

Abstract
The polyketide synthases responsible for the biosynthesis of the polyether antibiotics nanchangmycin (1) and salinomycin (4) harbor a number of redox-inactive ketoreductase (KR0) domains that are implicated in the generation of C2-epimerized (2S)-2-methyl-3-ketoacyl-ACP intermediates. Evidence that the natural substrate for the polyether KR0 domains is, as predicted, a (2R)-2-methyl-3-ketoacyl-ACP intermediate, came from a newly developed coupled ketosynthase (KS)-ketoreductase (KR) assay that established that the decarboxylative condensation of methylmalonyl-CoA with S-propionyl-N-acetylcysteamine catalyzed by the Nan[KS1][AT1] didomain from module 1 of the nanchangmycin synthase generates exclusively the corresponding (2R)-2-methyl-3-ketopentanoyl-ACP (7a) product. In tandem equilibrium isotope exchange experiments, incubation of [2-2H]-(2R,3S)-2-methyl-3-hydroxypentanoyl-ACP (6a) with redox-active, epimerase-inactive EryKR6 from module 6 of the 6-deoxyerythronolide B synthase and catalytic quantities of NADP+ in the presence of redox-inactive, recombinant NanKR10 or NanKR50, from modules 1 and 5 of the nanchangmycin synthase, or recombinant SalKR70 from module 7 of the salinomycin synthase, resulted in first-order, time-dependent washout of deuterium from 6a. Control experiments confirmed that this washout was due to KR0-catalyzed isotope exchange of the reversibly-generated, transiently-formed oxidation product [2-2H]-(2R)-2-methyl-3-ketopentanoyl-ACP (7a), consistent with the proposed epimerase activity of each of the KR0 domains. Although they belong to the superfamily of short chain dehydrogenase-reductases, the epimerase-active KR0 domains from polyether synthases lack one or both residues of the conserved Tyr-Ser dyad that has previously been implicated in KR-catalyzed epimerizations.
INTRODUCTION
Polyether ionophores, represented by the widely used and intensively studied nanchangmycin (1, dianemycin), monensin (2), nigericin (3), and salinomycin (4) (Figure 1) are a large class of complex, branched chain polyketide carboxylic acids characterized by the presence of two or more tetrahydrofuran, tetrahydropyran, or acetal rings that serve as potent and selective ligands for monovalent or divalent metal cations. Polyethers disrupt physiological ion gradients by allowing diffusion of the complexed cations across cell membranes, thereby accounting for their antibacterial action and leading to their use as commercially important coccidiostats and veterinary growth promoters.
Figure 1.

Representative polyethers and domain organization of the PKS modules that generate epimerized α-methyl ketones, highlighted in red.
A representative biosynthetic pathway is illustrated in Scheme 1 for the prototypical polyether nanchangmycin (1). Extensive isotopic labeling, enzymatic, molecular genetic, and protein structural evidence supports a general mechanism for polyether biosynthesis in which a modular polyketide synthase (PKS) first generates the parent branched-chain, unsaturated polyketide carboxylic acid, containing all E double bonds, from a combination of malonyl-CoA and methylmalonyl-CoA building blocks.1-7 Stereospecific epoxidations by flavin-dependent monooxygenases generate the corresponding polyepoxide which then undergoes a cascade nucleophilic cyclization, catalyzed by one or more epoxide hydrolases, resulting in the formation of the core, acyl carrier protein-bound polyether. Additional late-stage modifications such as oxidation, glycosylation, and/or O-methylation, are followed by thioesterase-catalyzed release of the mature polyether antibiotic from the polyketide synthase.
Scheme 1.

Nanchangmycin Biosynthesis. NanA1-NanA11 correspond to the 14-module nanchangmycin synthase. NanO is an epoxidase, NanI is an epoxide hydrolase, NanP is a P450-dependent hydroxylase, NanG5 is a glycosylase, and NanE is the thioesterase that releases the mature polyether from the ACP. Epimerized α-methyl ketones are highlighted in red.
The biosynthesis of the parent ACP-bound, branched-chain, unsaturated polyketide carboxylic acid for each polyether is controlled by a modular polyketide synthase (PKS), similar in composition, organization, and function to the well-studied modular polyketide synthases of macrolide and polyene antibiotic biosynthesis, typified by the extensively characterized 6-deoxyerythronolide B synthase of erythromycin biosynthesis.5a,8-11 According to the canonical organization of the prototypical modular PKS, each module consists of a core set of three domains – an acyl carrier protein (ACP) to which the growing polyketide is tethered by thioester linker to the flexible phosphopantetheinyl prosthetic group, an acyl transferase (AT) that strictly selects the appropriate malonyl-CoA, methylmalonyl-CoA, or related chain extender and transfers it to the pantetheinyl arm of the ACP, and a ketosynthase (KS) domain that catalyzes the polyketide chain-building decarboxylative condensation between the ACP-bound malonyl- or methylmalonyl unit and the partially elaborated polyketide chain provided by the upstream PKS module. The EryKS domains of the 6-deoxyerythronolide B synthase have been shown to utilize exclusively the (2S)-methylmalonyl chain extender12 in a reaction that proceeds with net inversion of configuration to generate exclusively a (2R)-2-methyl-3-ketoacyl-ACP intermediate.13 Most PKS modules also harbor NADPH-dependent ketoreductase (KR) domains that strictly control the stereochemistry at both C-2 and C-3 of the reduced intermediates.13-15 In some cases, inherently redox-inactive KR0 domains, such as EryKR30 from module 3 of the 6-deoxyerythronolide B synthase, as well as the homologous PicKR30 from module 3 of the picromycin/narbonolide synthase, possess an intrinsic epimerase activity that reversibly converts the KS-generated (2R)-2-methyl-3-ketoacyl-ACP intermediate to its (2S)-2-methyl-3-ketoacyl-ACP diastereomer that then is stereoselectively recognized as the substrate for polyketide extension by the KS domain of the proximal downstream module.16,17 In certain modules, additional dehydratase (DH) and enoylreductase (ER) domains further modify the growing chain to give unsaturated or saturated acyl-ACP chain elongation products that are then passed to the proximal downstream PKS module for a further round of polyketide chain elongation and functional group modification.
We have previously reported that the fully reducing nanchangmycin synthase module 2 (NanA2), which harbors the complete set of polyketide chain-processing domains, is stereo-selective for chain translocation and reductive elongation of the NanACP1-bound diketide substrate, (2S)-2-methyl-3-ketobutyryl-ACP1, which is converted to the diastereomerically pure triketide (2S,4R)-2,4-dimethyl-5-ketohexanoyl-ACP2 in the presence of methylmalonyl-CoA and NADPH (Scheme 1).5d,e NanKS2 therefore carries out in effect a dynamic kinetic resolution of the diastereomeric mixture of 2-methyl-3-ketobutyryl-ACP1 intermediates.
Since the NanKS1 domain is expected to generate a (2R)-2-methyl-3-ketoacyl-ACP intermediate, by analogy to the known stereochemistry of the homologous EryKS domains, the resultant diketide chain elongation product must be epimerized prior to translocation and processing by Nan module 2 (NanA2, Scheme 1). In fact, nanchangmycin module 1 harbors an apparently redox-inactive ketoreductase domain, NanKR10, that is likely responsible for the reversible intercon-version of (2R)- and (2S)-2-methyl-3-ketoacyl-ACP intermediates. Immediately upstream of NanKR10 is a DH domain of uncertain function (Figure 1). Indeed, this pairing is not uncommon, as polyether PKS modules that generate nominally epimerized (2S)-2-methyl-3-ketoacyl-ACP intermediates often harbor a DH domain adjacent to a redox-inactive KR0 domain, as is also the case for NanKR50 from module 5 of the nanchangmycin synthase as well as MonKR10 from module 1 of the monensin synthase (Figure 1).18 On the other hand, pairing with a DH domain is clearly not essential, since neither the presumptively redox-inactive SalKR70 domain from module 7 of the salinomycin PKS nor the NigKR90 from module 9 of the nigericin synthase are paired with a DH domain (Figure 1). Finally, some apparently epimerizing modules lack a KR0 domain altogether, containing instead only a DH domain, as seen for NigDH1 in module 1 of the nigericin synthase and SalDH10 in module 10 of the salinomycin synthase (not shown).
