

Abstract
Objective:
To investigate the feasibility, safety, and efficacy of a ketogenic diet (KD) for superrefractory status epilepticus (SRSE) in adults.
Methods:
We performed a prospective multicenter study of patients 18 to 80 years of age with SRSE treated with a KD treatment algorithm. The primary outcome measure was significant urine and serum ketone body production as a biomarker of feasibility. Secondary measures included resolution of SRSE, disposition at discharge, KD-related side effects, and long-term outcomes.
Results:
Twenty-four adults were screened for participation at 5 medical centers, and 15 were enrolled and treated with a classic KD via gastrostomy tube for SRSE. Median age was 47 years (interquartile range [IQR] 30 years), and 5 (33%) were male. Median number of antiseizure drugs used before KD was 8 (IQR 7), and median duration of SRSE before KD initiation was 10 days (IQR 7 days). KD treatment delays resulted from intravenous propofol use, ileus, and initial care received at a nonparticipating center. All patients achieved ketosis in a median of 2 days (IQR 1 day) on KD. Fourteen patients completed KD treatment, and SRSE resolved in 11 (79%; 73% of all patients enrolled). Side effects included metabolic acidosis, hyperlipidemia, constipation, hypoglycemia, hyponatremia, and weight loss. Five patients (33%) ultimately died.
Conclusions:
KD is feasible in adults with SRSE and may be safe and effective. Comparative safety and efficacy must be established with randomized placebo-controlled trials.
Classification of evidence:
This study provides Class IV evidence that in adults with SRSE, a KD is effective in inducing ketosis.
Status epilepticus (SE) is the second most common neurologic emergency worldwide.1 SE that continues despite appropriate first- and second-line antiseizure drugs is classified as refractory SE (RSE).1 Current treatment algorithms use intravenous anesthetic agents for ≥24 hours to suppress RSE. If RSE returns after anesthetics are withdrawn, patients are diagnosed with superrefractory SE (SRSE). RSE and SRSE carry a high risk of morbidity, and reported mortality rates range from 23% to 57%.2–4 Anesthetic agents may contribute to poor clinical outcomes,5 and determining optimal clinical management of SRSE is critical.
The ketogenic diet (KD) is a high-fat, low-carbohydrate diet that induces ketone body production through fat metabolism.6 KD is effective in drug-resistant epilepsy in both children and adults, and recent retrospective studies have illustrated the potential efficacy of KD in treating SRSE.7–9 KD is shown in case reports and small case series to be particularly effective in treating SRSE in patients with febrile-induced refractory epilepsy syndrome,8 new-onset refractory SE, and anti-NMDA receptor encephalitis.6,10 Furthermore, KD is also shown to have anti-inflammatory properties that may halt SRSE.11
This study examines the feasibility, safety, and efficacy of implementing a KD algorithm in adults with SRSE in a critical care setting.
METHODS
Study design.
This was a prospective, open-label, single-arm study with the primary research question of whether adults with SRSE can reach therapeutic ketosis using a standardized treatment algorithm (Class IV evidence). Secondary outcome measures included adverse and serious adverse events and response to treatment (Class IV evidence).
Standard protocol approvals, registrations, and patient consents.
The Johns Hopkins Hospital, Johns Hopkins Bayview Medical Center (Baltimore, MD), Mayo Clinic (Rochester, MN), Queen's Medical Center (Honolulu, HI), and Thomas Jefferson University Hospital (Philadelphia, PA) Institutional Review boards approved this study.
Population.
Consecutive patients 18 to 80 years of age diagnosed with SRSE (treated with medically induced coma for >24 hours with persistent SE or return after attempt to wean one or more anesthetics) from November 2014 to February 2016 were screened for eligibility (see the figure for inclusion and exclusion criteria). Written informed consent was obtained from each patient's legally authorized representative.
Figure. KD treatment algorithm for SRSE.

ALT = alanine transaminase; AST = aspartate transaminase; CBC = complete blood count; CMP = comprehensive metabolic panel; HCG = human chorionic gonadotropin; KD = ketogenic diet; MAD = modified Atkins diet; MAP = mean arterial pressure; SRSE = superrefractory status epilepticus*Patients were excluded on the basis of a prior report of fatal propofol infusion syndrome in a patient who received concomitant KD therapy for status epilepticus.13
Treatment.
