

Abstract
Nicotine dependence is defined by dopaminergic neuronal activation within the nucleus accumbens (ACB) and by affected neural projections from nicotine-stimulated neurons. Control of any subsequent neural activities would underpin any smoking cessation strategy. While extensive efforts have been made to study the pathophysiology of nicotine addiction, more limited works were developed to find imaging biomarkers. If such biomarkers are made available, addictive behaviors could be monitored noninvasively. To such ends, we employed manganese (Mn2+)-enhanced magnetic resonance imaging (MEMRI) to determine whether it could be used to monitor neuronal activities after acute and chronic nicotine exposure in rats. The following were observed. Mn2+ infusion identified ACB and hippocampal (HIP) neuronal activities following acute nicotine administration. Chronic exposure was achieved by week long subcutaneously implanted nicotine mini-pump. Here nicotine was shown to activate neurons in the ACB, HIP, and the prefrontal and insular cortex. These are all central nervous system reward regions linked to drug addiction. In conclusion, MEMRI is demonstrated to be a powerful imaging tool to study brain subregion specific neuronal activities affected by nicotine. Thus, we posit that MEMRI could be used to assess smoking-associated tolerance, withdrawal and as such serve as a pre-clinical screening tool for addiction cessation strategies in humans.
Keywords: Manganese-enhanced MRI, smoking, nicotine, addiction, neuronal activity
Introduction
Smoking is a major public health problem and a leading cause of death in the United States [1,2]. This is linked, in large measure, to the development of heart disease, peripheral and central vascular insufficiencies, and lung, pancreas, bladder, liver, stomach and throat cancer. All significantly affects healthcare delivery and costs for the person, family and society [1,2]. Long-term abstinence has proven difficult to achieve. Smoking cessation leads to number of aversive syndromes and relapse [2,3] as well as to hostility, agitation, mood fluctuations and in the most severe state-suicide [3,4]. Notably, only a handful of such strategies have proven successful [4,5]. One significant obstacle of effective cessation rests in the limited understanding of the neurobiological mechanisms underlying nicotine’s effects on brain function. This is best explained by physiologic brain subregion changes that occur as a consequence of chronic nicotine exposure [6-13]. Thus, to develop any effective dependence treatment strategies, the neural responses to nicotine must be considered and subsequently controlled. The principal question is how to monitor such drug effects as to date there is no developed blood test or any specific biomarker that can quantitate addiction of nicotine.
Investigative research developed during the past decades demonstrated that during smoking, nicotine acts broadly on distributed brain acetylcholine receptors (nAChRs) and excites dopaminergic neurons within the ventral tegmental area (VTA). These activities elevate dopamine release in the mesolimbic pathway that includes the nucleus accumbens (ACB), hippocampus (HIP), amygdala (AMY), and prefrontal cortex (PFC) [6-13]. The noted mesolimbic dopamine pathway underlies the reward and reinforcement effects for nearly all addictive drugs [8]. Repeated use of nicotine induces cellular adaptations which influence several neural pathways that underlie addictive behaviors. Studies suggest that subtypes of nAChRs have different effects on nicotine reinforcement and dependence [14,15]. Nicotine also modulates dopamine release by binding to nAChRs located on excitatory glutamatergic and inhibitory gamma aminobutyric acid (GABAergic) neurons [14,15]. The serotonergic pathways also appear to be involved in nicotine dependence [16], while the interaction between PFC and VTA is not as well defined [17]. There is little argument that biomarkers for nicotine addiction are desperately needed. Bioimaging is one as yet underdeveloped opportunity. If successful, the use of noninvasive imaging techniques to determine the activation and/or suppression within neurological networks could improve the understanding of nicotine effects on the central nervous system (CNS). This would provide a path towards success for smoking cessation strategies. As neuronal activation can be measured by manganese (Mn2+)-enhanced magnetic resonance imaging (MEMRI), this approach appears as a powerful first step analyses for nicotine addiction and withdrawal. Indeed, Mn2+ is a paramagnetic MRI contrast agent and calcium-analog intimately tied to metabolomic brain states. Moreover, Mn2+ enters neurons by calcium channels and is transported along the axons anterogradely [18,19]. In this manner, MEMRI can be used to directly map the neurons activated by nicotine. In fact, MEMRI has been used to study neurologic effects of recreational substances in animals including alcohol [20-22], cocaine [18,23,24], and methamphetamine [25,26]. Such studies found that MEMRI is sensitive to neuronal activities affected by drug abuse. To this end, MEMRI tests were developed to assess rat neuronal activity in response to acute and chronic nicotine exposure. Neuronal groups in brain subregions involved in nicotine abuse were activated in both acute and chronic exposure conditions. The MEMRI tests developed in this study can serve as an operative biomarker for brain region-specific neuronal activities affected by nicotine and perhaps other drugs of abuse.