Although the redox-inactive KR0 domains found in typical polyether PKS modules belong to the short chain dehydrogenase-reductase superfamily, as is evident from both their overall sequence similarity to typical KR domains (Figure 2) as well as their predicted protein structures (Figure S2), they nonetheless lack one, two, or sometimes all three amino acids residues of the otherwise highly conserved Tyr-Ser-Lys active site motif characteristic of the superfamily of short-chain dehydro-genase-reductases (Figure 2).16b,19 The absence of these conserved active site residues is intriguing, since we have shown that the epimerase activity of the redox-inactive macrolide synthase domains, EryKR30 and PicKR30, as well as that of the redox-active EryKR1, is in each case dependent on the paired active site Tyr and Ser residues.16b
Figure 2.

Alignment of conserved Tyr-Ser-Lys regions of redoxactive, epimerase-active KR domains (rows 1-4) and redox-inactive, epimerase-active KR0 domains (rows10-13) with presumptive redox-inactive, epimerase-active KR0 domains from polyether synthases (rows 5-9). (Lan, lankamycin). MAFFT server (v7, http://mafft.cbrc.jp/alignment/server/)
We report below the development of a general chemoenzymatic assay that allows the assignment of the 2-methyl configuration of any 2-methyl-3-ketoacyl-ACP and the use of this analytical method to confirm that NanKS1 from module 1 of the nanchangmycin synthase generates exclusively the (2R)-2-methyl-3-ketoacyl-ACP product, which must then undergo epimerization of the 2-methyl group. We then demonstrate that NanKR10, as well as the NanKR50 and SalKR70 domains, are all indeed redox-inactive and that each possesses intrinsic epimerase activity, as established by the recently developed tandem equilibrium isotope exchange assay. Finally, we demonstrate that these ketoreductase-inactive, epimerase-active domains do not have the same dependence on the active site Tyr-Ser dyad previously established for other epimerizing KR0 domains.
RESULTS
The decarboxylative condensation catalyzed by NanKS1 generates the (2R)-2-methyl-3-ketoacyl-ACP product
The KS domains of modules 1, 3, and 6 of the 6-deoxyerythonolide B synthase, as well as PicKS1 from the closely related picromycin (narbonolide) synthase, have all been shown to produce exclusively (2R)-2-methyl-3-ketoacyl-ACP intermediates by a decarboxylative condensation of (2S)-methylmalonyl-CoA with an electrophilic acyl-ACP partner that involves inversion of configuration.12,13 Since early speculation that some KS domains might also have an auxillary epimerase activity20 has not been borne out by experiment, the stereospecificity of all other KS domains that utilize methylmalonyl-CoA as the chain elongation substrate has to date simply been assumed on the basis of plausible biochemical analogy. We have previously determined the stereochemistry of a number of EryKS-catalyzed reactions using chemical quenching with aq. NaBH4 to reduce the enzyme-bound keto-acyl-ACP products, a laborious method had been unable to trap configurationally labile 2-methyl-3-ketoacyl-ACP diketides.13,21 In order to dissect the individual biochemical roles of the NanKS1 and NanKR10 domains, we therefore sought to develop a more sensitive, robust, and general chemo-enzymatic method that could be used to assign the configuration of any enzymatically-generated, configurationally labile 2-methyl-3-ketoacyl-ACP intermediate.
A variety of epimerase-inactive KR domains are known to be strictly specific for direct reduction of only (2R)-2-methyl-3-ketoacyl-ACP substrates. For example, EryKR6 from module 6 of the 6-deoxyerythronolide B synthase generates exclusively a (2R,3S)-2-methyl-3-hydroxyacyl-ACP product,13 while TylKR1 from module 1 of the tylactone synthase produces the diastereomeric (2R,3R)-2-methyl-3-hydroxyacyl-ACP.14 On the other hand, there have been no reports of naturally-occurring, epimerase-inactive KR domains that catalyze net reduction of only (2S)-2-methyl-3-ketoacylthioester substrates. Interestingly, while wild-type, epimerase-inactive AmpKR2 from module 2 of the amphotericin PKS reduces (±)-2-methyl-3-ketopentanoyl-SNAC (5) exclusively to (2R,3S)-2-methyl-3-hydroxypentanoyl-SNAC (9a), structure-based, rational engineering gave rise to a double mutant, AmpKR2-G355T/Q364H, that exhibited reversed stereospecificity for the 2-methyl configuration, reducing racemic 5 primarily (94%) to the diastereomeric (2S,3S)-2-methyl-3-hydroxypentanoyl-SNAC (9d) (Scheme 2), with a net kcat/Km 4 times that of the wild-type AmpKR2 (Table S5).15e Using the original EIX assay,22 we have now established that this AmpKR2-G355T/Q364H mutant has not acquired any detectable epimerase activity, thereby demonstrating that the preferred formation of 9d from racemic 5 by the double mutant is solely due to a change of the intrinsic diastereoselectivity of the parent AmpKR2 domain.
Scheme 2.
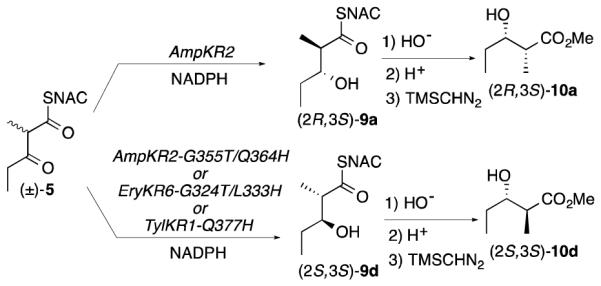
Targeted inversion of the stereoselectivity of KR domains by site-directed mutagenesis.
Guided by comparisons with the sequence and structure of AmpKR2 (Figure S9), we engineered homologous mutants of two additional KR domains in order to reverse their native reduction specificity from (2R)- to (2S)-2-methyl-3-ketoacylthioester substrates. Both the double mutant EryKR6-G324T/L333H and the single TylKR1-Q377H mutant reduced (±)-5 to >98% (2S,3S)-2-methyl-3-hydroxypentanoyl-SNAC (9d), of the derived methyl ester 10d (Scheme 2, Table S6).23 Activity assays established that the kcat/Km for reduction by EryKR6-G324T/L333H was essentially the same as that of wild-type EryKR6, while TylKR1-Q377H showed a ~16-fold decrease in kcat/Km compared to the parent TylKR1 (Table S5).25
The availability of a set of epimerase-inactive KR domains diastereospecific for reduction of either (2R)- or (2S)-2-methyl-3-ketoacylthioester substrates makes possible direct chemo-enzymatic determination of the condensation stereo-specificity of any KS domain. To this end, the Nan[KS1][AT1] didomain was expressed from the previously characterized S. nanchangensis NanA1 gene encoding module 1 of the nanchangmycin PKS (Figures 1, S3, and S6).5a A series of incubations was then carried out (Scheme 3), consisting of Nan[KS1][AT1] and recombinant holo-NanACP1 with the primer substrate propionyl-SNAC and chain extender methylmalonyl-CoA in the presence of NADPH and a set of individual epimerase-inactive KR domains of known diastere-ospecificity, comprised of a) (2R)-2-methyl-3-ketopentanoyl-ACP-specific EryKR6, AmpKR2, and TylKR1, b) (2S)-2-methyl-3-ketopentanoyl-ACP-specific AmpKR2-G355T/Q364H, EryKR6-G324T/L333H, and TylKR1-Q377H, and c) the epimerase-active EryKR1 and NysKR1 (from module 1 of the nystatin synthase),13,26 which generate the (2S,3R)- and (2S,3S)-2-methyl-3-hydroxypentanoyl-ACP products, respectively, from a (2R)-2-methyl-3-ketopentanoyl-ACP substrate. Aliquots from each incubation mixture were withdrawn and quenched after a period of 15, 30, and 60 min, and the derived diketide methyl esters (10) were analyzed by chiral GC-MS to establish the absolute configuration and relative yields of the respective products (Figures S14 and S15). A parallel control set of incubations was also performed using the previously described Ery[KS6][AT6] and holo-EryACP6 pair13 in place of Nan[KS1][AT1] and holo-Nan[ACP1]. For both incubation series, each of the three epimerase-inactive, (2R)-2-methyl-specific KR domains, as well as the two previously characterized epimerase-active KR domains, produced increasing amounts with time of reduced diketide 6 of the predicted configuration. Significantly, none of the three (2S)-2-methyl-specific mutant KR domains produced reduced diketides, even after a 60-min incubation time, consistent with their lack of epimerase activity (Scheme 3, Figure S15, and Table S7). It was thereby conclusively demonstrated that the decarboxylative condensation between propionyl-SNAC and methylmalonyl-CoA catalyzed by the polyether synthase didomain Nan[KS1][AT1] generates exclusively the corresponding (2R)-2-methyl-3-ketopentanoyl-NanACP1 (7a), identical to the known stereospecificity of macrolide synthase KS domains. This sensitive and robust coupled enzyme assay is in fact completely general and can be used to determine the 2-methyl configuration of any chemoenzymatically generated 2-methyl-3-ketoacyl-ACP intermediate.