A 4:1 (fat:carbohydrate and protein grams) ratio KD treatment algorithm is outlined in the figure.
Clinical data.
Data collected included patient demographics, history of epilepsy, etiology of SRSE, Glasgow Coma Scale (GCS) and modified Rankin Scale (mRS) scores, duration of SRSE before KD, and antiseizure drugs used before KD. Continuous video EEG data and urine and serum ketone levels were collected every 12 hours after KD initiation. Adverse events were recorded, and an independent Data and Safety Monitoring Board (DSMB) determined whether unanticipated serious adverse events were potentially related to KD.
Outcome variables.
Significant ketosis (defined as urine acetoacetate ≥40 mg/dL and/or serum β-hydroxybutyrate ≥2 mmol/L) was the primary outcome measure. Secondary outcome measures included resolution of SRSE, GCS and mRS scores at discharge, and disposition. Long-term (6 months) outcome measures included seizure frequency and continuation of KD therapy (either KD or modified Atkins Diet [MAD]).
Descriptive study statistics.
Proportions were calculated for categorical variables, and means, medians, and interquartile ranges (IQRs) were calculated for continuous variables.
RESULTS
Clinical characteristics.
Twenty-four adults met the inclusion criteria. After the exclusion criteria were applied, 15 patients were eligible to be included in the analysis. Patients were excluded because SRSE resolved before consent (3), patients died before consent (2), the legally authorized representatives declined patient participation (2), the patient was a prisoner (1), and the patient had liver failure (1). The median age was 47 years (IQR 30 years); 5 participants (33%) were male; and 6 (40%) had a history of epilepsy (table 1).
Table 1.
Patient characteristics and clinical management
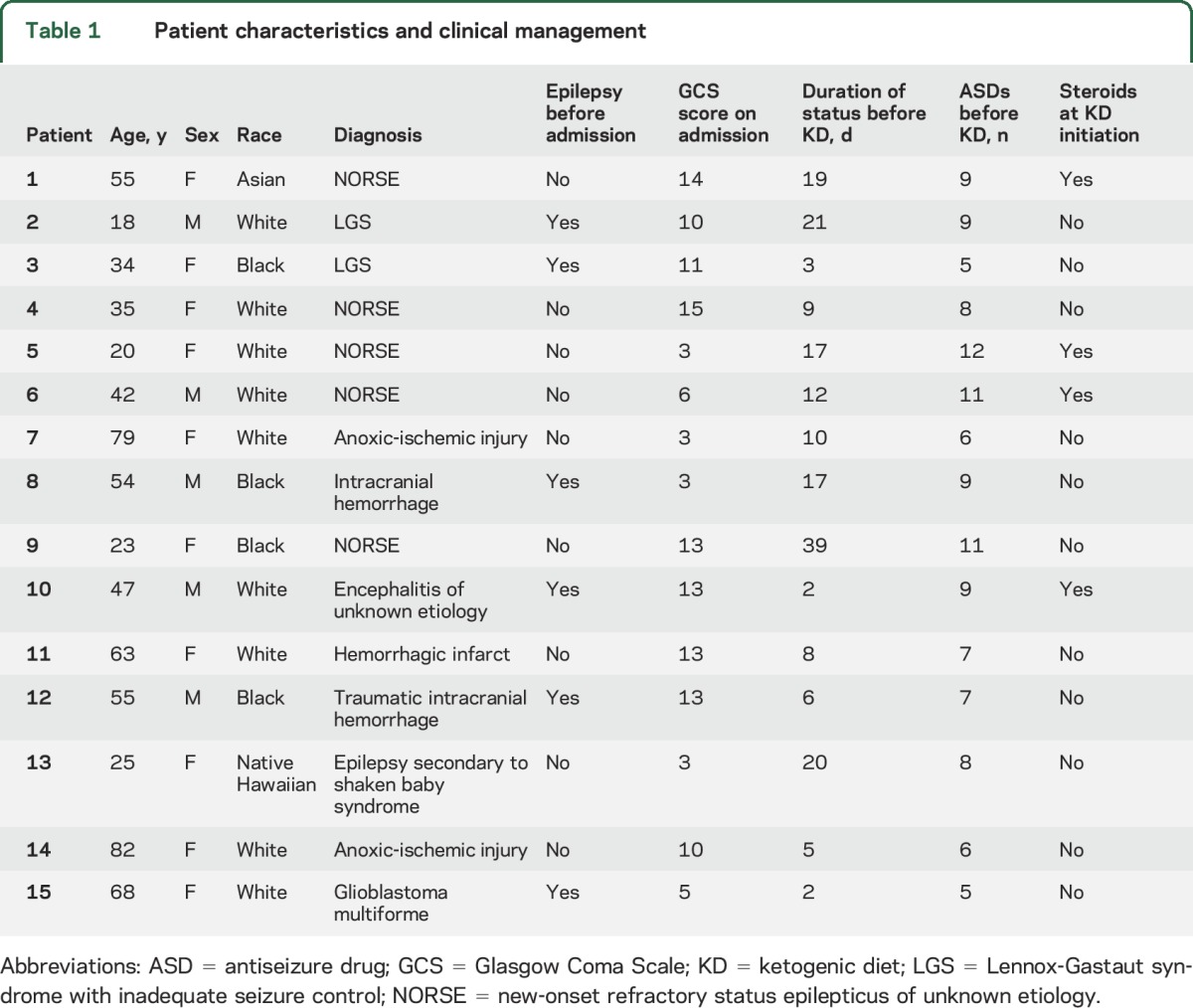
