

Abstract
Zika virus (ZIKV), a positive-sense RNA flavivirus, has attracted considerable attention recently for its potential to cause serious neurological problems, including microcephaly, cortical thinning, and blindness during early development. Recent findings suggest that ZIKV infection of the brain can occur not only during very early stages of development, but also in later fetal/early neonatal stages of maturation. Surprisingly, after peripheral inoculation of immunocompetent mice on the day of birth, the first cells targeted throughout the brain were isolated astrocytes. At later stages, more neurons showed ZIKV immunoreactivity, in part potentially due to ZIKV release from infected astrocytes. In all developing mice studied, we detected infection of retinal neurons; in many mice, this was also associated with infection of the lateral geniculate, suprachiasmatic nuclei, and superior colliculus, suggesting a commonality for the virus to infect cells of the visual system. Interestingly, in mature mice lacking a Type 1 interferon response (IFNR−/−), after inoculation of the eye, the initial majority of infected cells in the visual system were glial cells along the optic tract. ZIKV microinjection into the somatosensory cortex on one side of the normal mouse brain resulted in mirror infection restricted to the contralateral somatosensory cortex without any infection of midline brain regions, indicating the virus can move by axonal transport to synaptically coupled brain loci. These data support the view that ZIKV shows considerable complexity in targeting the CNS and may target different cells at different stages of brain development.
SIGNIFICANCE STATEMENT Zika virus (ZIKV) can cause substantial damage to the developing human brain. Here we examine a developmental mouse model of ZIKV infection in the newborn mouse in which the brain is developmentally similar to a second-trimester human fetus. After peripheral inoculation, the virus entered the CNS in all mice tested and initially targeted astrocytes throughout the brain. Infections of the retina were detected in all mice, and infection of CNS visual system nuclei in the brain was common. We find that ZIKV can be transported axonally, thereby enhancing virus spread within the brain. These data suggest that ZIKV infects multiple cell types within the brain and that astrocyte infection may play a more important role in initial infection than previously appreciated.
Keywords: astrocyte, behavior dysfunction, development, infection, neurotropic, virus
Introduction
Within the last 2 years, a virus of African origin, Zika virus (ZIKV), has become established in the Americas. The emergence of this virus has generated considerable alarm, particularly related to the potential for ZIKV to cause neurological complications in fetal humans, as first noted in Brazil (Kleber de Oliveira et al., 2016; Lessler et al., 2016). More recently ZIKV infection has expanded to a number of other countries within the Americas. In the United States, ZIKV has become a substantial concern as a growing number of infections are beginning to be reported in late summer 2016 (McCarthy, 2016) (http://www.cdc.gov/zika/geo/united-states.html). The most profound problem associated with ZIKV is the generation of permanent neurological dysfunction in infected fetuses. ZIKV-related brain dysfunction is found not only in obvious cases of microcephaly, which may be most commonly associated with ZIKV infection during the first trimester of human pregnancy (Brasil et al., 2016; Kleber de Oliveira et al., 2016), but also in neonates with a normal head size born from mothers infected during later stages of pregnancy (França et al., 2016; Hazin et al., 2016). The probability of fetal microcephaly in ZIKV-infected pregnant women ranges from 1% to 13%; there is a concern that other nervous system complications, although not as obvious as microcephaly, may be more prevalent (Cauchemez et al., 2016; Johansson et al., 2016; Trevathan, 2016). For instance, in addition to microcephaly, cortical thinning, abnormal limb postures, blindness and visual impairment, and auditory dysfunction have also been reported in neonates born from ZIKV-infected mothers (Calvet et al., 2016; de Carvalho Leal et al., 2016; de Fatima Vasco Aragoa et al., 2016; Driggers et al., 2016; França et al., 2016; Martines et al., 2016; van der Linden et al., 2016). ZIKV infections are also associated with an increase in Guillain-Barré syndrome (Dos Santos et al., 2016; Paixão et al., 2016; Niemeyer et al., 2017), an immune system-mediated motor dysfunction that can lead to paralysis that often dissipates over time (Hughes and Rees, 1997).
Models for studying ZIKV have been developed focusing in part on mice immunodeficient for Type 1 IFN responses (Lazear et al., 2016; Rossi et al., 2016) or on organoid brain-like cultures (Cugola et al., 2016; Dang et al., 2016; Garcez et al., 2016; Li et al., 2016) or E15 embryonic brain slices (Brault et al., 2016). However, within the developing brain, the types of cells infected and the progression of infection has not yet received much attention despite the importance of understanding ZIKV targeting in the brain. A number of papers have examined in utero infections of the mouse fetus (Aliota et al., 2016; Miner et al., 2016a, b; Yockey et al., 2016); in normal mice, ZIKV generally does not infect the fetus; in immunodeficient mice lacking a Type 1 IFN response, the pregnant mother usually shows a lethal response to the virus, but the fetal mice do get infected. Our focus here is to study the ontogeny of ZIKV movement into the brain in an animal model consisting of normal newborn neonatal mice to examine the progression of ZIKV infection within the developing CNS after peripheral inoculation. An important underlying rationale of our study is that the newborn mouse brain is substantially less developed than the newborn human fetal brain. Based on initial neurogenesis, axon extension, establishment and refinement of connections, myelin formation, increase in brain volume, and early behavioral milestones, the neonatal mouse CNS at birth approximately parallels a second-trimester human fetus (Clancy et al., 2001, 2007a, b; Workman et al., 2013), and therefore represents a viable animal model for studying potential nervous system complications associated with ZIKV infection in later phases of human gestation.
Materials and Methods
Zika virus.
ZIKV of the Asian lineage, from Cambodia (ZIKV FSS13025) (Heang et al., 2012), similar to the ZIKV that has entered the Americas was used. ZIKV was a gift from Dr. Brett Lindenbach (Yale University). ZIKV was harvested from infected cultures of Vero-E6 cells at 4 dpi, filtered, divided into aliquots and stored at −80°C. Harvested viral stocks were titered by plaque assay on Vero cells and typically had a concentration of 2 × 107 plaque forming units (pfu)/ml. We also used pseudorabies virus (PRV) expressing a GFP reporter (gift from Dr.Lynn Enquist, Princeton University) for one set of experiments using coinjection of both PRV+ZIKV into the left cortex: 150 nl of PRV (1.5 × 102 pfu) + 150 nl ZIKV (3 × 102 pfu), mixed together and injected simultaneously in the same volume.
Immunocytochemistry.
Antiserum against ZIKV was generated in adult male rats. Seven weeks after an initial subcutaneous and intraperitoneal inoculation with ZIKV, rats were inoculated a second time. Eight days later, serum was harvested. A goat anti-rat secondary antiserum was used for immunostaining (Invitrogen A11007).
Immunostaining was done on both cell cultures and histological sections from control and inoculated mice. Frozen or vibratome sections were cut from fixed mouse brain and after incubation in normal goat serum containing 0.3% Triton X-100, were incubated in primary rat anti-ZIKV serum. After multiple washes of the primary antiserum, goat-anti-rat conjugated to Alexa-594 was used at dilutions of 1:300 to 1:1000 for 1–2 h, and was then washed off. After immunostaining, some sections were labeled with DAPI or counterlabeled with immunostaining against GFAP (ThermoFisher, PA5–16291) or IBA1/microglia (Biocare Medical, CP290A) (Ito et al., 1998) using a different fluorophore.
The ZIKV antiserum only labeled cells that had been inoculated with ZIKV and not uninfected control cells. Absence of the primary antibody resulted in no staining. The primary anti-ZIKV serum was used for immunofluorescent labeling at a dilution between 1:1000 and 1:20,000. The antiserum labeled ZIKV-infected cells well; it worked poorly in immunolabeling a different flavivirus, Yellow Fever virus-17D.
In vitro neutralization of ZIKV infection.
To determine whether the antiserum would block ZIKV infection, a plaque reduction neutralization assay was performed, similar to that used for Dengue virus (Russell et al., 1967; Roehrig et al., 2008). Antiserum was heat-inactivated at 56°C for 30 min, then serial twofold dilutions were mixed with ZIKV and incubated for 1 h at room temperature. The dilutions were then plaque assayed in quadruplicate on Vero cells. A 50% reduction of ZIKV plaques was found at an antiserum dilution of 1:640.
Mice.
Several primary strains of mice were used: immunocompetent C57BL/6 and Swiss Webster mice and immunocompromised mice that lacked the Type 1 IFNR (IFNR−/−) and therefore showed no Type 1 IFN response. Immunocompetent neonatal mice (n = 51) were inoculated intraperitoneally on the day of birth (P0) with 3 μl (6 × 104 pfu) or 7 μl (4 × 105 pfu) ZIKV for survival and histological analyses, or with 103 or 2 × 103 pfu intraperitoneally for one survival study. Sample size was based on previous publications. Other virus concentrations are described in some of the figures. Some mice expressed GFP in the pro-opiomelanocortin (POMC) cells (gift from Dr. M.Low), and were used to identify amacrine cells in the infected retina. Mice intended for CNS immunocytochemistry were killed by anesthetic overdose at daily intervals after inoculation and perfused transcardially with saline followed by 4% paraformaldehyde.
Anesthetized mice lacking the Type 1 IFNR (IFNR−/−) 4 weeks old were given an intraocular injection of ZIKV (1 μl containing 2 × 104 pfu ZIKV). Work with ZIKV in mice and rats was approved by the university committee on animal use. With the exception of the use of pregnant female mice to investigate the possibility of ZIKV transfer to the fetus, all other experiments used both male and female mice randomly. In survival experiments, if a mouse showed substantial deterioration with difficulty in movement and trouble feeding, it was killed, as per recommendation of the university committee on animal use.
Cell culture.
A number of cell types were used in vitro including Vero-E6 African green monkey kidney epithelial cells obtained from C. Cepko (Harvard University), human brain primary astrocytes were described previously (Ozduman et al., 2008), and primary mouse brain cells harvested from C57BL/6 mice shortly after birth. Vero cells were grown in MEM supplemented with 10% FBS. Primary mouse and human brain cells were grown in DMEM with 10% FBS. All cultures were maintained in a Napco incubator with humidified atmosphere at 37°C with 5% CO2.
Virus release and plaque assay.