Materials and methods
Animals
Male Wistar rats (250-300 g) were obtained from Charles River Laboratories (Wilmington, MA). For acute nicotine exposure experiment, 12 rats were used. Six were administrated with nicotine and another 6 were used as controls that were injected with phosphate-buffered saline (PBS). In the chronic nicotine exposure study, 6 rats received continuous nicotine infusion for 7 days. A control group (n = 6) received PBS.
Animals were housed in the University of Nebraska Medical Center (UNMC) laboratory animal facility. All animal protocols were approved for use according to Association for Assessment and Accreditation of Laboratory Animal Care guidance and by the Institutional Animal Care and Use Committee (IACUC) of UNMC. All UNMC animal ethical guidelines set forth through the National Institutes of Health were met.
MRI
MRI was performed on a 7T/21 cm horizontal bore MRI scanner (Bruker, Billerica, MA) operating ParaVision 5.1 with a quadrature birdcage volume coil for RF signal transmission and reception. During scanning each rat was anesthetized with isoflurane mixed with oxygen. Anesthesia level was set during the experiment at ~1.0% isoflurane to maintain the breathing rate between 40-60 breaths per minute. Breathing rate was monitored using an MRI compatible physiological monitoring system (Model 1025, SA Instruments, Stony Brook, NY).
Acute nicotine exposure
Following acute nicotine exposure (Figure 1A), serial T1-weighted (T1-wt) MRI tests were performed using a RARE sequence with TR/TE = 440/17 ms, RARE factor = 4, and number of signal averages = 8, matrix size = 208 × 208, FOV = 32 × 32 mm2, and number of slices = 10. The slices were selected from the front part of the brain as shown in Figure 1B. Each T1-wt scan duration was 3.05 minutes. T2-weighted (T2-wt) MRI was first performed to acquire anatomyof the rat brain in sagittal direction. The anatomical image was used for the geometry prescription for the T1-wt scans (Figure 1B). Three baseline T1-wt images were acquired prior to manganese and drug (nicotine or PBS) administrations. Continuous injection of 50 mM MnCl2-4H2O (Sigma, St Louis, MO) via the left femoral vein was initiated by using an infusion pump (Harvard Apparatus, Holliston, MA) positioned outside of the scanner. After infusing 30 mg/kg Mn2+ over a period of 35 minutes, a bolus of 25% mannitol (5-7 ml/kg; Sigma) was injected through the right carotid artery to disrupt the brain-blood barrier (BBB). Mn2+ infusion was continued at a reduced infusion rate for another 25 minutes until the MRI signal was stabilized. The total amount of Mn2+ was 40 mg/kg. Twelve minutes after MnCl2 injection termination, either nicotine bitartrate (0.18 mg/kg free base concentration) or PBS saline was injected through the right femoral vein. The nicotine dose was chosen according to previous studies [17,27-29]. T1-wt MRI was then continuously acquired for 45 minutes.
Figure 1.