Etiologies of SRSE included new-onset refractory SE (5; etiology unknown despite extensive serologic and histopathologic analyses), intracranial hemorrhage (3), Lennox-Gastaut syndrome (2), anoxic-ischemic injury (2), prior diagnosis of encephalitis with recurrent SE (1), glioblastoma multiforme (1), and remote nonaccidental traumatic head injury (shaken-baby syndrome) with resultant drug-resistant epilepsy (1). The median duration of SRSE was 10 days (IQR 6 days) with a median of 8 (IQR 7) antiseizure drugs administered before KD. Delay in KD initiation resulted from use of intravenous propofol (1), ileus (1), and initial SRSE treatment received at an outside hospital before transfer to a participating center (8). Four patients (27%) were receiving corticosteroids at the time of KD initiation, and 5 (33%) received another form of immunotherapy (5 with plasmapheresis, 1 also with cyclophosphamide) during treatment with KD.
Outcomes after KD initiation.
All patients achieved ketosis in a median of 2 days (IQR 1 day). Three patients were fasting and in ketosis on the first day of KD because they received a feeding tube the previous day. Anesthetics were weaned before 72 hours on KD in 3 patients on the basis of clinical judgment. In one case (patient 7), the family elected to withdraw care after initiating KD, so she was withdrawn from the study and died. At the time that the decision was made to withdraw care, the patient had reached ketosis, and epileptiform activity was suppressed with general anesthetics (midazolam). Myoclonic SE returned on weaning anesthetics and stopping the KD. Eleven (79%) of 14 patients who completed the KD treatment algorithm had resolution of SRSE in a median of 5 days (IQR 3 days) after KD initiation; 8 (57%) had resolution of SRSE within 1 week. In an intention-to-treat analysis, SRSE resolved in 73% (11 of 15) of all participants who initiated KD. GCS score was equivalent to pre-SRSE baseline in 9 patients (60%) at the time of hospital discharge. Adverse events occurred in 67% (10 of 15) of all participants (table 2).
Table 2.
