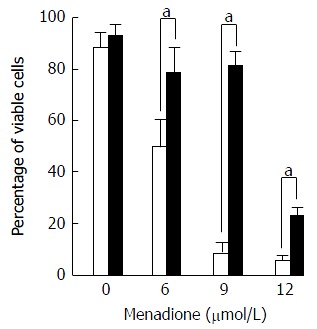
Protection of menadione-induced C2C12 cell death by L-carnitine. C2C12 cells were either untreated (white histogram) or pretreated with 500 μmol/L of L-carnitine (dark histogram) and incubated for 24 h with desired concentration of menadione (from 0 to 12 μmol/L). Cell survival was evaluated by the analysis of membrane permeability to PI measured by flow cytometry. Each value represents the average percentage of viable cells remaining in the total population ± standard values. Black histograms indicate the proportion (%) of C2C12 that was not PI positive. White histograms indicate the proportion (%) of C2C12 pretreated with L-carnitine that was not PI positive. A star on top of two histograms indicates a significant difference between untreated and L-carnitine treated cells (P < 0.05).
Noteworthy, cells treated with L-carnitine were less sensitive to menadione. Whatever the concentration of menadione was, the number of viable cells was always higher with L-carnitine treatment. At a concentration of 9 μmol/L of menadione, only 5% of C2C12 cells cultured in the absence of L-carnitine remained unstained by PI and in the presence of L-carnitine, 79% were unstained. In the presence of 12 μmol/L of menadione, 23% of cells treated with L-carnitine were viable and this percentage was only of 5 % in cells untreated with L-carnitine.
DISCUSSION
Due to the high rate of oxidative processes and the abundance of myoglobin, muscle cells are very susceptible to ROS damage which are responsible of various types of damages that can lead to cell death or to cell ineffectiveness[28]. It has been shown that ROS are implicated in the normal aging process but also in pathological processes. Muscle diseases like Duchenne muscular dystrophy make muscle cells more susceptible to ROS damages[29,30], leading to premature death or malfunction. Menadione (2-methyl-1, 4-naphthoquinone, vitamin K3) is a quinone-containing compound. Reductive enzymes such as microsomal NADPH-cytochrome P450 reductase and mitochondrial NADH-ubiquinone oxidoreductase are able to metabolize menadione with the subsequent formation of unstable semiquinones. These molecules can enter into a redox cycle with molecular oxygen leading to the formation of quinone and the generation of reactive oxygen species, including H2O2, O2-• and, in the presence of metal ions, to OH•[31]. Menadione is widely used to study the oxidative stress on various types of cells[29,31,32].
In this study, we showed that menadione induced an oxidative stress in C2C12 cells leading to cell death. The lethal dose 50 was calculated and found to be 6 μmol/L. In addition, menadione induced a significant increase in ROS generation in the mitochondria; at a dose of 6 μmol/L of menadione, more than 67% of the cells were over producing ROS. Menadione was also able to induce autophagy in C2C12 cells. The oxidative stress is also associated with a rearrangement in mitochondrial distribution inside the muscle cell. This aspect is also encountered in several diseases associated with a loss of myofibrillar organization (e.g., cells lacking desmin[33] or plectin[34]). It is very likely that the change in mitochondrial distribution impacts cell function.
The data presented in this paper are partly in agreement with those of Chiou et al[32] who described that menadione (used on C2C12 in a culture medium without FCS) was able to induce massive cell death. Privation of serum for C2C12 cells is known to initiate differentiation and deeply changes the phenotype of the cells[35]. In our experimental conditions, L-carnitine was added before the induction of the oxidative stress by menadione and appeared to protect C2C12 cells against ROS damages. For any concentration of menadione, L-carnitine treatment protected cells against menadione toxicity. This phenomenon is observed at all tested menadione concentrations and while the LD50 of untreated C2C12 was 6 μmol/L, in the presence of L-carnitine, the LD50 was calculated at 11 μmol/L. In C2C12 cells, L-carnitine supplementation was able to diminish cell death but also to reduce ROS production and to protect against mitochondrial transmembrane depolarization.