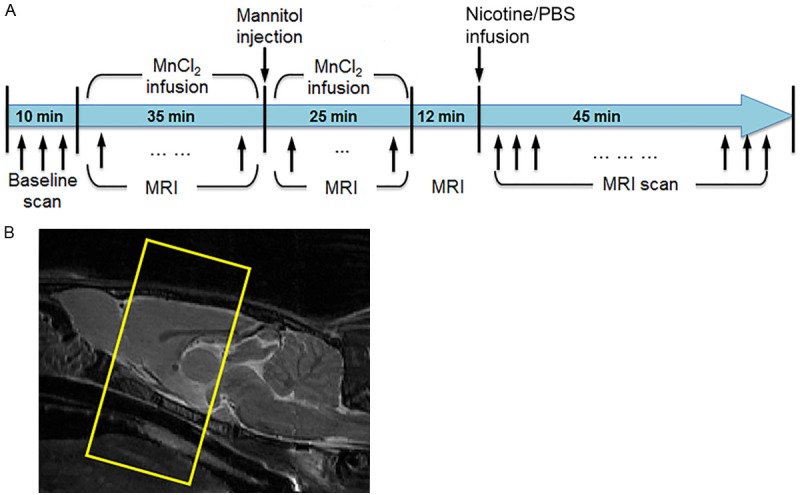
A. A schematic paradigm of the acute nicotine administration experiment. B. Location of brain slices of serial T1-wt MRI.
Chronic nicotine exposure
Each rat was implanted subcutaneously under isoflourane anesthesia with an Alzet osmotic minipump (Model 2ML1, Durect Corporation, Cupertino, CA) filled with nicotine bitartrate in physiological saline at concentrations adjusted to the animal’s weight. The infusion rate was set to deliver 3.0 mg/kg/day free base nicotine [28-30]. After 7 days of infusion, the pump was removed under anesthesia. MEMRI scanning was performed at 22 hours after the termination of nicotine infusion. PBS infused animals were used as controls. MnCl2 (50 mM) was administered i.p. at 48 and 24 hrs prior to MRI at a dose of 60 mg/kg. Rats were scanned using a T1 mapping sequence (RARE with variable repetition time (TR) from 0.5 s to 10 s, 12 slices, slice thickness = 1.0 mm, matrix size = 256 × 128, and FOV = 30 × 30 mm) and T1-wt MRI (FLASH, TR = 20 ms, flip angle = 20°, 3D isotropic resolution = 0.125 × 0.125 × 0.125 mm3). T1 mapping and T1-wt scans were also performed prior to nicotine/PBS infusion, and the acquired images served as baseline data for the calculation of Mn2+-induced signal enhancement.
MEMRI data analyses
In acute and chronic nicotine exposed rats time series analysis of the signal change due to Mn2+ infusion was calculated as: (SI-Sb)/Sb, where Sb is averaged signal intensity of the baseline scans, and SI is the signal intensity after MnCl2 administration. Brain regions assessed included the ACB, hippocampus (HIP), amygdala (AMY) and subregions of cortex. These were identified using the region of interest (ROI) function in MIPAV (http://mipav.cit.nih.gov/). The injection of mannitol through the right carotid artery suggests that the BBB opening on the contralateral left hemisphere of the brain might not be consistent. Thus, only the right side of the brain was used for the analysis. The regional brain activity reflected by manganese uptake was calculated using T1 weighted signal change. One-way ANOVA was used to compare the brain activity at each time point from controls versus nicotine injected rats. The nicotine effects were also determined by comparing time-averaged signals between pre- and post-injections. The time-series of signal intensities, reflecting neuronal activities, between MnCl2 termination and nicotine or PBS injection, were averaged and then normalized to baseline data. Similarly, signal increase relative to baseline after injection intervention with nicotine or PBS was calculated. The post-injection signal enhancement between nicotine injected rats and controls were compared using one-way ANOVA for ACB, HIP, AMY and cortical regions.