Scheme 3.
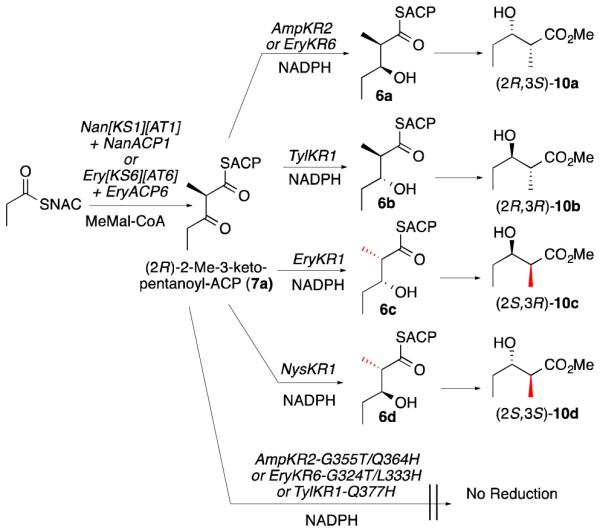
Chemoenzymatic determination of the stereochemistry of 2-methyl-3-ketoacyl-ACP intermediates.
Redox inactive KR domains from polyether PKS modules possess intrinsic epimerase activity
The prospective redox-inactive polyether KR domains, NanKR10, NanKR50, and SalKR70 were each expressed in Escherichia coli and purified as recombinant proteins carrying an N-terminal His6-tag using well-established modular PKS interdomain boundaries (Figures S3-S5). Consistent with the absence of the consensus nicotinamide cofactor binding sites in each KR0 domain (Figure S1), none of these three proteins was able to bind NADPH, as established by cofactor fluorescence enhancement assay (Figure S16), and all three were devoid of reductase activity, as determined by the standard KR assay using the N-acetylcysteamine thioester analog, (±)-2-methyl-3-ketopentanoyl-SNAC (5), as substrate in the presence of NADPH (Figure S17).16a,24
Strong evidence for the cryptic epimerase activity of each of these three redox inactive KR0 domains came from application of the recently developed tandem equilibrium isotope exchange (tandem EIX) assay.16a Thus the redox-active, epimerase-inactive ketoreductase domain, EryKR6 was used to reversibly oxidize the reduced, configurationally stable diketide [2-2H]-(2R,3S)-2-methyl-3-hydroxypentanoyl-EryACP6 ([2-2H]-6a) in the presence of a catalytic quantity of NADP+ so as to generate in situ a deuterated sample of the configurationally labile intermediate [2-2H]-(2R)-2-methyl-3-ketopentanoyl-EryACP6 ([2-2H]-7a) (Scheme 4). This transiently generated [2-2H]-(2R)-7a undergoes reversible isotope exchange and epimerization to (2R)-7b catalyzed by the redox-inactive, epimerase-active KR0 domain, after which the resultant unlabeled 7a is reduced back to nondeuterated 6a by EryKR6 and transiently generated NADPH. The reaction is conveniently monitored by periodic withdrawal of aliquots of the reaction mixture during the 60-min incubation and LC-ESI(+)-MS-MS analysis of the derived pantetheinate ejection fragments (8)27 at m/z 376 (d1) and m/z 375 (d0), as previously described (Scheme 4b).16a,22 By carrying out individual incubations with NanKR10, NanKR50, and SalKR70, we observed first-order, time-dependent loss of the deuterium label from ([2-2H]-6a in each case (Figure 3, Tables S9-S11), as previously observed for the redox-inactive, epimerase-active domains EryKR30 and PicKR30 as well as redox-inactive mutants of epimerase-active EryKR1.32 Control incubations with epimerase-inactive EryKR6 alone showed a maximum of 5-7% loss of label over 1 h, as expected, due simply to back-ground buffer-catalyzed exchange of the transiently generated labile intermediate [2-2H]-7a.
Scheme 4.
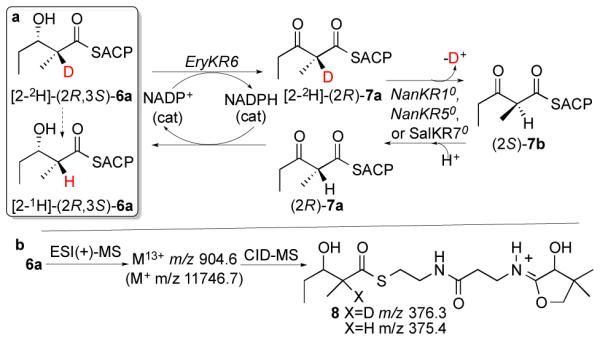
Tandem EIX assay.
Figure 3.

Time-dependent tandem EIX washout of deuterium from [2-2H]-6 by wild-type or mutant KR0 domains.
We have previously reported that the intrinsic epimerase activity of both the redox inactive EryKR30 and PicKR30 domains, as well as the redox active EryKR1 domain depends significantly on both of the conserved active site Ser and Tyr residues characteristic of short chain dehydrogenase/reductases.16b Intriguingly, NanKR10 lacks all three residues of the canonical Tyr-Ser-Lys triad, with Asp replacing the conserved Ser and Arg in place of the conserved active site Tyr (Figure 2). NanKR50 retains the conserved Tyr355 residue, but it carries Ala342 in place of the conserved Ser. Finally, although SalKR70 retains a conserved Ser349 residue, it carries a Phe362 in place of the conserved Tyr. Using site-directed mutagenesis, we found that the SalKR70/S349A mutant exhibited only 25% of the wild-type epimerase activity in the tandem EIX assay, while the SalKR70/F362Y mutant showed only a very modest 10% increase in the rate of deuterium exchange compared to wild-type (Figure 3b, Tables S10 and S11). Finally, the double mutant SalKR70-S349A/F362Y also exhibited a small increase in deuterium washout compared to the S349A single mutant. Control protein fluorescence quenching assays on wild-type and mutant SalKR70 established that none of the three mutations had more than a minimal effect on the binding affinity for the substrate analog (±)-2-methyl-3-ketopentanoyl-SNAC (5) (Table S8).