Patient feasibility, safety, and efficacy outcomes
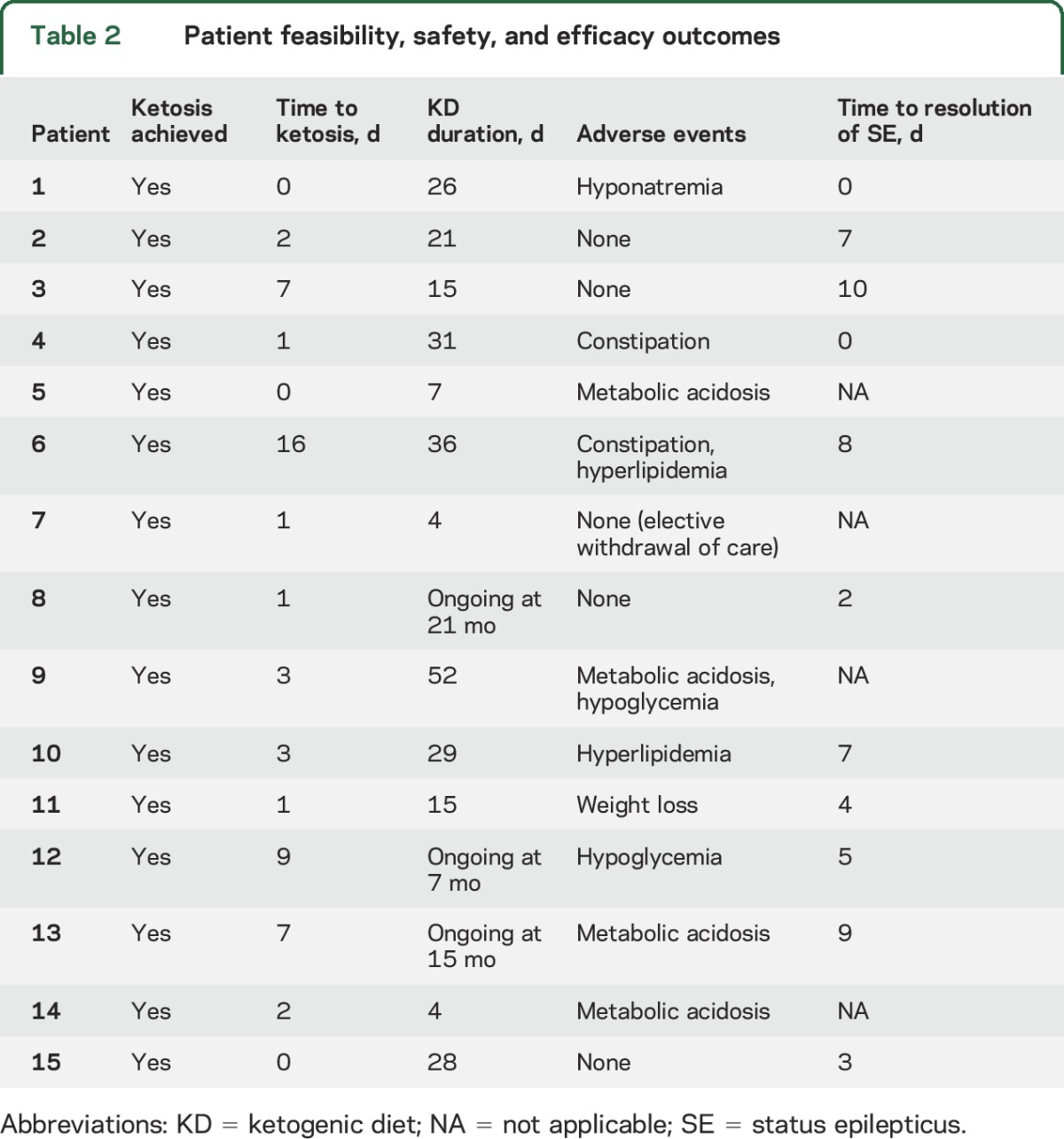
Five patients ultimately died, including the patient in whom care was withdrawn. Three patients did not have resolution of SRSE with KD and developed metabolic acidosis that did not resolve with bicarbonate supplementation, requiring KD discontinuation before death. In one patient (patient 10), SRSE resolved with KD, and he was transferred to a rehabilitation facility where KD was stopped and SRSE recurred. SRSE resolved after KD was restarted. However, he had a pulseless electrical activity cardiac arrest, failed resuscitation efforts, and died. The DSMB deemed the death unlikely to be related to KD given the short duration of KD before the event (29 total days).