For chronic nicotine exposure, brain volumes in the 3D T1-wt images were first extracted using an in-house MATLAB program [31] based on the level sets method. The brain images were then registered to the MEMRI-based rat brain atlas (http://www.nitrc.org/projects/memriratbrains) using affine transformation first, and then nonlinear transformation (DiffeoMap, Johns Hopkins University, Baltimore, MD). The baseline and manganese enhanced T1-wt images were calibrated using T1 values to minimize the MRI system variations between the baselines and post-MnCl2 injection scans as described [32]. Signal enhancement was calculated by: (SMn-Sb)/Sb, where SMn and Sb are T1-wt signal intensity of post- and pre-MnCl2 injection, respectively. A pixel-by-pixel comparison was performed between groups using one-way ANOVA.
Results
Acute nicotine paradigm
Neuronal activities in response to acute nicotine exposure were reflected as T1-wt signal change compared to time on right hemisphere of the ACB and HIP (Figure 2A and 2B). The signal increased on both brain subregions following MnCl2 infusion. The signal increase rate became higher after mannitol injection and was stable upon the termination of MnCl2. The signal increase was approximately 10 and 12% on ACB and HIP, respectively compared to baseline. Nicotine injection caused about 2% signal increase within the ACB and 4% in the HIP. The signal increase in rats with nicotine injection was significantly higher than in PBS controls (P < 0.05).
Figure 2.
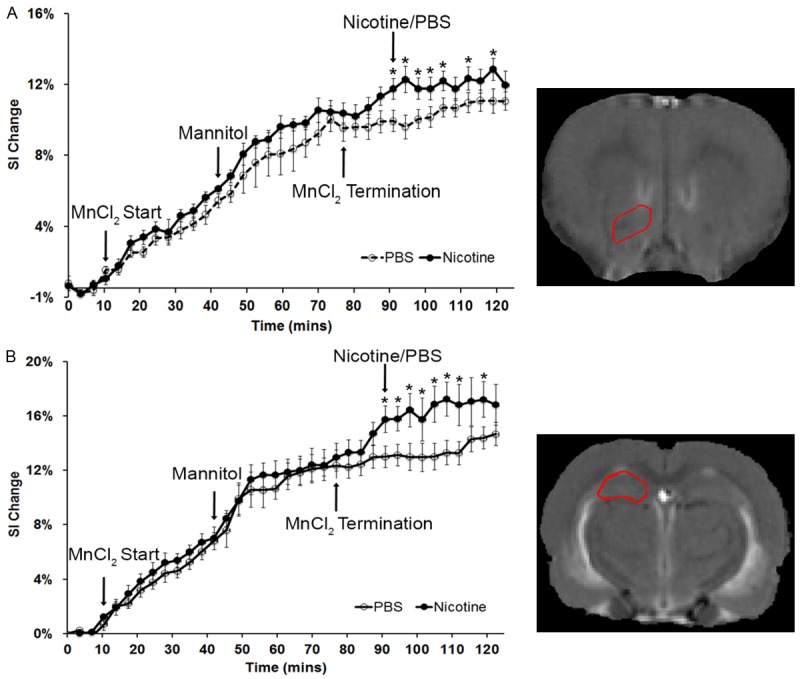
Signal alteration on nucleus accumbens (ACB) and hippocampus (HIP) in controls (PBS injected rats) and nicotine injected rats. A. Signal increase on ACB with time (*: P < 0.05). The MR image (right) illustrates the anatomy of ACB (enclosed by a red line). B. Signal increase on HIP with time (*: P < 0.05). MR image (right) illustrates the anatomy of HIP (enclosed by a red line).
Figure 3A and 3B shows the time-averaged signal increase maps of control and nicotine injected rats, respectively containing ACB and HIP regions. The signal increase was similar on ACB before nicotine/PBS injections. Nicotine injection led to higher signal increase on ACB compared to PBS injection (first row of Figure 3A and 3B). Similarly, the signal increase was comparable between the brain regions of rats prior to nicotine/PBS injections, and nicotine injection led to more signal enhancement on HIP compared to the control. t-test comparing the time-averaged signal increase on ACB and HIP between the controls and nicotine injected rats showed significant difference (P < 0.05).
Figure 3.