Currently, it is known that L-carnitine is a cofactor in the channeling of fatty acids inside the cell. It is involved in fatty acid oxidation by playing a role of cofactor in the transport of acyl groups across the mitochondrial membrane[36]. Beside its role in fatty acid oxidation, L-carnitine, added to the food, has been shown to counteract some effects associated with aging on mitochondria and on muscle[37-39]. Moreover, L-carnitine has been shown to prevent oxidative stress, to regulate nitric oxide production and to control the activity of enzymes involved in the defense against oxidative damage[40] and mitochondrial dysfunction[41]. Among the enzymes whose activity is protected by L-carnitine are found the catalase and the superoxide dismutase[42], two of three major enzymes involved in ROS detoxification. In this study we were able to prove that L-carnitine may act on the intracellular ROS status either by decreasing mitochondrial ROS production, either by increasing defense against these reactive molecules produced in mitochondria, or either by scavenging ROS at the mitochondrial level.
ACKNOWLEDGMENTS
The authors wish to thank the Association Française contre les Myopathies for financial support.
COMMENTS
Background
By controlling metabolism, L-carnitine seems to be able to limit reactive oxygen species (ROS) production under certain circumstances. In this paper, the protective effect of L-carnitine was assessed on muscle cells. L-carnitine appeared to limit ROS production in muscle cells and protecting them against oxidative stress.
Research frontiers
Muscle is a tissue with a high concentration of mitochondria and likely to produce high level of ROS. ROS may then induce oxidative stress and damage to muscle cells. Protecting muscle cells against oxidative stress was the authors’ objective.
Applications
Limiting oxidative stress may find many applications, especially in limiting damage to cells, organs and tissues.
Terminology
Oxidative stress theory: In this theory, highly reactive molecules, related to oxygen, interact and finally alter some vital molecules, leading to disease and sometimes to death. Carnitine: A small molecule that is involved in the metabolism of fat. Autophagy: Autophagy is a cellular mechanism consisting in the partial degradation of the cell cytoplasm by its own lysosomes. Depending on the circumstances, autophagy may lead to a large spectrum of effects: From cell repair to cell death.
Peer-review
The manuscript is interesting. The main objective study was to evaluate the beneficial effect that L-carnitine treatment may exert on muscle cells undergoing an oxidative stress.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Biochemistry and molecular biology
Country of origin: France
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: Not relevant no human or animal subjects were used in these experiments.
Institutional animal care and use committee statement: Not relevant no animal were used in these experiments.
Conflict-of-interest statement: Each author declares no conflict of interest.
Data sharing statement: All data are available on request.
Peer-review started: August 5, 2016
First decision: September 2, 2016
Article in press: December 14, 2016
P- Reviewer: Cui YP, Freire-De-Lima CG S- Editor: Song XX L- Editor: A E- Editor: Li D
References
- 1.Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol (1985) 2007;102:1677–1686. doi: 10.1152/japplphysiol.01145.2006. [DOI] [PubMed] [Google Scholar]
- 2.Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid Med Cell Longev. 2016;2016:3565127. doi: 10.1155/2016/3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y. Bulky DNA lesions induced by reactive oxygen species. Chem Res Toxicol. 2008;21:276–281. doi: 10.1021/tx700411g. [DOI] [PubMed] [Google Scholar]
- 4.Kahl R, Kampkötter A, Wätjen W, Chovolou Y. Antioxidant enzymes and apoptosis. Drug Metab Rev. 2004;36:747–62. doi: 10.1081/dmr-200033488. [DOI] [PubMed] [Google Scholar]