CONCLUSIONS
Modular polyketide synthases generate products containing numerous methyl-bearing centers whose configuration is controlled primarily by KR domains. Redox-active KR domains that are epimerase-inactive stereospecifically reduce the ketone moiety of the (2R)-2-methyl-3-ketoacyl-ACP intermediate produced by the paired KS domain of the same module to give the corresponding (2R,3R)- or (2R,3S)-2-methyl-3-hydroxyacyl-ACP product. By contrast, KR domains that are both redox-active and epimerase-active first catalyze epimerization of the 2-methyl substituent and then exclusively reduce the transiently-generated (2S)-2-methyl-3-ketoacyl-ACP diastereomer at the active site of the KR, performing in effect a kinetic resolution of the epimeric mixture of 2-methyl-3-ketoacyl-ACP esters.22 The specific protein structural factors that control this strict diastereoselection between the (2R)- and (2S)-methyl epimers are not yet understood. For KR0 domains that are redox-inactive but epimerase-active, the kinetic resolution of the resulting epimeric mixture of 2-methyl-3-ketoacyl-ACP intermediates is effected by the downstream KS domain of the proximal downstream module, which is strictly specific for chain elongation of the epimerized (2S)-2-methyl-3-ketoacyl-ACP. For example, NanKS2 in module 2 of the nanchangmycin synthase exclusively elongates (2S)-2-methyl-3-ketobutryl-NanACP1 when presented with a mixture of the corresponding NanACP1-bound (2S)- and (2R)-diastereomers.5e
Using a combination of previously characterized epimerase-inactive KR domains that are specific for reduction of (2R)-2-methyl-3-ketoacyl-ACP substrates as well as unique, epimerase-inactive mutant KR domains that had been engineered to be (2S)-2-methyl-3-ketoacyl-ACP-specific, we have developed a general chemoenzymatic assay for determination of the C2 configuration of any enzymatically-generated, configurationally labile 2-methyl-3-ketoacyl-ACP intermediate. Using this method we have established that the NanKS1 domain generates exclusively the (2R)-2-methyl-3-ketoacyl-ACP product, consistent with the known stereochemistry of KS-catalyzed decarboxylative condensation of macrolide synthase KS domains.21
We have also applied the tandem EIX assay16a to establish that the redox-inactive NanKR10 and NanKR50 domains from modules 1 and 5, respectively, of the nanchangmycin PKS and SalKR70 from module 7 of the salinomycin PKS each possess the intrinsic ability to exchange the H-2 protons of transiently generated (2R)-2-methyl-3-ketoacyl-ACP intermediates, an essential property of all epimerases. In distinction to numerous KR0 domains found in other modular PKS systems, but in common with many apparent KR0 domains from polyether modular PKSs, NanKR10 lacks the widely conserved active site Tyr-Ser-Lys triad characteristic of short-chain dehydrogenase reductases that we have previously shown to be important for the intrinsic epimerase activity of both redox-inactive and redox-active macrolide synthase KR domains,16b while NanKR50 only has the conserved Tyr355. Although SalKR70 retains the conserved Ser, it carries a Phe in place of the conserved Tyr. The Ser to Ala mutant of SalKR70 retained considerable activity in the tandem EIX assay, while the corresponding Phe->Tyr mutants showed only a minimal gain in activity. The specific biochemical mechanism for epimerization catalyzed by these polyether KR0 domains remains to be elucidated. Most likely as yet unidentified proximal amino acids serve the necessary H-bonding functions, while bound water molecules might also act as surrogates for the normally conserved Tyr and Ser residues.
Although we have confirmed the predicted epimerase activities of NanKR10, NanKR50, and SalKR70, the possible functional role of the DH domains that sometimes adjoin KR0 domains in redox-inactive, epimerase-active PKS modules remains unresolved. Thus although salinomycin module 7 contains only SalKR70 in addition to the core SalKS7, SalAT7, and SalACP7 domains, both nanchangmycin module 1 and nanchangmycin module 5 harbor a DH domain of unknown function immediately upstream of the epimerase-active domains, NanKR10 and NanKR50, respectively. Even more intriguingly, nigericin synthase module 1, which produces the same (2S)-2-methylbutyryl-ACP product as both nanchangmycin synthase module 1 and monensin synthase module 1, lacks a KR0 domain altogether, harboring only an auxiliary DH domain. Ongoing studies are aimed at the elucidation of the biochemical role of such cryptic DH domains.
EXPERIMENTAL METHODS
Materials
Cosmid 3C5 encoding Nan[KS1][AT1] and NanACP1 from Streptomyces nanchangensis has been previously described.5a Isopropylthio-β-D-galactopyranoside (IPTG), ampicillin, carbenicillin, chloramphenicol, kanamycin, and Phusion Flash High-Fidelity PCR Master Mix were purchased from Thermo Scientific. All other chemical reagents were purchased from Sigma-Aldrich and utilized without further purification. DNA primers were synthesized by Integrated DNA Technologies. Competent E. coli DH 5-alpha, DH10 beta and BL21 (DE3) cloning and expression strains were purchased from New England Biolabs Inc (NEB). Rosetta™ 2(DE3) Singles™ Competent Cells was purchased from Novagen. Restriction enzymes and T4 DNA ligase were purchased from NEB and used according to the manufacturer’s specifications. Pre-charged 5 mL HisTrapTM FF columns were purchased from GE Healthcare Life Sciences. Amicon Ultra Centrifugal Filter Units (Amicon Ultra-15 and Amicon Ultra-4, 30,000 MWCO) were purchased from Millipore. Slide-A-Lyzer Dialysis Cassettes was purchased from Thermo Scientific. Black Greiner Bio-One UV-Star® 96-well micro-plates were purchased from VWR Life Science. The expression plasmids for Ery[KS6][AT6], EryACP6, EryKR6, and TylKR1 have been described.14,28 The expression plasmid for AmpKR2-G355T/Q364H was a gift from Prof. A. Keatinge-Clay.15e Ery[KS6][AT6] and EryACP6 were expressed in E. coli BL21(DE3) or the E. coli BAP1 strain harboring the sfp gene for the surfactin phosphopantetheinyl tranferase and purified using the previously described protocols.28b,c The expression of apo-NanACP1 in E. coli BL21(DE3) using the pXG_NANS_ACP1_5 vector has been previously described.5e The substrate analogue (±)-2-methyl-3-ketopentanoyl-SNAC (5) was prepared as previously described.29 Reference standards of methyl (2R,3S)-2-methyl-3-hydroxypentanoate (10a), methyl (2R,3R)-2-methyl-3-hydroxypentanoate (10b), methyl (2S,3R)-2-methyl-3-hydroxypentanoate (10c), and methyl (2S,3S)-2-methyl-3-hydroxypentanoate (10d), prepared as previously described,13 were used as standards for chiral GC-MS analysis. Propionyl-SNAC was synthesized as previously described.13
Methods
General methods were as previously described.13,30 Growth media and conditions used for E. coli strains and standard methods for handling E. coli in vivo and in vitro were those described previously,30 unless otherwise noted. All DNA manipulations were performed following standard procedures. Plasmid DNA was purified using a Thermo Scientific GeneJET Plasmid mini-prep kit. DNA sequencing was carried out by Genewiz, South Plainfield, NJ. Synthetic genes, optimized for expression in E. coli, were prepared by DNA 2.0, Newark, California. All proteins were handled at 4 °C unless otherwise stated. Protein concentrations were determined according to the method of Bradford, using a Tecan Infinite M200 Microplate Reader with bovine serum albumin as standard.31 Protein purity and size were estimated using SDS-PAGE, visualized using Coomassie Blue stain, and analyzed with a Bio-Rad ChemiDoc MP System. Protein accurate molecular weight was measured using an Agilent 6530 Accurate-Mass Q-TOF LC/MS. Kinetic assays of KR-catalyzed reactions were carried out on the Tecan Microplate Reader. Chiral GC-MS analysis was performed on an Agilent 5977A Series GC-MSD instrument, 70 eV EI in positive ion mode with a Varian CP-Chirasil-DEX CB capillary column, 25 m × 0.32 mm, using the previously described temperature program.25 1H and 13C NMR spectra were obtained on a Bruker Avance III HD Ascend 600 MHz spectrometer. A Thermo LXQ equipped with Surveyor HPLC system and a Phenomenex Jupiter C4 column (150 mm×2 mm, 5.0 μm) was utilized for analysis of diketide-ACP compounds. HPLC-ESI-MS-MS analysis was carried out in positive ion mode for analysis of pantetheinate ejection fragments, as previously described.16a,22