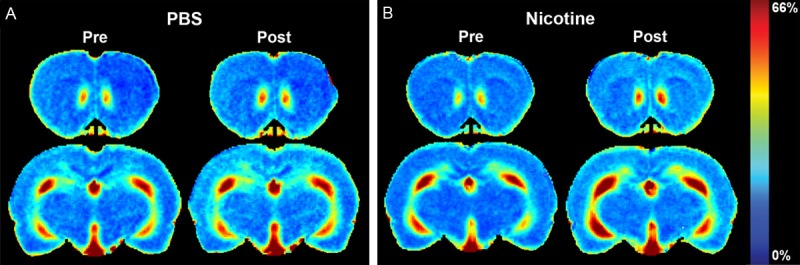
Time-averaged signal increase maps of a control and a nicotine injected rat. The color bar for the enhancement maps is at the right of the panel B. A. Time-averaged signal increase maps of controls containing ACB and HIP at pre- and post-PBS injection. B. Time-averaged signal increase maps of the nicotine injected rats containing ACB and HIP at pre- and post-nicotine injection.
Chronic nicotine paradigm
To determine the neuronal activity induced by chronic nicotine exposure, mini-pumps were implanted subcutaneously in rats. After 7 days of infusion, the pumps were removed. MEMRI acquisition was performed at 22 hours after the termination of nicotine infusion as previous studies demonstrated that abstinence signs are more likely to peak in rats around 18-22 hours [33,34]. The averaged MEMRI image of the control rats is shown on coronal brain slices as an anatomical reference in the Figure 4A. Positions of the coronal slices presented in the figure are depicted using a sagittal slice (top of the left column). The color-coded average enhancement maps of the control and nicotine exposed rat brains are illustrated in the second and third rows, respectively (Figure 4B and 4C, respectively). The enhancement represented the signal change induced by Mn2+ normalized to the MRI signal of pre-Mn2+ administration. Compared to controls, nicotine exposed rats showed higher MRI signal enhancement on striatum (1st column), ACB (2nd column), HIP (3rd column) and regions of cerebral cortex (1st through 3rd columns). Statistically significant increases in the signal enhancement are shown by pixels with P < 0.05 (Figure 5). Most pixels on ACB and HIP in nicotine exposed rats were significantly higher in enhancement than those in controls. The pixels showing significant difference on cortex were mostly on PFC and IC.
Figure 4.
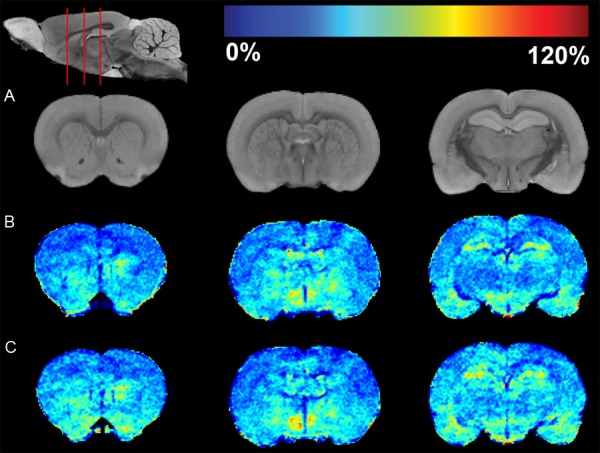
MRI enhancement maps of controls and rats with chronic nicotine exposure. A. The first row represents coronal slices of the averaged MEMRI of control rats as an anatomical reference. The sagittal slice (upper left) shows respective coronal positions (red lines). B. MRI enhancement map of controls. C. Enhancement map of rats with chronic nicotine exposure. The pseudo-colors of the enhancement maps are demonstrated using a color bar (at the top of figure). Dark blue color (0%) means no change in enhancement from Mn2+ compared to pre-injection signal intensity. Dark red color represents 120% signal increase compared to pre-injection.