- 5.Hibaoui Y, Roulet E, Ruegg UT. Melatonin prevents oxidative stress-mediated mitochondrial permeability transition and death in skeletal muscle cells. J Pineal Res. 2009;47:238–252. doi: 10.1111/j.1600-079X.2009.00707.x. [DOI] [PubMed] [Google Scholar]
- 6.Grundman M, Grundman M, Delaney P. Antioxidant strategies for Alzheimer's disease. Proc Nutr Soc. 2002;61:191–202. doi: 10.1079/PNS2002146. [DOI] [PubMed] [Google Scholar]
- 7.Littarru GP, Tiano L. Bioenergetic and antioxidant properties of coenzyme Q10: recent developments. Mol Biotechnol. 2007;37:31–37. doi: 10.1007/s12033-007-0052-y. [DOI] [PubMed] [Google Scholar]
- 8.Block KI, Koch AC, Mead MN, Tothy PK, Newman RA, Gyllenhaal C. Impact of antioxidant supplementation on chemotherapeutic toxicity: a systematic review of the evidence from randomized controlled trials. Int J Cancer. 2008;123:1227–1239. doi: 10.1002/ijc.23754. [DOI] [PubMed] [Google Scholar]
- 9.Demarquoy J. In: eLS. John Wiley & Sons Ltd, Chichester; 2011. L-carnitine: Structure and Function. Available from: http://www.els.net. [Google Scholar]
- 10.Demarquoy J, Georges B, Rigault C, Royer M, Clairet A, Soty M, Lekounougou S, Le Borgne F. Radioisotopic determination of -carnitine content in foods commonly eaten in Western countries. Food Chemistry. 2004;86:137–142. [Google Scholar]
- 11.Rigault C, Le Borgne F, Demarquoy J. Genomic structure, alternative maturation and tissue expression of the human BBOX1 gene. Biochim Biophys Acta. 2006;1761:1469–1481. doi: 10.1016/j.bbalip.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Rigault C, Le Borgne F, Tazir B, Benani A, Demarquoy J. A high-fat diet increases L-carnitine synthesis through a differential maturation of the Bbox1 mRNAs. Biochim Biophys Acta. 2013;1831:370–377. doi: 10.1016/j.bbalip.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Siliprandi N, Sartorelli L, Ciman M, Di Lisa F. Carnitine: metabolism and clinical chemistry. Clin Chim Acta. 1989;183:3–11. doi: 10.1016/0009-8981(89)90267-2. [DOI] [PubMed] [Google Scholar]
- 14.Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142:77–85. doi: 10.1002/ajmg.c.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Borgne F, Ben Mohamed A, Logerot M, Garnier E, Demarquoy J. Changes in carnitine octanoyltransferase activity induce alteration in fatty acid metabolism. Biochem Biophys Res Commun. 2011;409:699–704. doi: 10.1016/j.bbrc.2011.05.068. [DOI] [PubMed] [Google Scholar]
- 16.Huang H, Liu N, Guo H, Liao S, Li X, Yang C, Liu S, Song W, Liu C, Guan L, et al. L-carnitine is an endogenous HDAC inhibitor selectively inhibiting cancer cell growth in vivo and in vitro. PLoS One. 2012;7:e49062. doi: 10.1371/journal.pone.0049062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiMauro S, Hirano M, Schon EA. Approaches to the treatment of mitochondrial diseases. Muscle Nerve. 2006;34:265–283. doi: 10.1002/mus.20598. [DOI] [PubMed] [Google Scholar]
- 18.Westerblad H, Allen DG. Emerging roles of ROS/RNS in muscle function and fatigue. Antioxid Redox Signal. 2011;15:2487–2499. doi: 10.1089/ars.2011.3909. [DOI] [PubMed] [Google Scholar]
- 19.Zuo L, Pannell BK2. Redox Characterization of Functioning Skeletal Muscle. Front Physiol. 2015;6:338. doi: 10.3389/fphys.2015.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comporti M. Three models of free radical-induced cell injury. Chem Biol Interact. 1989;72:1–56. doi: 10.1016/0009-2797(89)90016-1. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Mizumoto K, Sato N, Ogawa T, Kusumoto M, Niiyama H, Tanaka M. Quantitative determination of apoptotic death in cultured human pancreatic cancer cells by propidium iodide and digitonin. Cancer Lett. 1999;142:129–137. doi: 10.1016/s0304-3835(99)00107-x. [DOI] [PubMed] [Google Scholar]
- 22.Rosiello AP, Essignmann JM, Wogan GN. Rapid and accurate determination of the median lethal dose (LD50) and its error with a small computer. J Toxicol Environ Health. 1977;3:797–809. doi: 10.1080/15287397709529614. [DOI] [PubMed] [Google Scholar]