Construction of Expression Plasmid Encoding Nan[KS1][AT1] from NANS Module 1
The Nan[KS1][AT1] didomain of S. nanchangensis NanAI (NANS module 1, GenBank accession number AF521085.1), is located on cosmid 3C5.5a The expression plasmid for the NANS [KS1][AT1] didomain was constructed in several steps, on the basis of the domain boundaries illustrated in Figure S3. The 5′-end of Nan[KS1][AT1], including the MluI site at nt-909, was amplified by PCR using the primer pairs Nan[KS1][AT1]-1-NdeI909f: ATCGTAATCCATATG-GAGGGGCAGCCCACCGCCGCCCCG (NdeI site under-lined) and NanKSAT1-1-EcoRI909r: TGATTCGAT-GAATTCAGCGTCGTGCCCGT-GCCATGGGCC (EcoRI site underlined) to amplify DNA from Cosmid 3C5. The resulting fragment was digested with NdeI and EcoRI and ligated into the corresponding site of doubly digested pET28a to give pXQX1. The 3′-end of the Nan[KS1][AT1] DNA fragment including the KpnI site at nt-2465 was amplified by PCR using primer pairs NanKSAT1-2-NdeI2372f: ATCGTAATCCATATGAGCCGACGGAGGAGCTGGA-CGCCG (NdeI site underlined) and NanKSAT1-2-EcoRI2811r: TGATCGATGAATTCACCCGGCCGG-CCGGGTTCCGACCCCG (EcoRI site underlined, stop codon bold), and the resultant amplicon was digested with NdeI and EcoRI before ligation into the corresponding site of digested pET28a to give pXQX2. Each of the two inserted DNA sequences was confirmed by sequencing. The fragment from nt-906 to nt-2465 of Cosmid 3C5 was obtained by stepwise restriction digestion. After treatment of Cosmid 3C5 with SgfI and PmaCI, the resulting 7238-bp fragment was purified and excised from an agarose gel. The 7238-bp fragment was then digested by MluI and KpnI and the resulting 1559-bp fragment was purified from an agarose gel. Plasmid pXQX1 was doubly digested by NdeI and MluI and the resulting 906-bp fragment was recycled by agarose gel. Plasmid pXQX2 containing the 3′-end of Nan[KS1][AT1] was doubly digested by KpnI and EcoRI and the 346-bp DNA fragment was recycled by agarose gel. Finally the 906-bp NdeI/MluI fragment, the 1559-bp MluI/KpnI fragment, and the 346-bp 3′-end of Nan[KS1][AT1] DNA fragment were co-ligated into NdeI/EcoRI-digested pET28a to give the protein expression plasmid pXXQ-Nan[KS1][AT1] encoding Nan[KS1][AT1].
Cloning of NanKR10
On the basis of the domain boundaries illustrated in Figure S3, the DNA sequence from Cosmid 3C5 encoding NanKR10 was amplified by PCR using the primers 5′-ATCGTAATCCATATGGCGGGTGGTCTC-TTCGCCC-3′ and 5′-TGATTCGATGAATTCAGCCG-GGCGCGGCGGCGGCG-3′ carrying NdeI and EcoRI restriction sites, respectively (underlined) and stop codon (bold). The resultant amplicon was ligated into the corresponding sites of pET28a to give the NanKR10 expression vector pXXQ-NanKR10.
Cloning of NanKR50
The DNA sequence from Cosmid 3C5 encoding NanKR50 was amplified by PCR using the primers 5′-ATCGTAATCCATATGACGGCCGCGCTG-TTCACGG-3′ and 5′-TGATTCGATGAATTCA-CGACTGCCGTTCCGCGGC-3’ carrying NdeI and EcoRI restriction sites, respectively, (underlined) and stop codon (bold). The resultant amplicon was ligated into the corresponding sites of pET28a to give the NanKR50 expression vector pXXQ-NanKR50.
Expression Vector for the SalKR70 Domain from Sal Module 7
Plasmid pXXQ-SalKR70 for expression of SalKR70 corresponding to the region from R982 to G1464 of Sal Module 7 (Figures S4 and S5) was constructed by subcloning the synthetic gene optimized for expression in E. coli into pET28a using the appended NdeI and XhoI restriction sites.
Mutagenesis of SalKR70
Plasmid pXXQ-SalKR70 harboring the SalKR70 domain served as the template for PCR mutagenesis, using outside primers SalKR7-NdeI: 5′-CGAAGTGAACATATGCGCGGTGAAGTGGATAGCTG-GCG-3′, NdeI restriction site underlined, and SalKR7-XhoI: 5′-ATTGCAATGCTCGAGTTAACCCGGATC-CGCACCAGTACCGG-3′, XhoI restriction site underlined, stop codon bold) in combination with the appropriate pairs of mutant primers (Table S3) to generate mutants of SalKR70 by two cycles of Overlap Extension PCR.32 The resulting mutant constructs were then doubly digested with NdeI and XhoI and ligated into the corresponding sites of pET28b (Novagen) using the same protocols previously described for mutants of EryKR1.16a The SalKR70-F362Y mutant plasmid DNA was also used as the template for two further rounds of Overlap Extension PCR amplification using two pairs of primers (SalKR7-NdeI/XhoI and SalKR7-S349A F/R) to construct the double mutant SalKR7-F362Y/S349A. Plasmid DNA was purified from cultures derived from a single colony, and DNA sequencing confirmed the sequences of all mutant plasmid inserts.
Mutagenesis of TylKR1 and EryKR6
The expression plasmids harboring the TylKR1 and EryKR6 domains were used as templates for the generation of mutants of the corresponding enzymes by Overlap Extension PCR, using the primers listed in Table S2.15c,28b,c Plasmid DNA was purified in each case from cultures derived from a single colony, and DNA sequencing confirmed the sequences of all mutant plasmid inserts.
Expression and Purification of Nan[KS1][AT1]
Since the expression plasmid pXXQ-Nan[KS1][AT1] encoding Nan[KS1][AT1] from Streptomyces nanchangensis contains more than 30 codons that are rare in E. coli (Table S1), the pRARE2 plasmid from Rosetta™ 2(DE3) Singles™ was cotransformed along with pXXQ-Nan[KS1][AT1] into competent cells of E. coli BL21(DE3). Single colonies of the resultant transformants were inoculated into LB media containing 50 mg/L kanamycin and 35 mg/L chloramphenicol and incubated overnight at 37 °C. The detailed expression and purification of the resulting Nan[KS1][AT1] protein was same as that used for KR proteins, as described below. SDS-PAGE analysis by Bio-Rad Image Lab Software indicated that the purity of Nan[KS1][AT1] protein was >90% (Figure S6). The molecular mass MD of Nan[KS1][AT1] was verified by Agilent Technologies Q-TOF LC-MS and matched the predicted values (observed MW, 99098; predicted MW, 99095.24; Figure S6).
Construction of Sfp Expression Plasmid Lacking an N-terminal His6-tag
The DNA sequence from the E. coli BAP1 strain harboring the gene for Sfp was amplified by PCR using the primers 5′-TGATTCGATCCATGG- CTAAGATTTACGGAATTTATATGG-3′ and 5′-TGATT-CGATGAATTCATAAAAGCTCTTCGTACGAGACC-3′ carrying NcoI and EcoRI restriction sites (underlined). The resultant amplicon was ligated into the corresponding sites of pET15b to generate the vector pXXQ-sfp for expression of Sfp without an N-terminal His-Tag.
Expression and Purification of holo-NanACP1
PXXQ-sfp was then used for production of holo-NanACP1 by co-transformation of E. coli BL21(DE3) along with pXG_NANS_ACP1_5, the previously described expression plasmid for apo-NanACP1 carrying an N-terminal His6-tag.5d Single colonies were inoculated into LB media containing 50 mg/L kanamycin and 100 mg/L ampicillin and incubated overnight at 37 °C. The detailed expression and purification of holo-NanACP1 proteins followed the same procedures as for purification of the apoNanACP1. SDS-PAGE analysis by Bio-Rad Image Lab Software indicated that the purity of holo-NanACP1 protein was >90% (Figure S7). The molecular mass MD of holo-NanACP1was verified by Q-TOF LC-MS and matched the predicted value (observed MW, 19000.55; predicted MW, 19001; Figure S7).