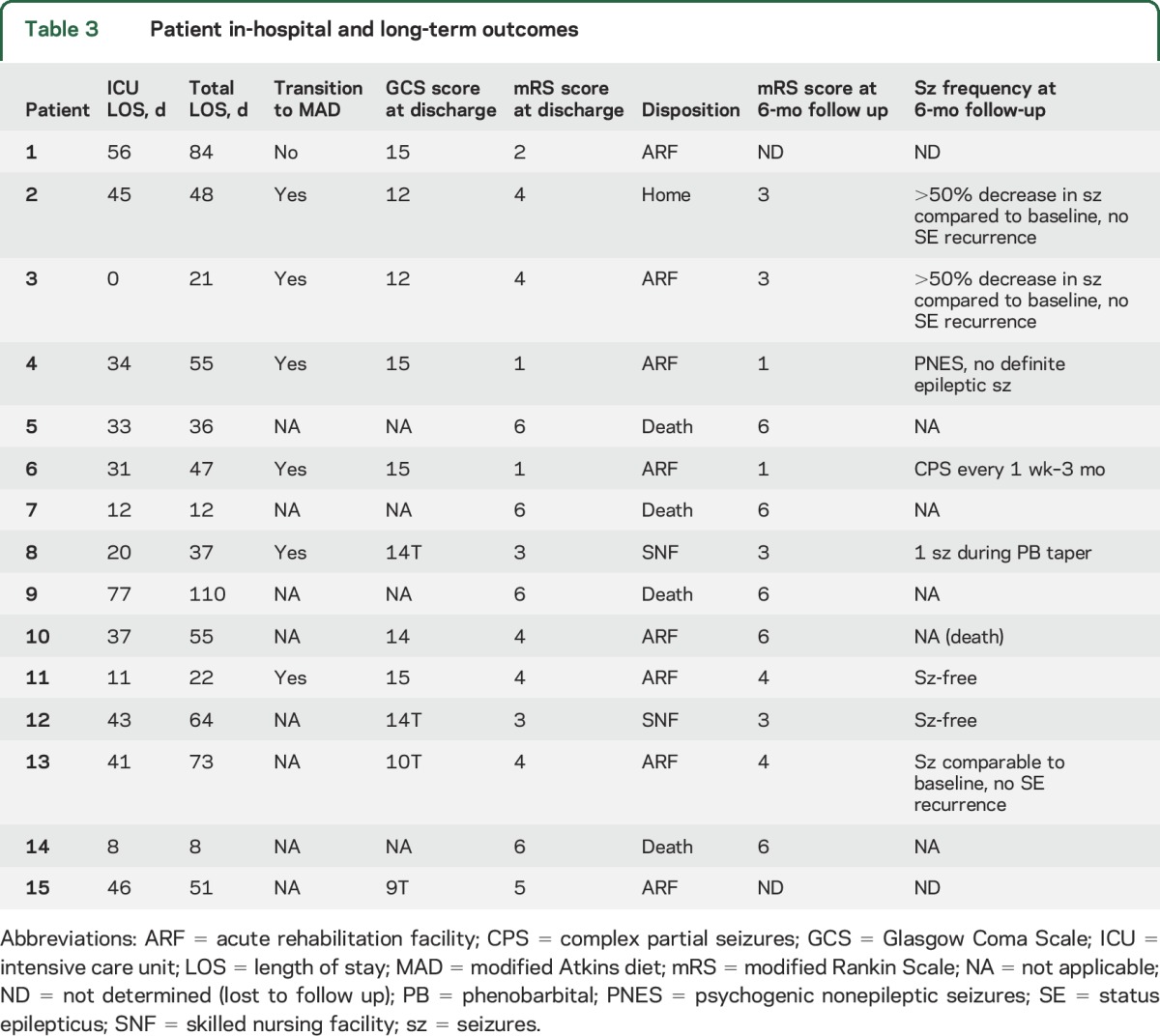
Once able to intake oral nutrition, 6 of 11 patients (55%) on KD transitioned to MAD (table 3). Four (67%) remained on MAD as of the most recent follow-up. Two patients (33%) who were seizure-free tapered off of MAD because of difficulty adhering and remained seizure-free after diet discontinuation. Two patients (18%) with inadequate oral nutrition remained on a 4:1 ratio KD by feeding tube. Of 8 patients in whom 6-month follow up outcomes were available, 2 (25%) were seizure-free, 2 (25%) with a history of Lennox-Gastaut syndrome had a >50% reduction in seizures from pre-SRSE baseline and no further hospitalizations for SRSE (multiple prior episodes), 2 (25%) had ongoing seizures, 1 (12%) had a single seizure during antiseizure drug tapering and none since, and 1 (12%) developed psychogenic nonepileptic seizures but no definite epileptic seizures.