- 23.Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- 24.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3:941–947. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- 25.Marella M, Seo BB, Matsuno-Yagi A, Yagi T. Mechanism of cell death caused by complex I defects in a rat dopaminergic cell line. J Biol Chem. 2007;282:24146–24156. doi: 10.1074/jbc.M701819200. [DOI] [PubMed] [Google Scholar]
- 26.Mancias JD, Kimmelman AC. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J Mol Biol. 2016;428:1659–1680. doi: 10.1016/j.jmb.2016.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 28.Mattiasson G. Analysis of mitochondrial generation and release of reactive oxygen species. Cytometry A. 2004;62:89–96. doi: 10.1002/cyto.a.20089. [DOI] [PubMed] [Google Scholar]
- 29.Rando TA, Crowley RS, Carlson EJ, Epstein CJ, Mohapatra PK. Overexpression of copper/zinc superoxide dismutase: a novel cause of murine muscular dystrophy. Ann Neurol. 1998;44:381–386. doi: 10.1002/ana.410440315. [DOI] [PubMed] [Google Scholar]
- 30.Disatnik MH, Dhawan J, Yu Y, Beal MF, Whirl MM, Franco AA, Rando TA. Evidence of oxidative stress in mdx mouse muscle: studies of the pre-necrotic state. J Neurol Sci. 1998;161:77–84. doi: 10.1016/s0022-510x(98)00258-5. [DOI] [PubMed] [Google Scholar]
- 31.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 32.Chiou TJ, Chu ST, Tzeng WF. Protection of cells from menadione-induced apoptosis by inhibition of lipid peroxidation. Toxicology. 2003;191:77–88. doi: 10.1016/s0300-483x(03)00189-6. [DOI] [PubMed] [Google Scholar]
- 33.Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–1298. doi: 10.1083/jcb.150.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winter L, Kuznetsov AV, Grimm M, Zeöld A, Fischer I, Wiche G. Plectin isoform P1b and P1d deficiencies differentially affect mitochondrial morphology and function in skeletal muscle. Hum Mol Genet. 2015;24:4530–4544. doi: 10.1093/hmg/ddv184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blau HM, Pavlath GK, Hardeman EC, Chiu CP, Silberstein L, Webster SG, Miller SC, Webster C. Plasticity of the differentiated state. Science. 1985;230:758–766. doi: 10.1126/science.2414846. [DOI] [PubMed] [Google Scholar]
- 36.Zammit VA, Ramsay RR, Bonomini M, Arduini A. Carnitine, mitochondrial function and therapy. Adv Drug Deliv Rev. 2009;61:1353–1362. doi: 10.1016/j.addr.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 37.Kumaran S, Subathra M, Balu M, Panneerselvam C. Supplementation of L-carnitine improves mitochondrial enzymes in heart and skeletal muscle of aged rats. Exp Aging Res. 2005;31:55–67. doi: 10.1080/03610730590882846. [DOI] [PubMed] [Google Scholar]
- 38.Le Borgne F, Guyot S, Logerot M, Beney L, Gervais P, Demarquoy J. Exploration of lipid metabolism in relation with plasma membrane properties of Duchenne muscular dystrophy cells: influence of L-carnitine. PLoS One. 2012;7:e49346. doi: 10.1371/journal.pone.0049346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernard A, Rigault C, Mazue F, Le Borgne F, Demarquoy J. L-carnitine supplementation and physical exercise restore age-associated decline in some mitochondrial functions in the rat. J Gerontol A Biol Sci Med Sci. 2008;63:1027–1033. doi: 10.1093/gerona/63.10.1027. [DOI] [PubMed] [Google Scholar]
- 40.Kremser K, Stangl H, Pahan K, Singh I. Nitric oxide regulates peroxisomal enzyme activities. Eur J Clin Chem Clin Biochem. 1995;33:763–774. doi: 10.1515/cclm.1995.33.11.763. [DOI] [PubMed] [Google Scholar]
- 41.Lohrke B, Xu J, Weitzel JM, Kruger B, Goldammer T, Viergutz T. N-acetylcysteine impairs survival of luteal cells through mitochondrial dysfunction. Cytometry A. 2010;77:310–320. doi: 10.1002/cyto.a.20873. [DOI] [PubMed] [Google Scholar]
- 42.Binienda ZK, Ali SF. Neuroprotective role of L-carnitine in the 3-nitropropionic acid induced neurotoxicity. Toxicol Lett. 2001;125:67–73. doi: 10.1016/s0378-4274(01)00415-5. [DOI] [PubMed] [Google Scholar]