Expression and Purification of Wild-type and Mutant Ketoreductase Domains
The expression plasmids for wild-type and mutant ketoreductase proteins were individually transformed into competent cells of E. coli BL21(DE3) and the resulting single colonies were inoculated into LB media containing 50 mg/L kanamycin and incubated overnight at 37°C. The 10-mL seed cultures were each inoculated into 500 mL Super Broth (SB) media containing 50 mg/L kanamycin at 37 °C, and the resultant culture was grown to an OD600 of 0.5-0.7. The broth was cooled to 16 °C for 1 h and induced with 0.2 mM ITPG. After 40 h at 16 °C, cells were harvested by centrifugation, then washed and resuspended in lysis buffer (50 mM sodium phosphate, 500 mM NaCl, 40 mM imidazole, pH 7.8). Following sonication and removal of cell debris by centrifugation (23,000g for 50 min), the supernatant was loaded on a lysis-buffer-equilibrated, pre-charged 5 mL HisTrapTM FF column (GE Healthcare). The column was washed with 25 mL lysis buffer and then 25 mL washing buffer (50 mM sodium phosphate, 500 mM NaCl, 60 mM imidazole, pH 7.6). The protein was eluted by elution buffer (50 mM sodium phosphate, 50 mM NaCl, 150 mM imidazole, 10% glycerol, pH 7.4). The protein was concentrated by ultrafiltration (Amicon, 30,000 MWCO), the buffer was exchanged with exchange buffer (50 mM sodium phosphate, 100 mM NaCl, 10% glycerol, pH 7.2), and concentrated and stored at −80 °C until use. SDS-PAGE analysis by Bio-Rad Image Lab Software indicated that the purity of all wild-type and mutant proteins was >90% (Figure S8). The molecular mass MD of each protein was verified by Q-TOF LC-MS and matched the predicted values (Table S4).
Assay of Reductase Activity of Wild-Type and Mutant KR Proteins
KR activity was assayed by reduction of (±)-5 using the UV microplate plate reader and Falcon polystyrene 96-well plates to monitor continuously NADPH consumption at 340 nm.24 The reductase activity of TylKR1, EryKR6, and AmpKR2 and each of their mutants was assayed using variable concentrations of 5 from 0 to 60 mM (for EryKR6 and TylKR1 and their mutants) or 0 to 150 mM (for AmpKR2 and its mutants). KR protein was diluted in assay buffer (50 mM sodium phosphate, 100 mM NaCl, pH 7.2) to give a stock solution of 50 μM protein. Substrate 5 was diluted with DMSO:H2O (1:1) to give substrate concentrations of between 0 and 60 mM in the final mixture. The total amount of DMSO was kept constant at 5% v/v. Incubations with 5 μM of each KR or mutant protein and 2 mM NADPH were initiated by addition of substrate with rapid mixing by pipetting up and down 3 times. The course of the reaction was followed over 30 min by UV monitoring of the change in absorbance at 340 nm at 1-min intervals. The steady-state kinetic parameters were calculated by fitting the observed rate and substrate concentration data to the Michaelis-Menten equation by nonlinear least squares regression using the SigmaPlot 12.5 program (Figure S10, Table S5). Each assay was conducted in duplicate. The stated errors are the statistical standard deviations calculated by the least squares fitting algorithm.
Chiral GC-MS Analysis of the KR-Catalyzed Reduction of (±)-2-Methyl-3-ketopentanoyl-SNAC (5)
The protocol for the analysis of the KR-catalyzed reduction of (±)-2-methyl-3-ketopentanoyl-SNAC (5) was based on the previously described procedure.15e A typical incubation was carried out in a total volume of 500 μL containing 1 mM (±)-2-methyl-3-ketopentanoyl-SNAC (5), 2 mM NADPH, 50 mM phosphate buffer (50 mM sodium phosphate, 100 mM NaCl, pH 7.2) containing tris-2-carboxyethyl-phosphine (TCEP, 2.5 mM) and 50 μM ketoreductase proteins, as well ~1% glycerol from protein solutions and DMSO from SNAC substrate solutions. The enzyme mixtures were incubated at room temperature for 2 h, and the reaction was then quenched by addition of 150 μL of 0.5 M NaOH solution and heated to 65 °C for 20 min. The basic mixture was treated with 200 μL of 1 M HCl solution and then extracted with ethyl acetate (3× 600 μL). After evaporation of the organic solvent, the concentrated organic extract was analyzed by Chiral GC-MS under the conditions described below (Figures S11-S13, Table S6).
Incubation of Recombinant KR domains with Reconstituted Nan[KS1][AT1] plus holo-NanACP1 or Ery[KS6][AT6] plus EryACP6
The protocol for GC-MS assay of the products of reconstituted mixtures of recombinant PKS components was based on that previously described.13 In a typical assay, 5 mM propionyl-SNAC was pre-incubated with 40 μM Nan[KS1][AT1] or Ery[KS6][AT6] didomain protein in a total volume of 800 μL of phosphate buffer (50 mM sodium phosphate, 100 mM NaCl, pH 7.2) containing tris-2-carboxyethyl-phosphine (TCEP, 2.5 mM). After 30 min, 200 μM of holo-NanACP1 or holo-EryACP6, 300 μM methylmalonyl-CoA, 2 mM NADPH and 300 μM ketoreductase were added. The enzyme mixture was incubated at room temperature and 200 μL samples were withdrawn at periodic intervals of 15 min, 30 min, and 1 h. The withdrawn samples were immediately quenched by mixing with 50 μL of 0.5 M NaOH and the mixture incubated at 65 °C for 20 min. Then the reaction mixture was acidified with 100 μL of 1 M HCL and extracted with ethyl acetate (4×600 μL). The concentrated organic extract was methylated and analyzed by Chiral GC-MS (Figures S14 and S15, Table S7).
Quantitative Chiral GC-MS and Stereochemical Analysis of the KR-Catalyzed Reduction of Chemoenzymatically-Generated 2-Methyl-3-ketopentanoyl-ACP Intermediates
The concentrated organic extracts of the above-described enzymatic incubations were dissolved in 200 μL of methanol and then treated with 5 μL of TMS-diazomethane (2 M in hexane) for 10 min at room temperature. The derived reduced 2-methyl-diketide acid methyl esters were directly analyzed by chiral GC-MS.13 GC-MS spectra were recorded on a GC-MS Agilent 5977A Series GC-MSD instrument, 70 eV EI in positive ion mode, using a Varian CP-Chirasil-DEX CB capillary column (0.25 mm inside diameter × 25 m length × 0.25 mm film, Agilent Technologies) and a temperature program of (1) initial temp 60 °C for 1 min, (2) increase at rate 0.3 °C/min up to 75 °C, (3) 5 °C/min up to 90 °C, and then 20 °C/min to final temp 200 °C. The extracted ion current (XIC) of the chromatogram was analyzed set on the base peak at m/z 88 for 2-methyl-3-hydroxypentanoic acid methyl ester. All assays were conducted in duplicate. The products were compared directly with authentic standards, as previously described (Figures S11-S15).13
Assay of NADPH Binding by Fluorescence Enhancement
To confirm that the NanKR10, NanKR50 and SalKR70 proteins lack NADPH binding activity, the cofactor fluorescence enhancement assay was performed in 96-well plates (UV-Star® 96-Well Microplates, Greiner Bio-One, black) with a Tecan microplate reader.16a,33 Each KR0 protein, as well as redox-active EryKR1 as positive control and NADPH-nonbinding EryKR30 as negative control, was prepared at 10 μM in 50 mM sodium phosphate, pH 7.2. Solutions of NADPH (10-μL) of increasing concentration were added to the microplate wells containing 90 μL of purified 10 μM KR protein solution or buffer alone. The final NADPH concentration in the assay ranged from 0 to 50 μM (0, 2, 4, 8, 12, 16, 20, 30, 40, 50 μM). The solution was incubated for 20 min at room temperature. NADPH fluorescence was excited at 340 nm (5 nm slit width) and monitored between 440 nm and 470 nm (6 nm slit width). NADPH fluorescence enhancement was calculated from the difference in NADPH fluorescence intensity in the presence and absence of proteins (Fen=Fpre-Fab, where Fen means fluorescence enhancement, Fpre means fluorescence intensity of NADPH in the presence of protein, Fab means fluorescence intensity of NADPH in the absence of protein; Fpre and Fab were measured at the same concentration of NADPH). If the protein can bind cofactor NADPH, the fluorescence enhancement will be measurably increased in the presence of protein as the concentration of NADPH is increased over the range used in the assay. In the positive control, the observed NADPH fluorescence increased in the presence of the positive control EryKR1 protein, but showed no increase in the presence of the negative control, redox-inactive EryKR30 protein, which is known not to bind NADPH (Figure S16). The NADPH fluorescence intensities showed no increase in the presence of NanKR10, NanKR50, and SalKR70 proteins. Each assay was conducted in duplicate using different preparations of each protein.