Table 3.
Patient in-hospital and long-term outcomes
DISCUSSION
We report the results of a prospective multicenter study of the feasibility of treatment of SRSE with a KD in adults. All patients reached ketosis in a median of 2 days after KD initiation, and SRSE stopped in 73% of patients, within 1 week of KD initiation in most patients. The KD was well tolerated and feasible in this clinical trial. At 6 months, 67% of patients were alive, and the majority recovered to their premorbid functional baseline. These findings are promising given reported mortality rates in SRSE of nearly 60%.2,3 These results are also comparable to a recent phase I/II platform presentation of 22 patients treated with 547-SSE-201 (allopregnanolone solution) in which SRSE was suppressed in 19 patients (77%) and 6 patients (27%) died during the trial.12 Barriers to KD initiation included use of propofol and ileus. Use of corticosteroids did not prevent patients from reaching ketosis. Metabolic acidosis was the most notable KD-related adverse effect, which occurred in 4 patients (27%) and necessitated stopping KD in 3. This highlights the importance of vigilant monitoring and aggressive bicarbonate supplementation. Other KD-related side effects included hypoglycemia, hyperlipidemia, constipation, hyponatremia, and weight loss.
This observational study provides preliminary evidence that inducing ketosis with a KD in adults with SRSE is feasible, despite their already complex treatment regimens. Randomized placebo-controlled trials are needed to determine whether KD is safe and effective in reducing morbidity and mortality in patients with SRSE.
ACKNOWLEDGMENT
The authors acknowledge the patients who participated in this study, their families, and members of the DSMB and the Johns Hopkins Encephalitis and Status Epilepticus Workgroup.
GLOSSARY
- DSMB
Data and Safety Monitoring Board
- GCS
Glasgow Coma Scale
- IQR
interquartile range
- KD
ketogenic diet
- MAD
modified Atkins diet
- mRS
modified Rankin Scale
- RSE
refractory status epilepticus
- SE
status epilepticus
- SRSE
superrefractory status epilepticus
AUTHOR CONTRIBUTIONS
Mackenzie C. Cervenka, MD, and Sara Hocker, MD: study design and conceptualization, data acquisition, analysis, and interpretation of the data, and drafting and revising of the manuscript. Matthew Koenig, MD: study design and conceptualization, data acquisition, analysis, and interpretation of the data, and revising of the manuscript. Barak Bar, MD: study data acquisition, analysis, and interpretation of the data, and revising of the manuscript. Bobbie Henry-Barron, RD: study design and conceptualization and revising of the manuscript. Eric Kossoff, MD, Adam L. Hartman, MD, and John C. Probasco, MD: study design and conceptualization and revising of the manuscript. David R. Benavides, MD: study data acquisition, analysis, and interpretation of the data, and revising of the manuscript. Arun Venkatesan, MD, PhD: study design and conceptualization. Eliza C. Hagen, MD, MBA, Denise Dittrich, RN, and Tracy Stern, RN: study data acquisition, analysis, and interpretation of the data. Batya Radzik, CRNP, Marie Depew, ACNP, and Filissa M. Caserta, MSN, ACNP-BC, CNRN: study design and conceptualization, study data acquisition. Paul Nyquist, MD, MPH: study design and conceptualization. Peter W. Kaplan, MB, FRCP: study design and conceptualization, study data acquisition. Romergryko Geocadin, MD: study design and conceptualization.
STUDY FUNDING
This study was funded by generous philanthropic support from Chris Garrod, Dawn Griffiths, and The Carson Harris Foundation.