A virus release assay for ZIKV progeny was performed using human brain astrocytes. Briefly, cells were plated in 35 mm dishes and inoculated the following day with ZIKV (multiplicity of infection = 20) and allowed to adsorb for 1 h at 37°C. After adsorption, the cells were washed with PBS and 3 ml of fresh media was added to the well. At the indicated time points, 60 μl samples of media were withdrawn and replaced with the same volume of fresh media. Samples were stored at −80°C for later viral titer determination by plaque assay. The plaque assay consisted of inoculation of Vero cells grown in 12 well plates using serial dilutions of ZIKV samples for 1 h at 37°C. Cells were then washed with PBS and an overlay of 1% carboxymethyl cellulose in MEM with FBS was applied. ZIKV-infected cultures were incubated 4 d to allow time for plaque development. Plaques were visualized with crystal violet after removal of the carboxymethyl cellulose overlay.
IFN experiments in human and mouse brain cells.
Nearly confluent primary cultures of human and mouse brain cells were grown in 24 well plates and pretreated for 12 h with IFN-αA/D (Sigma I4401) at the indicated concentrations. After IFN pretreatment, cultures were inoculated with ZIKV (6 × 105 pfu/well) or media (control) and incubated for 2 d. Cells were then fixed and ZIKV immunocytochemical labeling was done. ZIKV-infected cells were counted from triplicate wells for each condition.
Results
ZIKV invasion of developing brain
To study the natural progression of ZIKV infection in the developing brain, normal immune competent C57BL/6 mice were inoculated intraperitoneally on the day of birth with the Asian lineage of ZIKV (ZIKV FSS13025); this is the lineage of ZIKV that has spread to the Americas and has raised serious concerns about ZIKV-induced brain dysfunctions. Intraperitoneal inoculations in part model the potential movement of the virus transplacentally along the umbilical cord into the fetus better than subcutaneous application. Mice (n = 27) were killed by anesthetic overdose and fixative perfusion at daily intervals from 1 d post inoculation (1 dpi) to 10 dpi and at longer intervals after 10 dpi. We used a high-titer anti-ZIKV antiserum we raised in rats; the antiserum blocked ZIKV infection in vitro and was selective with immunocytochemistry for ZIKV-infected cells and did not label noninfected cells (see Fig. 4). We found no detectable ZIKV immunoreactivity in the brain at 1 and 2 dpi. At 3 dpi, we began to find ZIKV infection in muscles of the head, in the neural retina, and in a small number of cells within the brain. At 4 dpi (n = 6 of 6), infection in the brain was common, and consistently found in all mice (n = 18) after 4 dpi indicating a strong propensity for CNS infections after inoculation of the virus outside the brain (Fig. 1F–I).
Figure 4.
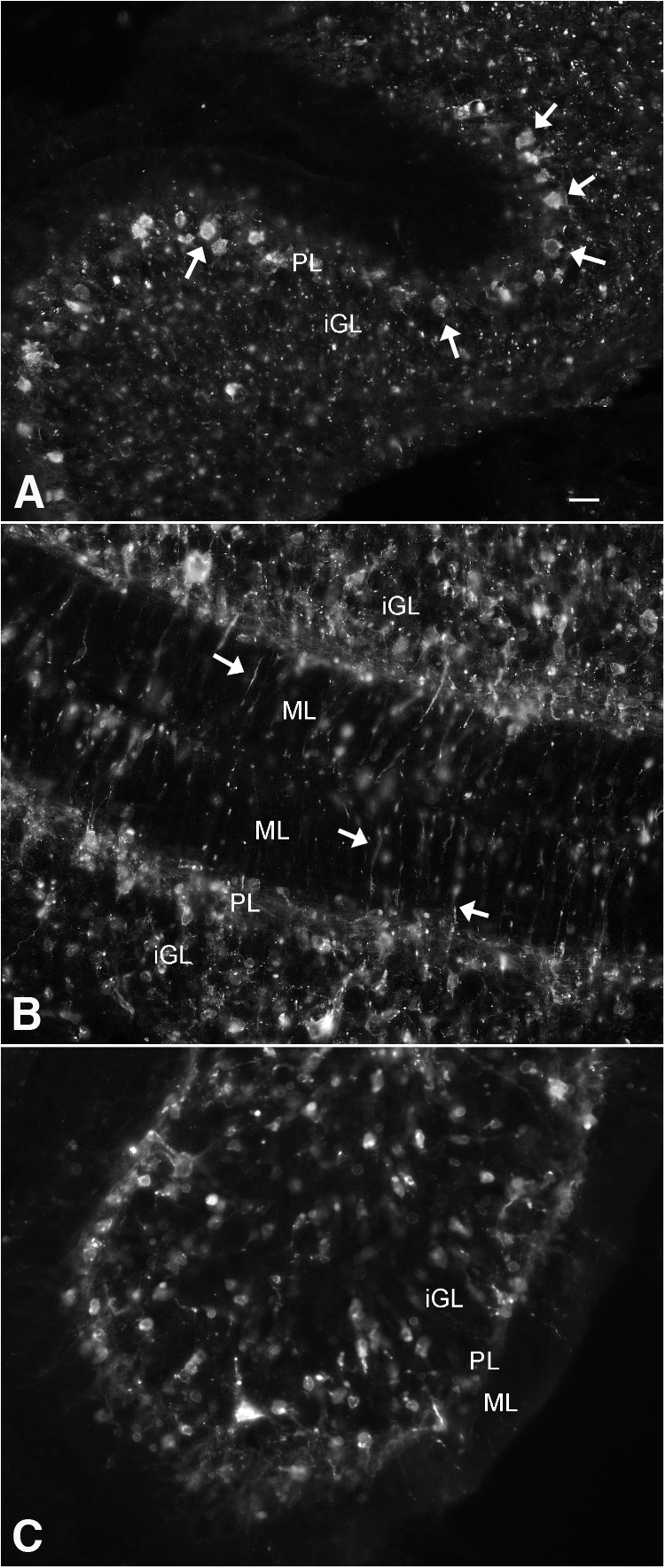
ZIKV heterogeneity of infection in cerebellum. At 7 dpi after intraperitoneal inoculation at P0, the cerebellum from the same mouse shows different stages and cell types of infection in different lobes of the cerebellar cortex in A–C. A, Arrows indicate Purkinje cells in Purkinje cell layer and punctate labeling suggestive of late-stage infection in the granule layer. B, Thin processes in the molecular layer are seen, and a large number of cells in the granule layer. C, Unlike in B, few processes or infected cells are detected in the molecular layer. Scale bar, 30 μm. iGL, Internal granule cell layer; PL, Purkinje cell layer containing cell bodies of Purkinje cells and Bergmann glia; ML, molecular layer.
Figure 1.

ZIKV enters brain after intraperitoneal inoculation. A, B, Confocal scanning microscope images of ZIKV-infected astrocytes. Scale bar, 8 μm. C, ZIKV-infected glial cell (red) contains GFAP immunoreactivity (green). The ZIKV immunoreactivity is found out to the tips of the glial processes, whereas the GFAP is confined more to the shaft of primary and secondary processes. Scale bar, 10 μm. D, No colocalization of ZIKV and Iba1 (a microglia marker) was detected. Scale bar, 12 μm. E, ZIKV-infected neuron with punctate immunoreactivity at 7 dpi after P0 inoculation. Scale bar, 15 μm. F, G, At 4 dpi after intraperitoneal inoculation, most infected cells are glia (blue dots); only rare neurons (red dots) are infected. G, More caudal midbrain region of the same mouse as in F. H, More cells, particularly astrocytes, are infected at 5 dpi. I, At 7 dpi after intraperitoneal inoculation, ZIKV has spread throughout the brain. At this stage of development, ZIKV infects glia (blue) and neurons (red) with little preference for brain regions, with the exception in this case of strong hippocampal neuron infection, including the dentate gyrus, CA3, and CA1.
Although most previous studies, in particular those based on brain organoids, have focused on potential infections of neuronal progenitor cells, surprisingly, initial infections targeted glial cells of the normal developing brain, particularly cells with an astrocyte morphology and a large number of short processes; both cell body and glial processes to their terminal endfeet showed robust ZIKV immunoreactivity as detected with fluorescence and confocal laser microscopy (Fig. 1A,B). ZIKV-infected glial cells with an astrocyte morphology expressed the astrocyte antigen GFAP immunoreactivity in many of the glial processes (Fig. 1C) but did not express microglia antigen Iba1 (Fig. 1D). One possible explanation for the initial astrocyte targeting may be the glial endfeet that wrap around the vasculature and present one of the first cellular targets for a virus leaving a blood vessel.
Neurons were infected soon after the glia, either from ZIKV released by infected glia or as primary infections via the vascular system. In initial stages of infection at 4 and 5 dpi, isolated infected glia far outnumbered neurons, as shown by the high number of cells with astrocyte morphology (Fig. 1F,G, blue) compared with those with a neuronal morphology (Fig. 1F–H, red). Over the next 2–3 d (6–8 dpi), the number of infected neurons increased (Fig. 1I) to the point that neurons began to outnumber infected glia in some brain areas. High densities of infected neurons were detected in different brain regions, sometimes initially on one side of the brain, suggestive of local release and infection. In some mice at 7 dpi, robust neuronal infection was seen in CA1 and CA3 regions of the hippocampus (Fig. 1I), raising concerns about long-term memory problems in ZIKV-infected human fetuses. We quantified the number of astrocytes and neurons in 4 brain regions from 4 to 10 dpi. All regions showed a predominant initial infection of astrocytes at 4 and 5 dpi. The hippocampus showed the greatest neuronal density at 7 dpi; and all regions studied, including cortex, hippocampus, thalamus, and hypothalamus, showed greater infection of neurons than of astrocytes by 10 dpi (Fig. 2).
Figure 2.