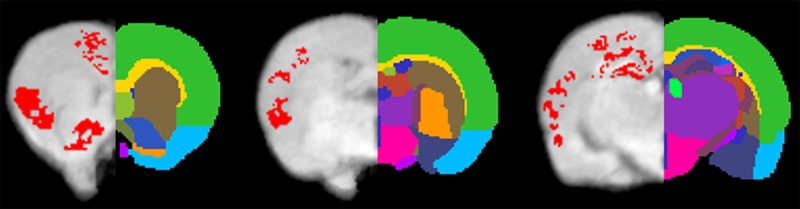
Figure 5.
Statistical comparison of MEMRI enhancement between controls and rats with chronic nicotine exposure. Pixels with significantly increased enhancement (P < 0.05) caused by nicotine infusion superimposed on brain anatomy. Brain regions were labeled using different colors on the right hemisphere. Brain regions: Mark 1 Isocortex; Mark 2 Striatum; Mark 3 Nucleus accumbens; Mark 4 Preoptic area; Mark 5 Corpus callosum; Mark 6 Thalamus; Mark 7 Dentate gyrus; Mark 8 Hypothalamus; Mark 9 Pallidum; Mark 10 Amygdala; Mark 11 CA1 of hippocampus; Mark 12 CA3 of hippocampus; Mark 13 Fimbria fornix.
Discussion
We now demonstrate that MEMRI can be used as a noninvasive biomarker to uncover brain subregions affected in response to acute and chronic nicotine exposures. These results are of immediate importance in evaluating the neurobiological mechanism underlying cigarette smoking addiction and the means now employed to prevent addiction to nicotine. Without a doubt, cigarette smoking is foremost amongst the leading causes of preventable deaths and disability worldwide. It is responsible for up to six million premature deaths/year [35]. The principal addictive agent delivered by cigarette smoke is nicotine [35]. Underlying its effects rests in its strong abilities to be a psychostimulant and affect reward behavior [6,12,35]. While the understanding of the molecular mechanisms of nicotine addiction have been realized through behavioral and neurochemical studies of glutamatergic, dopaminergic and γ-aminobutyric acidergic systems in the mesocorticolimbic system there have been few discovered means to assess such neuronal networks noninvasively [5-7,12,35,36]. This makes the abilities to use MEMRI testing to precisely pinpoint neuronal activities in regions of the brain involved in reward behavior; a significant step forward in studies to dissociate and compare effects of acute and chronic nicotine and the means to reverse it.
Indeed, increased ACB and HIP neuronal activity in rats were seen after acute nicotine exposure. After chronic nicotine exposures the ACB, HIP, PFC and IC were affected. Importantly, these brain subregions are well known to be part of CNS reward system involved in drug addiction [8,35,36]. ACB is a part of the mesolimbic dopamine pathway, and receives projections from VTA. ACB is directly involved in the immediate perception of the motivation [37]. The present study demonstrates that both acute and chronic nicotine exposure induced neuronal activity within the ACB. HIP is a key component in memory, emotion and reward. We found that neurons were activated by acute and chronic nicotine administrations in HIP. It needs to be noted that even though both acute and chronic nicotine exposure caused neuronal activation on ACB and HIP, the brain regions, cell types, and inter-cellular mechanism involved in the activation could be different between the administration schemes; thus mechanisms require further investigation.
Besides ACB and HIP, PFC and IC regions were also activated by chronic nicotine exposure. PFC is a VTA output in the mesocortical dopamine pathway. In humans, PFC is known for forming associations between the rewarding experiences of drug use and cues in the environment [38]. The cues are strong mediators of drug-seeking behavior, and can trigger relapse even after months or years of abstinence [39]. Studies found that IC plays an important role in the mechanisms underlying addiction to nicotine and other drugs [40]. Our imaging results of IC activity induced by chronic nicotine exposure corroborated these findings. The brain regions activated by chronic nicotine exposure but not by acute exposure could be responsible for abstinence symptoms and subsequently to the relapse of smoking. Nevertheless, our results are in agreement with previous MEMRI assessment on neuronal activity by alcohol [21,22], cocaine [23,24], and methamphetamine [25,26].