DISCLOSURE
M. Cervenka receives grants from Nutricia, Vitaflo, BrightFocus Foundation, and Army Research Laboratory and honoraria from American Epilepsy Society, New York University, The Neurology Center, and LivaNova. S. Hocker performs consulting activities for SAGE therapeutics for which she receives compensation. M. Koenig and B. Bar report no disclosures relevant to the manuscript. B. Henry-Barron receives grants from Johns Hopkins Institute for Clinical and Translational Research, which is funded in part by grant UL1 TR 001079 from the National Center for Advancing Translational Sciences, a component of the NIH, and NIH Roadmap for Medical Research, Nutricia, and Vitaflo. E. Kossoff receives a grant from Nutricia and consulting fees from Atkins Nutritionals, Inc. A. Hartman receives funding for travel from American Academy of Pediatrics; honoraria from American Epilepsy Society, Miami Children's Hospital, Epigenix Foundation, Uniformed Services University of Health Science, and George Washington University; royalties from Wiley, LWW, and Taylor & Francis; and grants from National Institute of Neurological Disorders and Stroke/NIH and Technology Development Corporation (State of Maryland) (ended in 2015); has been a board member for Hemispherectomy Foundation, Epilepsy Foundation of the Chesapeake Region/Abilities Network, and Carson Harris Foundation and on the Editorial Board for Epilepsia, Faculty of 1000, F1000 Research, Journal of Pediatric Epilepsy, Rare Disease Report; and has a patent pending for a novel therapeutic molecule for seizure treatment. J. Probasco reports no disclosures relevant to the manuscript. D. Benavides receives fellowship support from Mallinckrodt Pharmaceuticals. A. Venkatesan receives grants from the NIH, Maryland Stem Cell Research Foundation, and National Multiple Sclerosis Society. E. Hagen, D. Dittrich, T. Stern, B. Radzik, M. Depew, F. Caserta, P. Nyquist, and P. Kaplan report no disclosures relevant to the manuscript. R. Geocadin receives grants from NIH RO1 HL071568, NIH RO1 NS074425, and KeyTech Inc. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hocker SE, Britton JW, Mandrekar JN, Wijdicks EF, Rabinstein AA. Predictors of outcome in refractory status epilepticus. JAMA Neurol 2013;70:72–77. [DOI] [PubMed] [Google Scholar]
- 2.Ferlisi M, Shorvon S. The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain 2012;135:2314–2328. [DOI] [PubMed] [Google Scholar]
- 3.Hocker S, Tatum WO, LaRoche S, Freeman WD. Refractory and super-refractory status epilepticus: an update. Curr Neurol Neurosci Rep 2014;14:452. [DOI] [PubMed] [Google Scholar]
- 4.Young GB, Jordan KG, Doig GS. An assessment of nonconvulsive seizures in the intensive care unit using continuous EEG monitoring: an investigation of variables associated with mortality. Neurology 1996;47:83–89. [DOI] [PubMed] [Google Scholar]
- 5.Sutter R, Marsch S, Fuhr P, Kaplan PW, Ruegg S. Anesthetic drugs in status epilepticus: risk or rescue? A 6-year cohort study. Neurology 2014;82:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McNally MA, Hartman AL. Ketone bodies in epilepsy. J Neurochem 2012;121:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thakur KT, Probasco JC, Hocker SE, et al. Ketogenic diet for adults in super-refractory status epilepticus. Neurology 2014;82:665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nabbout R, Mazzuca M, Hubert P, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010;51:2033–2037. [DOI] [PubMed] [Google Scholar]
- 9.Wusthoff CJ, Kranick SM, Morley JF, Christina Bergqvist AG. The ketogenic diet in treatment of two adults with prolonged nonconvulsive status epilepticus. Epilepsia 2010;51:1083–1085. [DOI] [PubMed] [Google Scholar]
- 10.Amer S, Shah P, Kommineni V. Refractory status epilepticus from NMDA receptor encephalitis successfully treated with an adjunctive ketogenic diet. Ann Indian Acad Neurol 2015;18:256–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dupuis N, Curatolo N, Benoist JF, Auvin S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 2015;56:e95–e98. [DOI] [PubMed] [Google Scholar]
- 12.Kanes S, Rosenthal E, Vaitkevicius H, et al. 547-SSE-201 for super-refractory status epilepticus: response and relationship to underlying patient characteristics (S14.003). Neurology 2016;86(suppl S14.003). [Google Scholar]
- 13.Baumeister FA, Oberhoffer R, Liebhaber GM, et al. Fatal propofol infusion syndrome in association with ketogenic diet. Neuropediatrics 2004;35:250–252. [DOI] [PubMed] [Google Scholar]