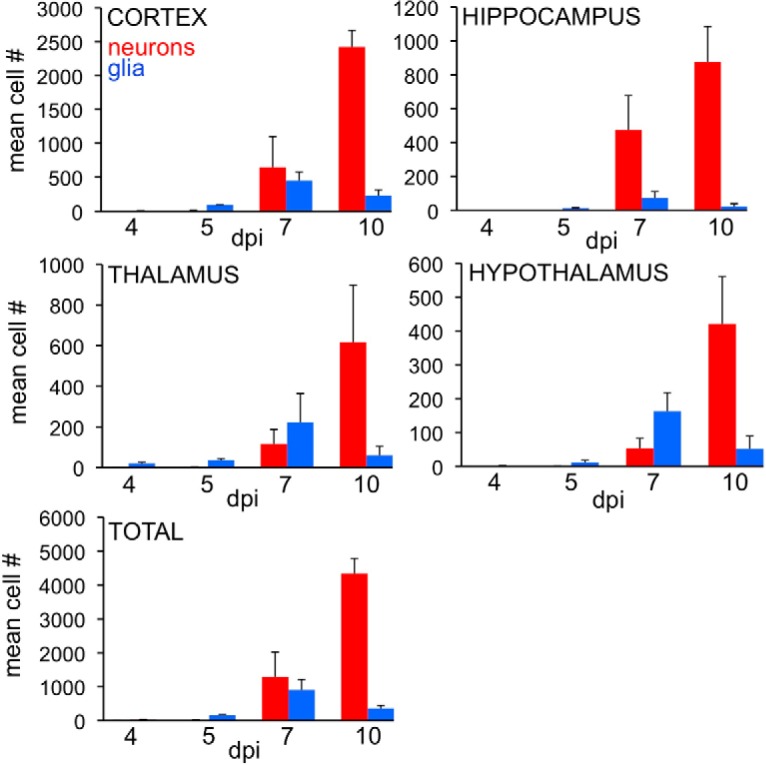
Relative number of infected astrocytes and neurons/section during development. The number of immunoreactive astrocytes (blue) and neurons (red) was counted at 4, 5, 7, and 10 dpi (n = 3/time point). Bar indicates SD. Initially at 4 and 5 dpi, most of the cells had the morphology of astrocytes in all 4 areas studied. By 7 dpi, astrocytes were still more numerous than neurons in thalamus and hypothalamus, whereas neurons were more numerous in cortex and hippocampus. By 10 dpi, infected neurons were more prevalent in all areas studied.
We found no propensity for infections to develop around the ventricular system in developing mice (Fig. 1F–I), arguing against a hypothesis that ZIKV initially enters the brain via the CSF at this stage of development. ZIKV infection was characterized by granular immunoreactivity, typical of the ZIKV “factories” that have been described (Bell et al., 1971). ZIKV was found not only in the cell body, but immunoreactive granules could be found far out in distal dendrites (Fig. 1E). In cortical pyramidal cells, ZIKV granular immunoreactivity was seen in both the primary large apical dendrite extending toward the cortical surface (Fig. 3) and also in smaller secondary basal dendrites ramifying closer to the cell body. In cortical interneurons, ZIKV granules were found in the cell body and multiple dendrites. Thin immunoreactive axons were detected at later stages of neuronal infection.
Figure 3.

Cortical neurons infected with ZIKV 7 dpi after intraperitoneal inoculation at P0. Substantive infection is seen in the primary dendrites extending toward the right to the cortical surface. The beaded dendrites are typical of the neuronal deterioration in late stages of ZIKV infection. Scale bar, 30 μm.
One striking finding was the initial widespread but sparse infection throughout multiple brain regions seen in all 4 dpi mice (n = 6) after intraperitoneal inoculation (Fig. 1F,G). In many cases, only a few cells were infected in any given region. Although not a common initial target of the virus, the cerebellum showed very strong infection by 7–10 dpi. The cerebellum is of particular interest during this period because it is one of the few areas of the brain in which (granule) neurons are still being generated from neural precursor cells during the postnatal day 6–10 period of development. Considerable heterogeneity of infection was noted, particularly in the early phase of infection. Even in a single cerebellum, different cells, including granule cells, Purkinje cells, and Bergmann glia, at different stages of infection appeared in different lobes of the developing cerebellar cortex (Fig. 4). ZIKV immunoreactivity was seen in cells and processes in different layers, including the molecular, Purkinje, and inner granule cell layer with different levels of infection and cell deterioration in different regions of the same cerebellum (Fig. 4).
To examine the spinal cord of developing mice, P0 mice (n = 6) received intraperitoneal inoculations of ZIKV. As we found motor dysfunction involving the hind limbs (see below), we focused on the lumbar spinal cord, a region of the cord that innervates the hind legs. Similar to the brain, small numbers of infected cells were seen at 4 dpi in the gray matter of the spinal cord. Astrocytes were often infected (Fig. 5B), and neurons were also found (Fig. 5A). By 10 dpi, the entire gray matter was heavily infected with cells in all spinal cord lamina. An image of the high infection rate in the ventral horn of lumbar cord is shown in Fig. 5C. All 6 mice examined from 4 to 10 dpi showed ZIKV infection in the lumbar spinal cord after intraperitoneal inoculation.
Figure 5.
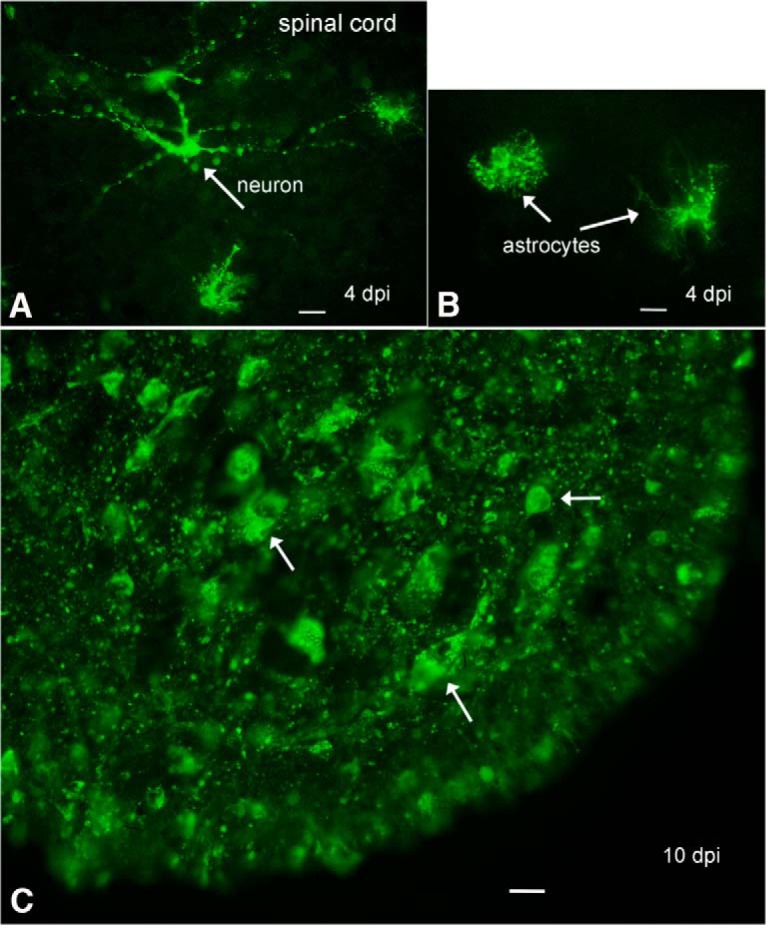
ZIKV in spinal cord. A, In the lumbar spinal cord gray matter, an immunoreactive degenerating neuron is seen (arrow) along with some immunoreactive glia, 4 dpi. Scale bar, 20 μm. B, Two immunoreactive astrocytes are shown (arrows), 4 dpi. Scale bar, 20 μm. C, By 10 dpi, the ventral horn of the spinal cord is filled with ZIKV-immunoreactive cells and processes. Scale bar, 15 μm.
To determine the time course of ZIKV infection and detection in brain cells, we inoculated human brain cultures consisting mostly of astrocytes, and examined these at multiple intervals after inoculation (Fig. 6A–G). ZIKV immunoreactivity was first detected at 12 hpi, and stronger staining at 1–3 dpi; by 4 dpi, infected cells showed substantial degeneration and cell death as determined with phase contrast microscopy and dead-cell ethidium homodimer labeling. Based on an in vitro progeny virus release assay, glia showed a productive infection and began to release new progeny ZIKV by 24 h after inoculation as determined by plaque assay of the culture medium (Fig. 6H). These data suggest that ZIKV may begin infecting cells in the brain 24 h earlier than we detect infection, and that astrocyte release of new ZIKV progeny may account for at least part of the increase in subsequent neuronal infection. Flaviviruses in general are often cytolytic but in some cells can establish a chronic infection (Lindenbach and Rice, 2001). In the current study, we found multiple indications that ZIKV infection led to cell death, including a reduced cell number in vitro as infection continued, labeling of infected cultured cells with the dead cell stain ethidium homodimer, the appearance of cells in the brain at late stages of infection with beaded processes and degenerating cell body, and the loss of neurons from some brain regions, such as the hippocampus in later stages of infection.
Figure 6.

Time course of ZIKV infection of brain cells. Primary human brain cells, mostly astrocytes, were inoculated at time 0, then fixed and immunostained at the indicated intervals. Immunoreactivity was not seen in uninfected control cultures (A) or at 2 h (B) post inoculation (hpi). C, D, At 12 hpi, faint immunoreactivity was detected in granules (D, enlarged region shown by arrow). Scale bar, 2.5 μm. Immunoreactivity became stronger up to 2 d (E, F) post inoculation (dpi). G, By 4 dpi, many of the immunoreactive cells were dead or dying. Scale bars: A–C, E–G, 25 μm. H, Viral release was measured using additional cultures infected after ZIKV (multiplicity of infection 20) inoculation for 1 h, then washed and supplied with fresh media. Media samples were harvested at the indicated time points, and viral concentration was measured by plaque assay. I, ZIKV antiserum harvested from inoculated rats and used for immunolabeling was tested for the ability to neutralize ZIKV infection in vitro. Top, Counterstained cultures show the decline in viral plaque number after exposure to increasing concentrations of antiserum, corroborating antibody selectivity. Bottom, Bar graph indicates that a 50% reduction in ZIKV plaque number was obtained at 1:640 antiserum dilution. Error bars indicate SEM (n = 4).