A number of human and non-primate studies have examined and showed that different regions of the brain have variable vulnerabilities to acute and chronic nicotine exposure [36,41-48]; the current study provides a unique opportunity for unbiased mapping of subregion specific neuronal activity in both experimental conditions. Studies in humans have found that the ACB was affected in both acute and chronic exposure; whereas PFC and other cortical regions were affected only in chronic nicotine exposure [36,41,42,44,45,47,48]. Such regions specific changes were also detected in the current study. We acknowledge that a direct comparison of the findings in humans and nonhuman primates with rats is difficult due to differences in anatomy, physiology, and neurochemistry, but our results are consistent with these human and non-human primate studies.
Functional MRI (fMRI) has been used to study the neural bases of nicotine dependence and to develop smoking cessation strategies [36,49-53]. fMRI is an indirect measure of brain function by detecting the local blood oxygenation level dependent (BOLD) signal changes coupled with the neuronal activity [54,55]. Nicotine may act not only upon nAChRs but also potentially upon the cerebral vascular system [56-60]. The hemodynamic changes are superimposed on the nicotine induced neuronal activity, confounding the interpretation of the fMRI signals. In contrast, MEMRI directly maps neurons involved in electrical transmission without relying on hemodynamic coupling. Therefore, MEMRI has advantage over fMRI to monitor nicotine induced neuronal activation. We plan to acquire fMRI and perfusion MRI data in the future studies, and investigate the hemodynamic changes caused by nicotine exposure by comparing fMRI results with perfusion MRI and MEMRI data. The results will be helpful in the design of imaging studies of nicotine in humans.
Only naïve rats were used in this study to investigate the effects of acute and chronic nicotine exposure on neurons. In the future, we will study the acute neuronal responses of rats that have been chronically exposed to nicotine. By comparing to the naïve rats with acute exposure presented here, we will be able to gain insights of brain functional changes by long-term smoking. The acute and chronic cellular toxicity of Mn2+ repressed the application of MEMRI in humans. When used in animals, Mn2+ toxicity needs to be minimized. In this study, we used administration schemes that have been proven safe for the MnCl2 administration [61,62]. The animals were also closely monitored after MnCl2 administration, and we did not observe any Mn2+ induced toxic clinical signs and symptoms.
Conclusion
MEMRI can be employed successfully to monitor acute and chronic nicotine exposure induced neuronal activities in cortical and subcortical regions involving nicotine addiction. While disparities were observed on brain regions affected by acute and chronic nicotine exposure such differences serve to highlight the process of addiction within the affected brain. To our knowledge, this is the first MEMRI study observing neuronal responses to nicotine exposure. The study demonstrates that MEMRI is a powerful imaging tool to study neuronal responses to nicotine exposure, tolerance, withdrawal, and relapse in animals. MEMRI will also provide a preclinical screening tool for smoking cessation strategies.
Acknowledgements
This study is supported by NE DHHS LB506 Cancer and Smoking Disease Research Program, P01 DA028555, and a grant from the Nebraska Research Initiative. The authors would like to thank Melissa Mellon, Lirong Xu, and Ahmad Tanwir for their technical assistance.
Disclosure of conflict of interest
None.
