
Fig. (2).
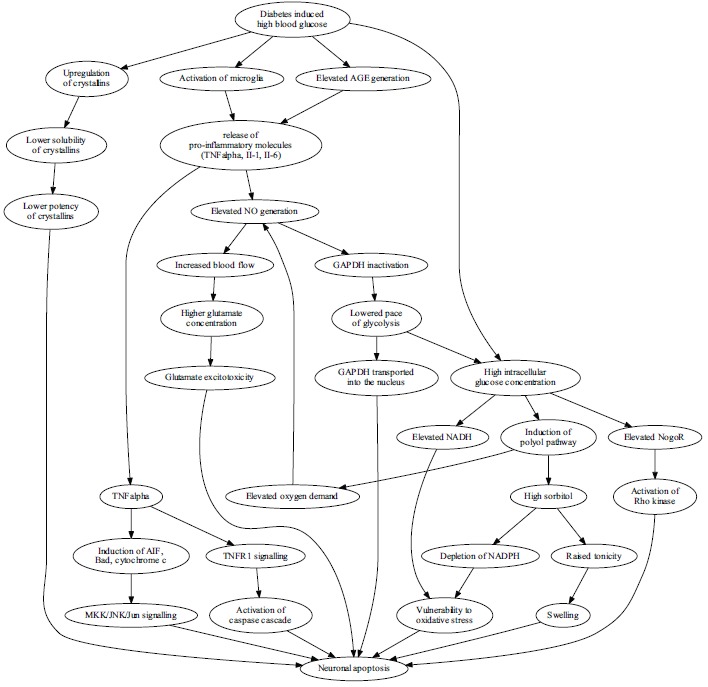
The high blood glucose leads to neuron vulnerability and apoptosis in diabetes through many mechanisms, with recently explored pathways illustrated. Hyperglycemia affects the expression and function of crystallins, a group of chaperones. Crystallins are upregulated, however their solubility is diminished with the net effect of lowered protection. Activated microglia and elevated levels of AGEs induce prolonged inflammation with pro-apoptotic effects on retinal ganglion cells through pathways dependent on TNFα and nitric oxide. Disruption of TNFα balance induces apoptosis via caspase cascade and via Jun kinase, while elevated NO generation leads to vasodilation, increased blood flow, higher glutamate concentrations and results in glutamate excitotoxicity. NO also lowers the pace of glycolysis by inactivating GAPDH, a process that leads to accumulation of GAPDH in the nucleus, which is an apoptosis-associated effect and increases the burden of high intracellular glucose concentration resulting mostly from high blood glucose concentration. Neurons are forced to accommodate the excess glucose by the polyol pathway which uses NADPH to generate sorbitol from glucose. High intracellular glucose concentration additionally leads to NADH (nicotinamide adenine dinucleotide, reduced) accumulation, which, collectively with NADPH (nicotinamide adenine dinucleotide phosphate, reduced) depletion, leads to vulnerability to oxidative stress. Finally, excess intracellular glucose activates the receptor for Nogo, and leads to apoptosis induction via the Rho kinase.