Previous reports based in part on in vitro brain organoid cultures have shown that ZIKV infects neural precursors (Cugola et al., 2016; Dang et al., 2016; Li et al., 2016) consistent with our detection of strong cerebellar infection during the period of granule cell generation during P7–P10 cerebellar development (Fig. 4). In neonatal mice, neither the subventricular zone nor the rostral migratory stream between the subventricular zone and the olfactory bulb, both sites containing neural progenitor cells, showed any preferential early infection (Fig. 1F–I). In older P18 brains of mice surviving P0 ZIKV inoculation, areas were identified containing sporadic infected cells in addition to groups of dead cells that lacked detectable active ZIKV infection, suggesting local elimination of the virus in the maturing brain.
Neurological/behavioral dysfunction
Neonatal infection, either by intraperitoneal or subcutaneous inoculation (n = 84 for i.p. and s.c.), was often lethal within 2–3 weeks (Fig. 7B) for higher doses (6 × 104 to 1.4 × 105 pfu). With lower doses (1–2 × 103 pfu), approximately one-fourth of the animals survived past 3 weeks (Fig. 7B). Subsequent to ZIKV inoculation, behavioral and developmental disturbances were noted indicating neurological deterioration, including reduced body weight gain (Fig. 7C) and reduced growth with reduced body length and attenuated tail length (Fig. 7D,E). We also studied neurological dysfunction from the time of infection. Infected mice showed a progressive increase in motor dysfunction particularly involving the hind legs (Fig. 7F,G). ZIKV-mediated neurological disturbances were first seen at 3 and 4 dpi with intraperitoneal inoculation (Fig. 7G), and slightly later with subcutaneous inoculation (Fig. 7F), consistent with the first detection of ZIKV in the brain. The ongoing behavioral and neurological deterioration of infected mice suggests that the ZIKV lethality may in large part be due to the spread of ZIKV within the brain over time.
Figure 7.

Zika virus in newborn mice induces neurological disease and death. A, Survival for P0 mice inoculated with 1.4 × 105 (black line, n = 13) or 6 × 104 pfu intraperitoneally (green line, n = 46). Total, n = 59. C57BLand Swiss Webster mice were used; because we found no statistical difference between the two strains, data were combined. B, Survival for slightly older P1 mice with 103 pfu intraperitoneally (n = 17) or 2 × 103 subcutaneously (n = 8). Noninfected controls, n = 10; total, n = 35. ***p < 0.001, survival at P28 (Log-rank, Mantel-Cox) test. The 25% survival in ZIKV subcutaneously, 22.2% survival in ZIKV intraperitoneal controls (CTR). C–E, Somatic parameters of postnatal development. Data are mean ± SEM. Two-way ANOVA with postnatal day as repeated measures, Holm-Sidak's multiple-comparison test: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. ZIKV subcutaneously versus CTR shown above CTR line; ZIKV intraperitoneally versus CTR shown below ZIKV intraperitoneal line. F, Neurological symptoms were assessed for 20 d for P1 mice inoculated subcutaneously with 2 × 103 pfu ZIKV similar to the observations of Lazear et al. (2016) in older mice. Chart shows that neurological symptoms occur in greater numbers of mice over time. G, P1 mice were infected intraperitoneally with 103 pfu with ZIKV and signs of neurological dysfunction assessed for 20 d. The percentage of each group of mice displaying the indicated motor dysfunction is shown. These are from the same mice evaluated for lethality and somatic development.
ZIKV in normal and IFNR deficient adult mice
In contrast to developing mice, normal adult mice receiving ZIKV intraperitoneally (n = 10) showed no lethal response to the virus and no long-term symptoms, as previously noted (Dang et al., 2016; Rossi et al., 2016). We examined the brains of adult mice inoculated intraperitoneally with ZIKV and found no infection within the CNS at 7–10 dpi (n = 7), suggesting that the normal immune system is sufficient to keep the virus out of the brain after early development. However, direct intracranial microinjection of ZIKV (0.5 μl/104 pfu) was lethal in 3 of 6 normal mice. Similar to adult mice, adult rats (n = 3) inoculated with ZIKV peripherally showed no obvious adverse symptoms over a period of 2 months.
Because one critical factor in the developing brain is a reduced IFN response (Lazear et al., 2016; Rossi et al., 2016), we tested adult IFNR−/− mice lacking the Type I IFNR. ZIKV was lethal in 6 of 6 adult IFNR−/− mice after intracerebral injection (0.5 μl/104 pfu). Immunocytochemical analysis of adult IFNR−/− mice at 6 d after intraperitoneal inoculation revealed widespread ZIKV infection throughout the brain with both astrocytes and neurons showing strong virus immunoreactivity, indicating that IFN plays an important role in attenuating ZIKV infection in the CNS. During development, Type 1 IFN responses increase with age to provide a first line of defense against viral infections of the brain, and in the adult can upregulate antiviral gene expression even at some distance from the initial site of virus infection (van den Pol, 2006; van den Pol et al., 2007, 2014). However, during early development, IFN responses to virus presence may be weaker than in the adult (van den Pol et al., 2007), potentially allowing virus spread in the immature brain.
In vitro experiments showed that both mouse and human brain cells are protected against ZIKV by Type 1 IFN (Fig. 8), similar to the ability of IFN to attenuate ZIKV infection in skin cells (Hamel et al., 2015). Maturation of the IFN responses within the brain may be one important factor that reduces the likelihood of problematic ZIKV infection in later development and in adults.
Figure 8.
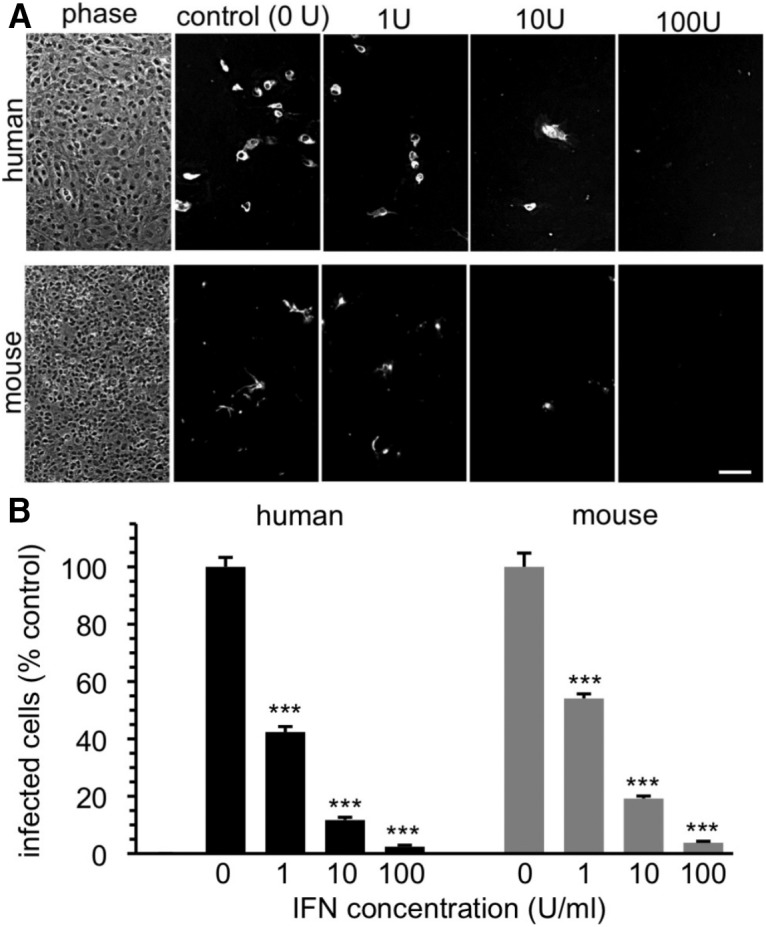
Type 1 IFN blocks ZIKV infection of human and mouse brain cells. A, Human (top) and mouse (bottom) brain cells were inoculated with ZIKV in the absence (0 U) or presence of 1, 10, and 100 U/ml IFN (Sigma I4401). Left, Phase shows typical cell density. Cells were fixed and immunostained at 2 dpi. Scale bar, 100 μm. B, Bar graph represents percentage infected cells, with controls set to 100%. IFN reduces infection in a dose-dependent manner. Error bars indicate SEM (n = 3). ***p < 0.01 (ANOVA with Bonferroni post-test).
Similar to previous reports (Cugola et al., 2016), intraperitoneal inoculation of pregnant normal mice (n = 7) with ZIKV from gestational day 6–14 showed no evidence of transplacental virus transfer to the fetus (n = 14 from 7 pregnancies), as determined with immunocytochemistry. Newborn mice (n = 22 mice from 5 litters) of ZIKV-infected mothers tended to be slightly smaller than controls (n = 10 mice from 2 litters) (p < 0.05 ANOVA) for the first 2–3 weeks of development, but over time returned to normal size, further arguing against ZIKV infection in these neonates. We attribute the slower initial neonatal growth to transient ZIKV-mediated debilitation in the mothers, all of whom recovered with no long-term symptoms.
ZIKV infection of the visual system
A substantial number of human cases of microcephaly associated with ZIKV infections also show ocular dysfunction and pathological disturbances to the retina as well as optic nerve abnormalities. In addition, some cases of retinal dysfunction have been associated with ZIKV infection in the absence of microcephaly (Miranda et al., 2016; Ventura et al., 2016a, b; de Paula Freitas et al., 2016). With intraperitoneal inoculations at P0, all 14 mice studied showed some retinal infection by 4 dpi and later; similarly, 2 of 2 mice inoculated at P0 subcutaneously showed retinal infection. Cells in both the retinal ganglion cell layer and in the inner nuclear layer were commonly infected (Fig. 9A–C). Optic nerves leaving the retina contained ZIKV-immunoreactive axons. The brains of mice studied with retinal infection also showed infection (5 of 5) of at least some part of the CNS visual system, including retina, optic chiasm (Fig. 9E), suprachiasmatic nucleus, lateral geniculate nucleus, and/or superior colliculus (Fig. 9D). Infection of glia in the median eminence and hypothalamic arcuate nucleus, a region of the brain with a weak blood–brain barrier outside and just caudal to the optic chiasm, commonly showed infection (Fig. 9F). In transgenic mice expressing GFP in retinal GABAergic amacrine cells under control of the POMC promoter (Gallagher et al., 2010), ZIKV-infected a number of these amacrine cells, indicated by coexpression of GFP and ZIKV immunoreactivity; ZIKV also infected many cells that were negative for POMC-GFP by 7 dpi (Fig. 9G).