Absence of Ketoreductase Activity of NanKR10, NanKR50, and SalKR70
The reductase activity of NanKR10, NanKR50, and SalKR70 proteins was tested in 96-well plates using the standard assay substrate (±)-2-methyl-3-ketopentanoyl-SNAC (5).24 Each KR0 protein (5 μM) was incubated with 1 mM NADPH in 50 mM sodium phosphate, pH 7.2, at 30 °C for 20 min, after which 5 (final concentration 8.0 mM) was added in a total volume of 100 μL and the consumption of NADPH was measured with the Tecan plate reader by monitoring the decrease in the absorbance at 340 nm for 30 min at 30 °C (Figure S17). Redox-active EryKR1 served as a positive control and redox-inactive EryKR30 as the negative control. None of the NanKR10, NanKR50 and SalKR70 proteins exhibited detectable ketoreductase activity.
Assay of Binding of (±)-2-Methyl-3-ketopentanoyl-SNAC to NADPH-free Ketoreductase Domains by Protein Fluorescence Quenching
All fluorescence measurements were carried using a microplate reader at 30 °C. Binding of (±)-2-methyl-3-ketopentanoyl-SNAC (5) to wild-type or mutant, redox-inactive KR0 domains was monitored by following the quenching of protein fluorescence intensity, as previously described.16b,34 Successive 5-μL portions of (±)-2-methyl-3-ketopentanoyl-SNAC solution (5) were added to 96-well plates (UV-Star® 96-Well Microplates, Greiner Bio-One, black) containing 95 μL of 1 μM protein solution or buffer alone. The final concentration of 5 ranged from 0 to 50 mM (0, 0.2, 0.5, 1.5, 2, 2.5, 3, 3.5, 4, 15, 20, 25, 35, 40, 45, 50 mM). The plate was incubated for 20 min at room temperature. The protein fluorescence was excited at 280 nm (5 nm bandwidth) and monitored at 338 nm (20 nm bandwidth). From the measured fluorescence intensity F at a given final concentration of 5, the quenched fluorescence intensity Fq was calculated by using Fq=Fapo−Fobs, where the Fapo represents the fluorescence intensity of the apoenzyme in the absence of 5, and Fobs the fluorescence intensity of apoenzyme at a given concentration of 5. The binding affinity (KD) was calculated by fitting the observed quenched fluorescence intensity and substrate concentration data to the Ligand Binding Equation (one site saturation, Eq 1) using the SigmaPlot 12.5 program (Table S8).
| (1) |
Tandem Equilibrium Isotope Exchange Assay of Redox-Inactive KR proteins
The Tandem EIX assay was carried out in duplicate as previously described.16a In a typical assay, chemoezymaticlly-prepared [2-2H]-(2R,3S)-2-methyl-3-hydroxypentanoyl-EryACP6 (90 μL of 500 μM solution, 45 nmol, final conc 300 μM), EryKR6 (11.25 nmol, 75 μM), NanKR10, NanKR50 or SalKR70 or its mutant protein (11.25 nmol, 75 μM), and NADP+ (1.5 μL of 1.5 mM soln, 2.25 nmol, final conc 15 μM) were incubated in 50 mM phosphate buffer (pH 7.2) (tot vol 150 μL) at room temp. Samples were withdrawn at periodic intervals up to 60 min and frozen in liq N2, before direct analysis of the pantetheinate ejection fragments by LC-ESI(+)-MS-MS, as previously described (Tables S9-S11).16a,22,27
Supplementary Material
ACKNOWLEDGMENT
We thank Prof. Adrian Keatinge-Clay of the University of Texas, Austin, for critical reading of the manuscript and valuable suggestions. This work was supported by grants from the U. S. National Institutes of Health, GM022172, to D.E.C., and GM087934, to C.K.
Footnotes
Supporting Information. Sequence alignments, protein structure comparisons, PKS domain boundaries, mutagenic primers, SDS-PAGE and LC-MS analysis of recombinant proteins, analysis of substrate and cofactor binding by recombinant KR domains, tandem EIX assays, steady-state kinetics, chiral GC-MS analyses, kinetic assays of coupled KS and KR incubations This material is available free of charge via the Internet at http:/pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES and NOTES
- 1.a) Cane DE, Liang TC, Hasler H. J. Am. Chem. Soc. 1982;104:7274–7281. [Google Scholar]; b) Cane DE, Celmer WD, Westley JW. J. Am. Chem. Soc. 1983;105:3594–3600. [Google Scholar]; c) Sood GR, Robinson JA, Ajaz AA. J. Chem. Soc. Chem. Commun. 1984:1421–1423. [Google Scholar]
- 2.Liu T, Cane DE, Deng Z. Methods Enzymol. 2009;459:187–214. doi: 10.1016/S0076-6879(09)04609-6. [DOI] [PubMed] [Google Scholar]
- 3.a) Bhatt A, Stark CB, Harvey BM, Gallimore AR, Demydchuk YA, Spencer JB, Staunton J, Leadlay PF. Angew. Chem. Int. Ed. Engl. 2005;44:7075–7078. doi: 10.1002/anie.200501757. [DOI] [PubMed] [Google Scholar]; b) Gallimore AR, Stark CB, Bhatt A, Harvey BM, Demydchuk Y, Bolanos-Garcia V, Fowler DJ, Staunton J, Leadlay PF, Spencer JB. Chem. Biol. 2006;13:453–460. doi: 10.1016/j.chembiol.2006.01.013. [DOI] [PubMed] [Google Scholar]; c) Oliynyk M, Stark CB, Bhatt A, Jones MA, Hughes-Thomas ZA, Wilkinson C, Oliynyk Z, Demydchuk Y, Staunton J, Leadlay PF. Mol. Microbiol. 2003;49:1179–1190. doi: 10.1046/j.1365-2958.2003.03571.x. [DOI] [PubMed] [Google Scholar]; d) Leadlay PF, Staunton J, Oliynyk M, Bisang C, Cortes J, Frost E, Hughes-Thomas ZA, Jones MA, Kendrew SG, Lester JB, Long PF, McArthur HA, McCormick EL, Oliynyk Z, Stark CB, Wilkinson CJ. J. Ind. Microbiol. Biotechnol. 2001;27:360–367. doi: 10.1038/sj.jim.7000204. [DOI] [PubMed] [Google Scholar]
- 4.a) Shichijo Y, Migita A, Oguri H, Watanabe M, Tokiwano T, Watanabe K, Oikawa H. J. Am. Chem. Soc. 2008;130:12230–12231. doi: 10.1021/ja8040543. [DOI] [PubMed] [Google Scholar]; b) Hotta K, Chen X, Paton RS, Minami A, Li H, Swaminathan K, Mathews II, Watanabe K, Oikawa H, Houk KN, Kim CY. Nature. 2012;483:355–358. doi: 10.1038/nature10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Sun Y, Zhou X, Dong H, Tu G, Wang M, Wang B, Deng Z. Chem. Biol. 2003;10:431–441. doi: 10.1016/s1074-5521(03)00092-9. [DOI] [PubMed] [Google Scholar]; b) Liu T, You D, Valen-zano C, Sun Y, Li J, Yu Q, Zhou X, Cane DE, Deng Z. Chem. Biol. 2006;13:945–955. doi: 10.1016/j.chembiol.2006.07.006. [DOI] [PubMed] [Google Scholar]; c) Liu T, Lin X, Zhou X, Deng Z, Cane DE. Chem Biol. 2008;15:449–458. doi: 10.1016/j.chembiol.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Guo X, Liu T, Valenzano CR, Deng Z, Cane DE. J. Am. Chem. Soc. 2010;132:14694–14696. doi: 10.1021/ja1073432. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Guo X, Liu T, Deng Z, Cane DE. Biochemistry. 2012;51:879–887. doi: 10.1021/bi201768v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harvey BM, Mironenko T, Sun Y, Hong H, Deng Z, Leadlay PF, Weissman KJ, Haydock SF. Chem. Biol. 2007;14:703–714. doi: 10.1016/j.chembiol.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 7.a) Jiang C, Wang H, Kang Q, Liu J, Bai L. Appl. Environ. Microbiol. 2012;78:994–1003. doi: 10.1128/AEM.06701-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yurkovich ME, Tyrakis PA, Hong H, Sun Y, Samborskyy M, Kamiya K, Leadlay PF. Chembiochem. 2012;13:66–71. doi: 10.1002/cbic.201100590. [DOI] [PubMed] [Google Scholar]