Figure 9.

Infection of visual system and other brain loci after intraperitoneal inoculation. A–C, Retina at 4 dpi after P0 inoculation; intraperitoneal ZIKV infects the ganglion cell layer (GCL) and the inner nuclear layer (INL). Immunoreactive processes are found in the internal plexiform layer (IPL). Red represents ZIKV immunoreactivity. Blue represents DAPI counterstain. Scale bar, 8 μm. D, ZIKV in superior colliculus. E, ZIKV in optic chiasm (OC). F, Directly caudal to the optic chiasm is the median eminence (ME), which also showed infection. Scale bar, 15 μm. G, Transgenic mouse expressing GFP in retinal POMC cells was inoculated at P0. By 7 dpi, both GFP-expressing amacrine cells (double arrowhead) and GFP-negative cells showed ZIKV infection. Green represents POMC amacrine cells. Orange represents ZIKV. Scale bar, 15 μm.
To examine infection of the visual system further, ZIKV was applied by intraocular inoculation to mice (n = 4, 4–6 weeks old) lacking a Type 1 IFN response due to the absence of the IFNR (IFNR−/−); the lack of Type 1 IFN response in these mice parallels the weak IFN response found during early development (van den Pol et al., 2002, 2007). At 3 and 4 dpi, infection was found in the optic nerve and visual system loci within the brain, including the lateral geniculate, superior colliculus, and suprachiasmatic nuclei (Fig. 10A–E). Surprisingly, the infections seen in the optic nerve often included glial cells within the optic chiasm and optic tract (Fig. 10C). Groups of infected optic nerve glia were found along the optic tract from the optic chiasm to the lateral geniculate nucleus, an unusual mechanism of virus spread. In addition, both astrocytes and neurons were infected in the visual system nuclei. A number of infected cells within the optic tract and optic chiasm expressed the astrocyte antigen GFAP (Fig. 11). Associated with the glial labeling was infection of the meninges at the surface of the brain, particularly adjacent to the infected cells. To corroborate the finding of glial cells along infected nerves within the brain, we also examined the sciatic nerve after intramuscular injection in the hind leg of IFNR−/− mice (4–6 weeks old). Again, we found infected glial cells associated with the sciatic nerve (Fig. 10F). It is notable that both the normal neonates and near-adult and adult IFNR−/− mice showed strong initial infection of astrocytes.
Figure 10.
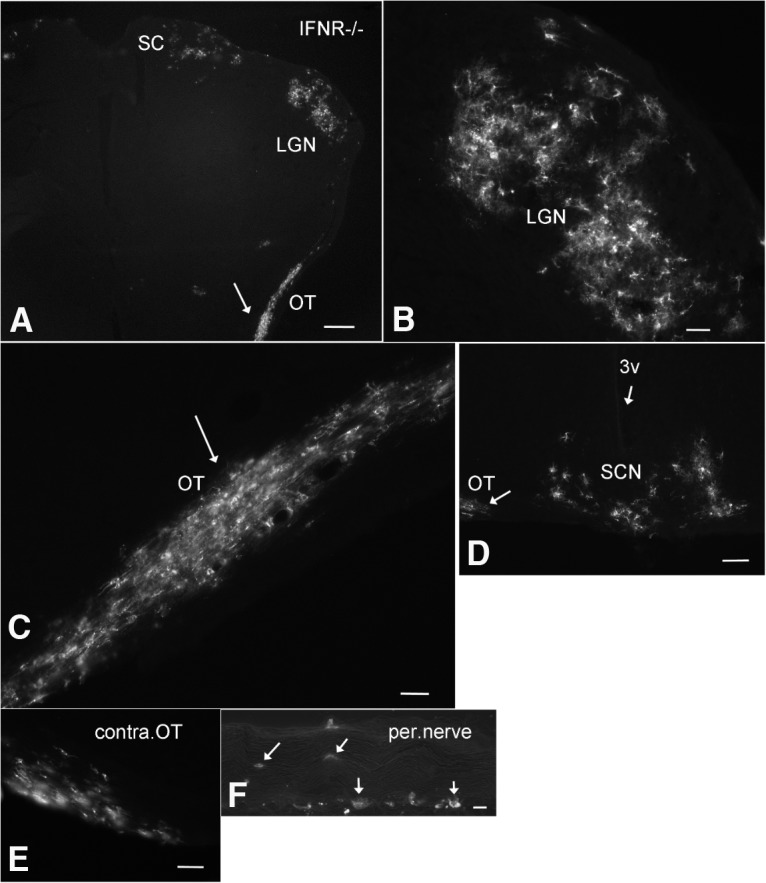
ZIKV infects the visual system in IFNR−/− mouse. A–E, After intraocular inoculation of 4-week-old IFNR−/− mice, ZIKV was identified at 4 dpi in visual system regions (A), including the optic tract (OT), lateral geniculate nucleus (LGN), superior colliculus (SC), and the suprachiasmatic nucleus (SCN). Contralateral to C, the other optic tract also showed ZIKV infection. F, In another IFNR−/− mouse, intramuscular injection into the hind leg led to ZIKV infection of cells in and around the peripheral (per.) nerve innervating the leg. Scale bars: A, 200 μm; B, 60 μm; C, 40 μm; D, 60 μm; E, 40 μm; F, 30 μm.
Figure 11.
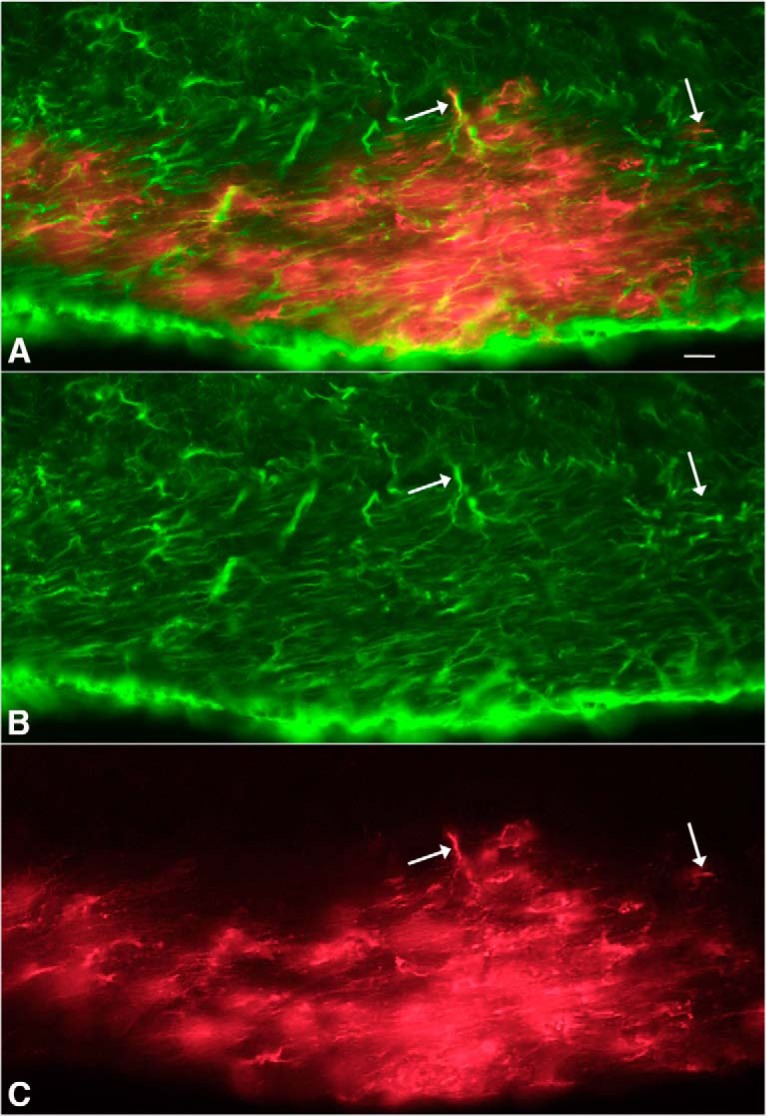
ZIKV-infected cells (red) in optic chiasm colabeled with GFAP. A, Merged image from red ZIKV infections (C) and green immunostaining for GFAP (B) after intraocular inoculation in IFNR−/− mouse, 4 dpi. A–C, Same microscope field. Scale bar, 5 μm.
Axonal transport of ZIKV
In the course of examining brains of mice inoculated intraperitoneally on the day of birth, in later stages of infection, in some mice we found infection in mirror image on opposite sides of the brain. One possible explanation for this is axonal transport from a common area of innervation, or axonal projections between corresponding regions on opposite sides of the brain. To determine whether ZIKV is transported intra-axonally to distant brain regions, we made microinjections of ZIKV into the left cortex of normal mice (4 weeks old, n = 4), and killed mice at 3 and 4 dpi. Here we used 300 or 500 nl, a volume 100 times smaller than that used in classical work showing that ZIKV does infect the brain (Bell et al., 1971); further, unlike the early work, we did not use virus harvested from developing brain inoculations. At 3 dpi, we found infected neurons not only in different cortical layers at the injection site, but also in the contralateral cortex. By 4 dpi, we detected robust infection at the injection site (Fig. 12A,F), and a growing number of infected neurons in the contralateral cortex (Fig. 12B,F) and ipsilateral and contralateral striatum (Fig. 12D,E); both regions receive axonal innervation from the cortex (Molyneaux et al., 2007). Importantly, in the middle region of the brain between the two sets of infected cortical neurons, there were no detectable infected cells of any sort, arguing against virus diffusion from one side of the brain to the distant contralateral side (Fig. 12C,F).
Figure 12.
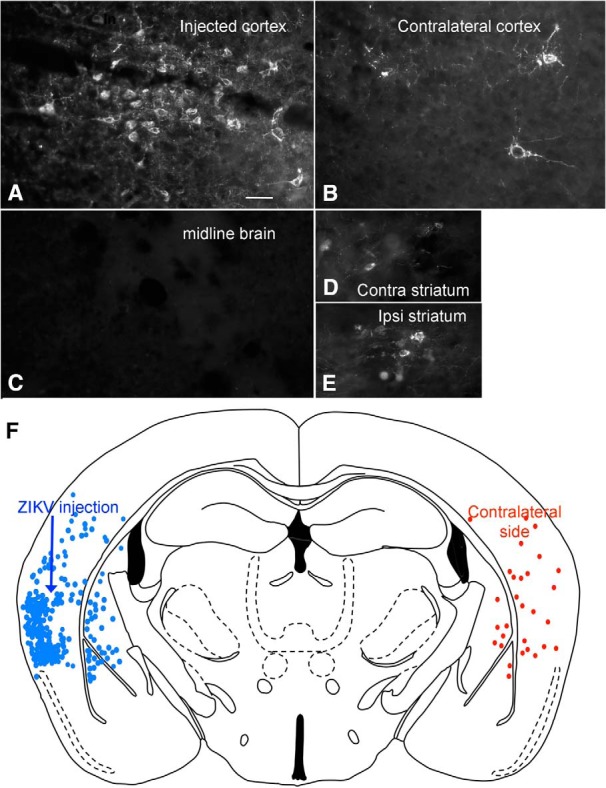
Axonal transport of ZIKV from one brain region to another. After intracortical microinjection (300 nl) into the left cortex (A, F), 4 d later strong infection was found in the contralateral right cortex (B, F) and in the contralateral (contra) (D) and ipsilateral (ipsi) (E) striatum. A few cells were also detected in the amygdala. All these regions are synaptically connected with the cortex. No detectable infection was found in the middle region of the brain (C). F, Composite image with all infected cells drawn from two sections of the same brain. Scale bar, 30 μm.
To corroborate the results above, we used coinjections of ZIKV with the Bartha strain of PRV that serves as a viral axonal tracer (Card et al., 1993, 1995). Both the PRV GFP reporter and red ZIKV immunofluorescence was found in the same region of the injected side of the cortex, and in the contralateral cortex showing a mirror image of the injected side (Fig. 13). In the cortex contralateral to the injected side, some neurons expressed the PRV reporter only (Fig. 13B), others expressed ZIKV immunoreactivity only (Fig. 13A), and a third group expressed both PRV GFP reporter together with ZIKV immunoreactivity. The coinjections corroborated our initial interpretation because cells on the side of the brain contralateral to the injection showed both green GFP reporter (from PRV) and red immunofluorescence indicating ZIKV. Together, these data suggest that at least some axonal pathways within the brain can transport ZIKV to infect neurons in a distant brain site.
Figure 13.
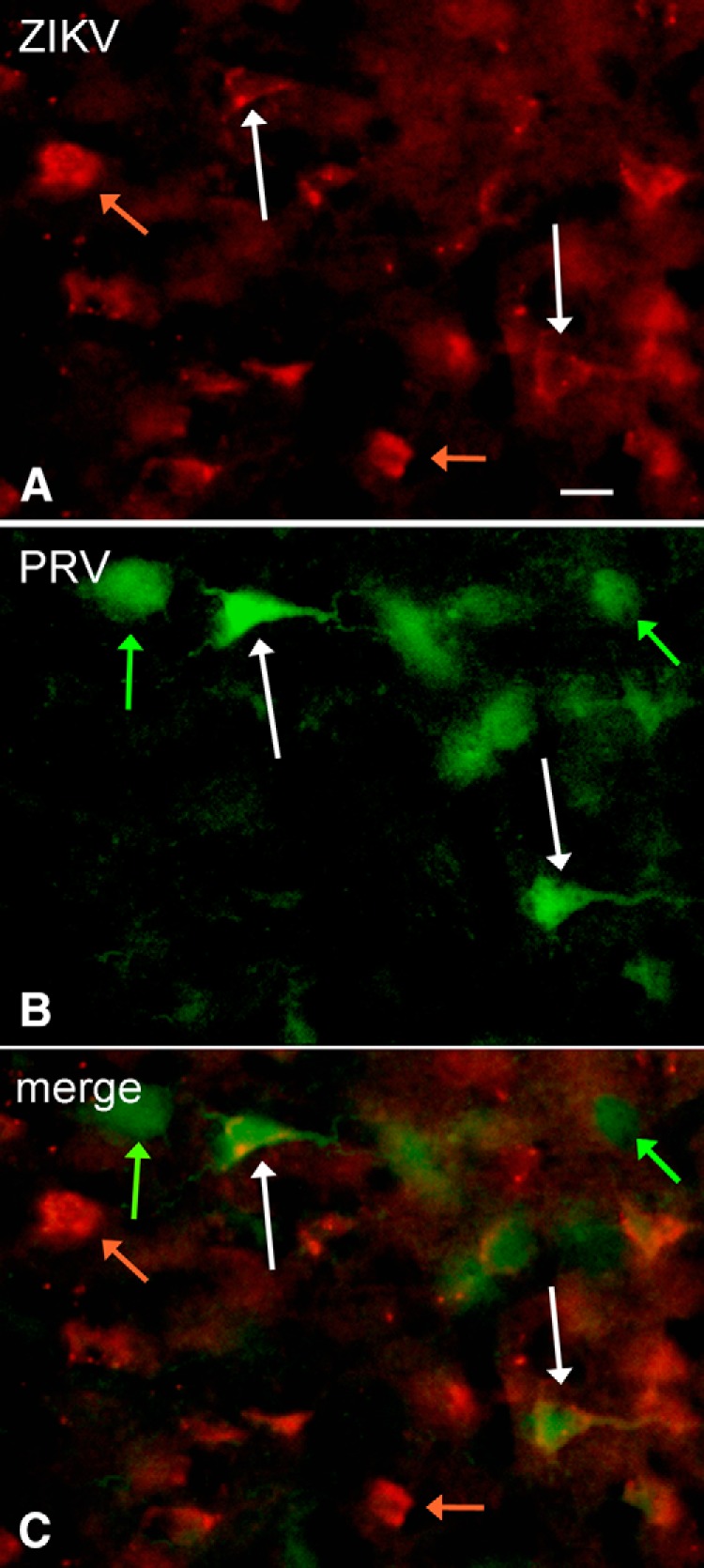
Axonal transport of PRV and ZIKV to contralateral cortex. After comicroinjection (300 nl) of PRV and ZIKV to the left cortex, both viruses are carried by axonal transport to the contralateral right cortex by 3 dpi. A, ZIKV immunoreactivity. Scale bar, 18 μm. B, PRV GFP green reporter expression. C, Merged image showing that some cells are infected only with ZIKV (orange arrows), only with PRV (green arrows), or with both PRV and ZIKV (white arrows).
Discussion
A number of recent papers have examined ZIKV infections in different mouse models, particularly in immunodeficient mice (Lazear et al., 2016; Rossi et al., 2016). In addition, 3D organoid in vitro brain cultures have been elegantly used to describe a propensity of ZIKV to infect neuronal progenitor cells in early brain development (Cugola et al., 2016; Dang et al., 2016; Garcez et al., 2016; Li et al., 2016); a potential limitation of organoid cultures is the lack of a vascular system and the absence of the normal types of immune cells.
Our animal model of early ZIKV infection emulates the early second trimester of human brain development; during the second trimester of human brain development and P0 mouse CNS development, there are a number of parallels, including cortical layer II/III and IV neurogenesis, onset of retinal waves of action potential propagation, peak of optic nerve axon number, and peak of subventricular zone expansion in the developing cortex (Clancy et al., 2007a, b; Workman et al., 2013). A key difference between our neonatal model and an in utero model is the absence of placental virus inhibition in the neonate. Both the structure and the immune components of the placenta constitute a biological barrier that blocks microorganisms in the pregnant mother from easily accessing the fetus (Mor and Cardenas, 2010; Robbins and Bakardjiev, 2012).
Another virus that can generate many of the same neurological symptoms as ZIKV if infections occur in the fetal period is the unrelated cytomegalovirus, which uses a double-stranded DNA genome; CMV is often considered the most common infectious agent causing permanent neurological dysfunction in the developing human, and these problems can include sensory, motor, memory, and other complications. Importantly, CMV can continue to induce neurological dysfunction even during early neonatal development (Bray et al., 1981; Perez-Jimenez et al., 1998; Gaytant et al., 2002; Dollard et al., 2007; Mocarski et al., 2007; Tsutsui, 2009), raising the question of whether ZIKV can similarly evoke neurological problems during the same neonatal developmental period in humans; these later neurological problems would be more subtle and difficult to diagnose than microcephaly, a current focus of ZIKV concern.
Here we studied developing newborn normal mice. Our data show, for the first time, that after peripheral inoculation, there is a substantial initial infection of glial cells within the brain, particularly cells with a morphology and GFAP antigen expression consistent with astrocytes. Infected isolated astrocytes were found throughout the brain, indicating that this was a widespread occurrence and suggesting a large number of ZIKV penetrations into the developing brain. Over the next few days of development, the number of infected neurons showed a substantial increase such that the relative number of infected neurons exceeded the number of infected astrocytes by P10. Consistent with our in vitro demonstration of ZIKV progeny release from infected glia, astrocytes may not only show the first signs of infection but may also serve to further amplify and distribute infectious virus to nearby neurons and glia. Similar to ZIKV, CMV also tends to target astrocytes (van den Pol et al., 2007). That glia are not necessarily a common cell target of other viruses in the developing brain is shown by vesicular stomatitis virus, which targets neurons rather than glia (van den Pol et al., 2002, 2014).
Interestingly, in the early stages of brain infection, microglia showed little ZIKV immunoreactivity, whereas astrocytes in the same brain were commonly infected. Our finding of the initial selective infection of astrocytes does not argue against a perspective that in earlier stages of brain development, macrophages that can be infected by ZIKV (Quicke et al., 2016) may migrate into the brain carrying the virus in a Trojan-horse mechanism of spreading infection. Although infected astrocytes were previously found after large-volume brain-derived ZIKV injections directly into the brain (Bell et al., 1971), we show, for the first time, that the native virus initially selects and targets astrocytes after peripheral inoculation during the period of mouse development immediately after birth.