- 8.Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. Nature. 1990;348:176–178. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- 9.a) Donadio S, Staver MJ, Mcalpine JB, Swanson SJ, Katz L. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]; b) Donadio S, Katz L. Gene. 1992;111:51–60. doi: 10.1016/0378-1119(92)90602-l. [DOI] [PubMed] [Google Scholar]
- 10.Cane DE. J. Biol. Chem. 2010;285:27517–27523. doi: 10.1074/jbc.R110.144618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopwood DA. Methods in Enzymology. Complex enzymes in microbial natural product biosynthesis, part B: polyketides, amino-coumarins and carbohydrates. Vol. 459. Academic Press; United States: 2009. [DOI] [PubMed] [Google Scholar]
- 12.Marsden AF, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF. Science. 1994;263:378–380. doi: 10.1126/science.8278811. [DOI] [PubMed] [Google Scholar]
- 13.Valenzano CR, Lawson RJ, Chen AY, Khosla C, Cane DE. J. Am. Chem. Soc. 2009;131:18501–18511. doi: 10.1021/ja908296m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castonguay R, Valenzano CR, Chen AY, Keatinge-Clay A, Khosla C, Cane DE. J. Am. Chem. Soc. 2008;130:11598–11599. doi: 10.1021/ja804453p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Keatinge-Clay AT. Nat. Prod. Rep. 2016;33:141–149. doi: 10.1039/c5np00092k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Keatinge-Clay AT, Stroud RM. Structure. 2006;14:737–748. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]; c) Keatinge-Clay AT. Chem. Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]; d) Zheng J, Taylor CA, Piasecki SK, Keatinge-Clay AT. Structure. 2010;18:913–922. doi: 10.1016/j.str.2010.04.015. [DOI] [PubMed] [Google Scholar]; e) Zheng J, Piasecki SK, Keatinge-Clay AT. ACS Chem. Biol. 2013;8:1964–1971. doi: 10.1021/cb400161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Garg A, Xie X, Keatinge-Clay A, Khosla C, Cane DE. J. Am. Chem. Soc. 2014;136:10190–10193. doi: 10.1021/ja5056998. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xie X, Garg A, Keatinge-Clay AT, Khosla C, Cane DE. Biochemistry. 2016;55:1179–1186. doi: 10.1021/acs.biochem.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. The superscript "0" designates a redox-inactive KR domain.
- 18.Schwecke T, Aparicio JF, Molnár I, König A, Khaw LE, Haycock SF, Oliynyk M, Caffrey P, Cortes J, Lester JB, Böhm GA, Staunton J, Leadlay PF. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7839–7843. doi: 10.1073/pnas.92.17.7839. Such a DH-KR0 pair associated with formation of an epimerized (2S)-2-methyl-3-ketoacyl-ACP intermediate is also found in module 6 of the rapamycin synthase. Cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Kallberg Y, Oppermann U, Jornvall H, Persson B. Eur. J. Biochem. 2002;269:4409–4417. doi: 10.1046/j.1432-1033.2002.03130.x. [DOI] [PubMed] [Google Scholar]; b) Kallberg Y, Oppermann U, Jornvall H, Persson B. Protein Sci. 2002;11:636–641. doi: 10.1110/ps.26902. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Opper-mann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jornvall H. Chem. Biol. Interact. 2003;143-144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 20.a) Bohm I, Holzbaur IE, Hanefeld U, Cortes J, Staunton J, Leadlay PF. Chem. Biol. 1998;5:407–412. doi: 10.1016/s1074-5521(98)90157-0. [DOI] [PubMed] [Google Scholar]; b) Holzbaur IE, Ranganathan A, Thomas IP, Kearney DJ, Reather JA, Rudd BA, Staunton J, Leadlay PF. Chem. Biol. 2001;8:329–340. doi: 10.1016/s1074-5521(01)00014-x. [DOI] [PubMed] [Google Scholar]
- 21.Castonguay R, He W, Chen AY, Khosla C, Cane DE. J. Am. Chem. Soc. 2007;129:13758–13769. doi: 10.1021/ja0753290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garg A, Khosla C, Cane DE. J. Am. Chem. Soc. 2013;135:16324–16327. doi: 10.1021/ja408944s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Although wild-type EryKR6 generates exclusively the (2R,3S)-2-methyl-3-hydroxypentanoyl-ACP product when incubated with in situ-generated (2R)-2-methyl-3-ketopentanoyl-ACP, (cf. Valenzano, ref. 13), the native stereospecificity is completely lost when the corresponding SNAC derivative(±)-5 is utilized as a surrogate substrate. (cf. Siskos, ref. 24)
- 24.Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, Leadlay PF. Chem. Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 25.Bailey CB, Pasman ME, Keatinge-Clay AT. Chem Commun (Camb) 2016;52:792–795. doi: 10.1039/c5cc07315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.You YO, Khosla C, Cane DE. J. Am. Chem. Soc. 2013;135:7406–7409. doi: 10.1021/ja4014776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Bioorg. Med. Chem. Lett. 2008;18:3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a) Kim CY, Alekseyev VY, Chen AY, Tang Y, Cane DE, Khosla C. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]; b) Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. J. Am. Chem. Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen AY, Cane DE, Khosla C. Chem. Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piasecki SK, Taylor CA, Detelich JF, Liu J, Zheng J, Komsoukaniants A, Siegel DR, Keatinge-Clay AT. Chem. Biol. 2011;18:1331–1340. doi: 10.1016/j.chembiol.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y.: 1989. [Google Scholar]
- 31.Bradford M. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 32.Bryksin AV, Matsumura I. Biotechniques. 2010;48:463–465. doi: 10.2144/000113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witkowski A, Joshi AK, Smith S. Biochemistry. 2004;43:10458–10466. doi: 10.1021/bi048988n. [DOI] [PubMed] [Google Scholar]
- 34.a) He XM, Thorson JS, Liu HW. Biochemistry. 1996;35:4721–4731. doi: 10.1021/bi952706p. [DOI] [PubMed] [Google Scholar]; b) Menon S, Stahl M, Kumar R, Xu GY, Sullivan F. J. Biol. Chem. 1999;274:26743–26750. doi: 10.1074/jbc.274.38.26743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.