We found behavioral problems in ZIKV-inoculated neonatal mice, often involving hind limbs. Motor deficits may relate to the common infection of the cerebellum, motor cortex, or spinal cord.
Another striking finding here was the consistent infection of the retina in all mice inoculated intraperitoneally or subcutaneously at birth. This differs from previous work in both younger and older periods of development. Eye infections were very rare in fetal infections, occurring in only 5% of those infected (Miner et al., 2016b). ZIKV subcutaneous inoculations at older postnatal day 8 showed only a subset (50%) of mice with eye infections; P8 is approximately equivalent to the third trimester near-term fetus in humans, whereas our P0 inoculation parallels the second trimester (Clancy et al., 2001, 2007a, b; Workman et al., 2013). The retinal infections from P0 inoculation were not restricted to a single-cell type but rather were found in a number of different cells in the ganglion cell layer and inner nuclear layer. In addition to retinal infection, in many mice the CNS visual system showed signs of infection. Consistent with our finding of early astrocyte infection throughout the brain, astrocytes were commonly infected in visual system pathways.
An increase in caspase-3 immunoreactivity was reported in the brain after P8 inoculations (Miner et al., 2016b), suggesting a response to virus or to degeneration of the optic nerve following ZIKV retinal infection. Our data show that the virus itself displays an early preference for infecting regions of the brain subserving vision in the developing mouse brain at a developmental stage equivalent to mid-gestation in human CNS development. ZIKV infection of the regions of the brain involved in sight suggests that visual problems arising from ZIKV fetal infection in humans (Ventura et al., 2016b; de Paula Freitas et al., 2016) may not only arise from retinal infections, which were very prevalent in our studies in developing mice, but also from infection of the optic nerve or regions of the brain involved in vision.
After ZIKV microinjections into one side of the brain, 3–4 d later, the opposite mirror image region of the cortex showed infection, whereas the middle of the brain between the two cortices showed no infection of any cells. These data suggest axonal transport of the virus from one side of the brain to the synaptically connected contralateral cortex. We also used a coinjection of ZIKV with the retrogradely transported herpes Type 1 porcine PRV expressing a GFP reporter. In the cortex, contralateral to the site of coinjection, we found both ZIKV immunoreactivity and PRV reporter gene expression, consistent with the view that both viruses were axonally transported. Our data on ZIKV transport in the cortex are consistent with previous reports showing that axonally transported tracers label parallel groups of cells on opposite sides of the cortex after unilateral injection (Wise and Jones, 1976), and support the hypothesis that ZIKV can be transported within axons to infect distant sites within the brain. Contralateral axonal transport of ZIKV in the cortex is also consistent with our data from ocular injections of the virus that resulted in infection of the CNS visual system. Axonal transport of another flavivirus, West Nile Virus, has previously been described (Samuel et al., 2007).
In conclusion, the robust and consistent early infection of astrocytes before neurons was unexpected and suggests the infection of astrocytes merits more attention in brain infections in humans, particularly given the important roles of astrocytes in maintenance of the blood–brain barrier, enhancement of myelination, CNS repair and inflammation, development and migration of neurons, guidance of growing axons, and neurotransmitter modulation (Khakh and Sofroniew, 2015). Attenuating ZIKV infection of astrocytes may reduce subsequent infection of nearby neurons. The common infection of retina and central visual nuclei in our studies suggests that visual problems found in newborns from ZIKV-infected mothers could arise from both peripheral (retina) and CNS complications. That ZIKV can be transported axonally appears to constitute one mechanism underlying the spread of the virus within the brain.
Footnotes
References
- Aliota MT, Caine EA, Walker EC, Larkin KE, Camacho E, Osorio JE (2016) Characterization of lethal zika virus infection in AG129 Mice. PLoS Negl Trop Dis 10:e0004682. 10.1371/journal.pntd.0004682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TM, Field EJ, Narang HK (1971) Zika virus infection of the central nervous system of mice. Arch Gesamte Virusforsch 35:183–193. 10.1007/BF01249709 [DOI] [PubMed] [Google Scholar]
- Brasil P, Pereira JP Jr, Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M, Rabello RS, Valderramos SG, Halai UA, Salles TS, Zin AA, Horovitz D, Daltro P, Boechat M, Raja Gabaglia C, Carvalho de Sequeira P, Pilotto JH, Medialdea-Carrera R, Cotrim da Cunha D, Abreu de Carvalho LM, et al. (2016) Zika virus infection in pregnant women in Rio de Janeiro-preliminary report. N Engl J Med 375:2321–2334. 10.1056/NEJMoa1602412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault JB, Khou C, Basset J, Coquand L, Fraisier V, Frenkiel MP, Goud B, Manuguerra JC, Pardigon N, Baffet AD (2016) Comparative analysis between flaviviruses reveals specific neural stem cell tropism for Zika virus in the mouse developing neocortex. EBioMedicine 10:71–76. 10.1016/j.ebiom.2016.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray PF, Bale JF, Anderson RE, Kern ER (1981) Progressive neurological disease associated with chronic cytomegalovirus infection. Ann Neurol 9:499–502. 10.1002/ana.410090513 [DOI] [PubMed] [Google Scholar]
- Calvet G, Aguiar RS, Melo AS, Sampaio SA, de Filippis I, Fabri A, Araujo ES, de Sequeira PC, de Mendonça MC, de Oliveira L, Tschoeke DA, Schrago CG, Thompson FL, Brasil P, Dos Santos FB, Nogueira RM, Tanuri A, de Filippis AM (2016) Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 16:653–660. 10.1016/S1473-3099(16)00095-5 [DOI] [PubMed] [Google Scholar]
- Card JP, Rinaman L, Lynn RB, Lee BH, Meade RP, Miselis RR, Enquist LW (1993) Pseudorabies virus infection of the rat central nervous system: ultrastructural characterization of viral replication, transport, and pathogenesis. J Neurosci 13:2515–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, Dubin JR, Whealy ME, Enquist LW (1995) Influence of infectious dose upon productive replication and transynaptic passage of pseudorabies virus in rat central nervous system. J Neurovirol 1:349–358. 10.3109/13550289509111024 [DOI] [PubMed] [Google Scholar]
- Cauchemez S, Besnard M, Bompard P, Dub T, Guillemette-Artur P, Eyrolle-Guignot D, Salje H, Van Kerkhove MD, Abadie V, Garel C, Fontanet A, Mallet HP (2016) Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet 387:2125–2132. 10.1016/S0140-6736(16)00651-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL (2001) Translating developmental time across mammalian species. Neuroscience 105:7–17. 10.1016/S0306-4522(01)00171-3 [DOI] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB, Anand KJ (2007a) Extrapolating brain development from experimental species to humans. Neurotoxicology 28:931–937. 10.1016/j.neuro.2007.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy B, Kersh B, Hyde J, Darlington RB, Anand KJ, Finlay BL (2007b) Web-based method for translating neurodevelopmental from laboratory species to humans. Neuroinformatics 5:79–94. 10.1385/NI:5:1:79 [DOI] [PubMed] [Google Scholar]
- Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimarães KP, Benazzato C, Almeida N, Pignatari GC, Romero S, Polonio CM, Cunha I, Freitas CL, Brandão WN, Rossato C, Andrade DG, Faria Dde P, Garcez AT, Buchpigel CA, Braconi CT, et al. (2016) The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534:267–271. 10.1038/nature18296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, Rana TM (2016) Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19:258–265. 10.1016/j.stem.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho Leal M, Ferreira Muniz L, da Silva Caldas Neto S, van der Linden V, Ferreira Ramos RC (2016) Sensorineural hearing loss in a case of congenital Zika virus. Braz J Otorhinolaryngol 30:piiS1808–8694(16)30127–6. 10.1016/j.bjorl.2016.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Fatima Vasco Aragoa M, van der Linden V, Brainer-Lima AM, Coeli RR, Rocha MA, Sobral da Silva P, Durce Costa Gomes de Carvalho M, van der Linden A, Cesario de Holanda A, Valenca MM (2016) Clinical features and neuroimaging (CT and MRI) findings in presumed Zika virus related congenital infection and microcephaly: retrospective case series study. Br Med J 353:i1901. 10.1136/bmj.i1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paula Freitas B, de Oliveira Dias JR, Prazeres J, Sacramento GA, Ko AI, Maia M, Belfort R Jr (2016) Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in Salvador, Brazil. JAMA Ophthalmol 134:529–535. 10.1001/jamaophthalmol.2016.0267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollard SC, Grosse SD, Ross DS (2007) New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol 17:355–363. 10.1002/rmv.544 [DOI] [PubMed] [Google Scholar]
- Dos Santos T, Rodriguez A, Almiron M, Sanhueza A, Ramon P, de Oliveira WK, Coelho GE, Badaró R, Cortez J, Ospina M, Pimentel R, Masis R, Hernandez F, Lara B, Montoya R, Jubithana B, Melchor A, Alvarez A, Aldighieri S, Dye C, et al. (2016) Zika virus and the Guillain-Barré syndrome: case series from seven countries. N Engl J Med 375:1598–1601. 10.1056/NEJMc1609015 [DOI] [PubMed] [Google Scholar]
- Driggers RW, Ho CY, Korhonen EM, Kuivanen S, Jääskeläinen AJ, Smura T, Rosenberg A, Hill DA, DeBiasi RL, Vezina G, Timofeev J, Rodriguez FJ, Levanov L, Razak J, Iyengar P, Hennenfent A, Kennedy R, Lanciotti R, du Plessis A, Vapalahti O (2016) Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 374:2142–2151. 10.1056/NEJMoa1601824 [DOI] [PubMed] [Google Scholar]
- França GV, Schuler-Faccini L, Oliveira WK, Henriques CM, Carmo EH, Pedi VD, Nunes ML, Castro MC, Serruya S, Silveira MF, Barros FC, Victora CG (2016) Congenital Zika virus syndrome in Brazil: a case series of the first 1501 livebirths with complete investigation. Lancet 388:891–897. 10.1016/S0140-6736(16)30902-3 [DOI] [PubMed] [Google Scholar]
- Gallagher SK, Witkovsky P, Roux MJ, Low MJ, Otero-Corchon V, Hentges ST, Vigh J (2010) β-Endorphin expression in the mouse retina. J Comp Neurol 518:3130–3148. 10.1002/cne.22387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK (2016) Zika virus impairs growth in human neurospheres and brain organoids. Science 352:816–818. 10.1126/science.aaf6116 [DOI] [PubMed] [Google Scholar]
- Gaytant MA, Steegers EA, Semmekrot BA, Merkus HM, Galama JM (2002) Congenital cytomegalovirus infection: review of the epidemiology and outcome. Obstet Gynecol Surv 57:245–256. 10.1097/00006254-200204000-00024 [DOI] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau VM, Choumet V, Briant L, Desprès P, Amara A, Yssel H, Missé D (2015) Biology of Zika virus infection in human skin cells. J Virol 89:8880–8896. 10.1128/JVI.00354-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazin AN, Poretti A, Turchi Martelli CM, Huisman TA, Huisman TA, Di Cavalcanti Souza Cruz D, Tenorio M, van der Linden A, Pena LJ, Brito C, Gil LH, de Barros Miranda-Filho D, Marques ET, Alves JG (2016) Computed tomographic findings in microcephaly associated with Zika virus. N Engl J Med 374:2193–2195. 10.1056/NEJMc1603617 [DOI] [PubMed] [Google Scholar]
- Heang V, Yasuda CY, Sovann L, Haddow AD, Travassos da Rosa AP, Tesh RB, Kasper MR (2012) Zika virus infection, Cambodia, 2010. Emerg Infect Dis 18:349–351. 10.3201/eid1802.111224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RAC, Rees JH (1997) Clinical and epidemiologic features of Guillain-Barré Syndrome. J Infect Dis 176 [Suppl 2]:S92–S98. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localization of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57:1–9. 10.1016/S0169-328X(98)00040-0 [DOI] [PubMed] [Google Scholar]
- Johansson MA, Mier-y-Teran-Romero L, Reefhuis J, Gilboa SM, Hills SL (2016) Zika and the risk of microcephaly. N Engl J Med 375:1–4. 10.1056/NEJMp1605367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Sofroniew MV (2015) Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18:942–952. 10.1038/nn.4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber de Oliveira W, Cortez-Escalante J, De Oliveira WT, do Carmo GM, Henriques CM, Coelho GE, Araújo de França GV (2016) Increase in reported prevalence of microcephaly in infants born to women living in areas with confirmed Zika virus transmission during the first trimester of pregnancy-Brazil, 2015. MMWR Morb Mortal Wkly Rep 65:242–247. 10.15585/mmwr.mm6509e2 [DOI] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS (2016) A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730. 10.1016/j.chom.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessler J, Chaisson LH, Kucirka LM, Bi Q, Grantz K, Salje H, Carcelen AC, Ott CT, Sheffield JS, Ferguson NM, Cummings DA, Metcalf CJ, Rodriguez-Barraquer I (2016) Assessing the global threat from Zika virus. Science 353:aaf8160. 10.1126/science.aaf8160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin CF, Xu Z (2016) Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19:120–126. 10.1016/j.stem.2016.04.017 [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Rice CM (2001) Flaviviridae: the viruses and their replication. In: Fields virology (Knipe DM, Howley PM, eds), pp 991–1041. Philadelphia: Lippincott Williams Wilkins. [Google Scholar]
- Martines RB, Bhatnagar J, Keating MK, Silva-Flannery L, Muehlenbachs A, Gary J, Goldsmith C, Hale G, Ritter J, Rollin D, Shieh WJ, Luz KG, Ramos AM, Davi HP, Kleber de Oliveria W, Lanciotti R, Lambert A, Zaki S (2016) Notes from the field: evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses—Brazil, 2015. MMWR Morb Mortal Wkly Rep 65:159–160. 10.15585/mmwr.mm6506e1 [DOI] [PubMed] [Google Scholar]
- McCarthy M. (2016) US officials issue travel alert for Miami area as Zika cases rise to 15. Br Med J 354:i4298. [DOI] [PubMed] [Google Scholar]
- Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, Mysorekar IU, Diamond MS (2016a) Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165:1081–1091. 10.1016/j.cell.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JJ, Sene A, Richner JM, Smith AM, Santeford A, Ban N, Weger-Lucarelli J, Manzella F, Rückert C, Govero J, Noguchi KK, Ebel GD, Diamond MS, Apte RS (2016b) Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep 16:3208–3218. 10.1016/j.celrep.2016.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda HA 2nd, Costa MC, Frazão MA, Simão N, Franchischini S, Moshfeghi DM (2016) Expanded spectrum of congenital ocular findings in microcephaly with presumed Zika infection. Ophthalmology 123:1788–1794. 10.1016/j.ophtha.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Mocarski E, Shenk T, Pass R (2007) Cytomegaloviruses. In: Fields virology (Knipe DM, Howley PM, eds), pp 2701–2772. Philadelphia: Lippincott Williams Wilkins. [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8:427–437. 10.1038/nrn2151 [DOI] [PubMed] [Google Scholar]
- Mor G, Cardenas I (2010) The immune system in pregnancy: a unique complexity. Am J Reprod Immunol 63:425–433. 10.1111/j.1600-0897.2010.00836.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer B, Niemeyer R, Borges R Marchiori E (2017) Acute disseminated encephalomyelitis following Zika virus infection. Eur Neurol 77:45–46. 10.1159/000453396 [DOI] [PubMed] [Google Scholar]
- Ozduman K, Wollmann G, Piepmeier JM, van den Pol AN (2008) Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J Neurosci 28:1882–1893. 10.1523/JNEUROSCI.4905-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paixão ES, Barréto F, Teixeira Mda G, Costa Mda C, Rodrigues LC (2016) History, epidemiology, and clinical manifestations of Zika: a systematic review. Am. J Public Health 106:606–612. 10.2105/AJPH.2016.303112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Jiménez A, Colamaria V, Franco A, Grimau-Merino R, Darra F, Fontana E, Zullini E, Beltramello A, Dalla-Bernardina B (1998) Epilepsy and disorders of cortical development in children with congenital cytomegalovirus infection. Rev Neurol 26:42–49. [PubMed] [Google Scholar]
- Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O'Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, Schinazi RF, Chakraborty R, Suthar MS (2016) Zika virus infects human placental macrophages. Cell Host Microbe 20:83–90. 10.1016/j.chom.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JR, Bakardjiev AI (2012) Pathogens and the placental fortress. Curr Opin Microbiol 15:36–43. 10.1016/j.mib.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrig JT, Hombach J, Barrétt AD (2008) Guidelines for plaque-reduction neutralization testing of human antibodies to Dengue viruses. Viral Immunol 21:123–132. 10.1089/vim.2008.0007 [DOI] [PubMed] [Google Scholar]
- Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ, Langsjoen RM, Paessler S, Vasilakis N, Weaver SC (2016) Characterization of a novel murine model to study Zika virus. Am J Trop Med Hyg 94:1362–1369. 10.4269/ajtmh.16-0111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell PK, Nisalak A, Sukhavachana P, Vivona S (1967) A plaque reduction test for Dengue virus neutralizing antibodies. J Immunol 99:285–290. [PubMed] [Google Scholar]
- Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS (2007) Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc Natl Acad Sci U S A 104:17140–17145. 10.1073/pnas.0705837104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevathan E. (2016) Editorial brain malformation surveillance in the Zika era. Birth Defects Res A Clin Mol Teratol 106:869–874. 10.1002/bdra.23582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui Y. (2009) Effects of cytomegalovirus infection on embryogenesis and brain development. Congenit Anom (Kyoto) 49:47–55. 10.1111/j.1741-4520.2009.00222.x [DOI] [PubMed] [Google Scholar]
- van den Pol AN. (2006) Viral infections in the developing and mature brain. Trends Neurosci 29:398–406. 10.1016/j.tins.2006.06.002 [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Dalton KP, Rose JK (2002) Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J Virol 76:1309–1327. 10.1128/JVI.76.3.1309-1327.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Robek MD, Ghosh PK, Ozduman K, Bandi P, Whim MD, Wollmann G (2007) Cytomegalovirus induces interferon-stimulated gene expression and is attenuated by interferon in the developing brain. J Virol 81:332–348. 10.1128/JVI.01592-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Ding S, Robek MD (2014) Long-distance interferon signaling within the brain blocks virus spread. J Virol 88:3695–3704. 10.1128/JVI.03509-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden V, Filho EL, Lins OG, van der Linden A, Aragão Mde F, Brainer-Lima AM, Cruz DD, Rocha MA, Sobral da Silva PF, Carvalho MD, do Amaral FJ, Gomes JA, Ribeiro de Medeiros IC, Ventura CV, Ramos RC (2016) Congenital Zika syndrome with arthrogryposis: retrospective case series study. Br Med J 354:i3899. 10.1136/bmj.i3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura CV, Maia M, Dias N, Ventura LO, Belfort R Jr (2016a) Zika: neurological and ocular findings in infant without microcephaly. Lancet 387:2502. 10.1016/S0140-6736(16)30776-0 [DOI] [PubMed] [Google Scholar]
- Ventura CV, Maia M, Travassos SB, Martins TT, Patriota F, Nunes ME, Agra C, Torres VL, van der Linden V, Ramos RC, Rocha MǍ, Silva PS, Ventura LO, Belfort R Jr (2016b) Risk factors associated with the ophthalmoscopic findings identified in infants with presumed Zika virus congenital infection. JAMA Ophthalmol 134:912–918. 10.1001/jamaophthalmol.2016.1784 [DOI] [PubMed] [Google Scholar]
- Wise SP, Jones EG (1976) The organization and postnatal development of the commissural projection of the rat somatic sensory cortex. J Comp Neurol 168:313–343. 10.1002/cne.901680302 [DOI] [PubMed] [Google Scholar]
- Workman AD, Charvet CJ, Clancy B, Darlington RB, Finlay BL (2013) Modeling transformations of neurodevelopment sequences across mammalian species. J Neurosci 33:7368–7383. 10.1523/JNEUROSCI.5746-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yockey LJ, Varela L, Rakib T, Khoury-Hanold W, Fink SL, Stutz B, Szigeti-Buck K, van den Pol A, Lindenbach BD, Horvath TL, Iwasaki A (2016) Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166:1247–1256. 10.1016/j.cell.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]