References
- 1.National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. 2014 [Google Scholar]
- 2.Centers for Disease Control and Prevention (CDC) Quitting smoking among adults--United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2011;60:1513–9. [PubMed] [Google Scholar]
- 3.Torre GL. Smoking prevention and cessation. New York: Springer; 2013. [Google Scholar]
- 4.Cahill K, Stevens S, Perera R, Lancaster T. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev. 2013:CD009329. doi: 10.1002/14651858.CD009329.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Souza MS, Markou A. Neuronal mechanisms underlying development of nicotine dependence: implications for novel smoking-cessation treatments. Addict Sci Clin Pract. 2011;6:4–16. [PMC free article] [PubMed] [Google Scholar]
- 6.Balfour DJ. The neuronal pathways mediating the behavioral and addictive properties of nicotine. Handb Exp Pharmacol. 2009:209–33. doi: 10.1007/978-3-540-69248-5_8. [DOI] [PubMed] [Google Scholar]
- 7.De Biasi M, Dani JA. Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci. 2011;34:105–30. doi: 10.1146/annurev-neuro-061010-113734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–13. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 9.Kolokotroni KZ, Rodgers RJ, Harrison AA. Effects of chronic nicotine, nicotine withdrawal and subsequent nicotine challenges on behavioural inhibition in rats. Psychopharmacology (Berl) 2012;219:453–68. doi: 10.1007/s00213-011-2558-z. [DOI] [PubMed] [Google Scholar]
- 10.Lester RA. Cognitive mechanisms underlying relapse to nicotine. Rev Neurosci. 2011;22:467–70. doi: 10.1515/RNS.2011.038. [DOI] [PubMed] [Google Scholar]
- 11.Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–17. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- 12.Markou A. Review. Neurobiology of nicotine dependence. Philos Trans R Soc London B Biol Sci. 2008;363:3159–68. doi: 10.1098/rstb.2008.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paolini M, De Biasi M. Mechanistic insights into nicotine withdrawal. Biochem Pharmacol. 2011;82:996–1007. doi: 10.1016/j.bcp.2011.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penton RE, Lester RA. Cellular events in nicotine addiction. Semin Cell Dev Biol. 2009;20:418–31. doi: 10.1016/j.semcdb.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Lukas RJ. Naturally-expressed nicotinic acetylcholine receptor subtypes. Biochem Pharmacol. 2011;82:800–7. doi: 10.1016/j.bcp.2011.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kenny PJ, Markou A. Neurobiology of the nicotine withdrawal syndrome. Pharmacol Biochem Behav. 2001;70:531–49. doi: 10.1016/s0091-3057(01)00651-7. [DOI] [PubMed] [Google Scholar]
- 17.Zhang D, Gao M, Xu D, Shi WX, Gutkin BS, Steffensen SC, Lukas RJ, Wu J. Impact of prefrontal cortex in nicotine-induced excitation of ventral tegmental area dopamine neurons in anesthetized rats. J Neurosci. 2012;32:12366–75. doi: 10.1523/JNEUROSCI.5411-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu H, Yang S, Zuo Y, Demny S, Stein EA, Yang Y. Real-time animal functional magnetic resonance imaging and its application to neuropharmacological studies. Magn Reson Imaging. 2008;26:1266–1272. doi: 10.1016/j.mri.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pautler RG. Biological applications of manganese-enhanced magnetic resonance imaging. Methods Mol Med. 2006;124:365–386. doi: 10.1385/1-59745-010-3:365. [DOI] [PubMed] [Google Scholar]
- 20.Dudek M, Hyytia P. Alcohol preference and consumption are controlled by the caudal linear nucleus in alcohol-preferring rats. Eur J Neurosci. 2016;43:1440–1448. doi: 10.1111/ejn.13245. [DOI] [PubMed] [Google Scholar]
- 21.Dudek M, Canals S, Sommer WH, Hyytia P. Modulation of nucleus accumbens connectivity by alcohol drinking and naltrexone in alcohol-preferring rats: A manganese-enhanced magnetic resonance imaging study. Eur Neuropsychopharmacol. 2016;26:445–455. doi: 10.1016/j.euroneuro.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Dudek M, Abo-Ramadan U, Hermann D, Brown M, Canals S, Sommer WH, Hyytia P. Brain activation induced by voluntary alcohol and saccharin drinking in rats assessed with manganese-enhanced magnetic resonance imaging. Addict Biol. 2015;20:1012–1021. doi: 10.1111/adb.12179. [DOI] [PubMed] [Google Scholar]
- 23.Perrine SA, Ghoddoussi F, Desai K, Kohler RJ, Eapen AT, Lisieski MJ, Angoa-Perez M, Kuhn DM, Bosse KE, Conti AC, Bissig D, Berkowitz BA. Cocaine-induced locomotor sensitization in rats correlates with nucleus accumbens activity on manganese-enhanced MRI. NMR Biomed. 2015;28:1480–1488. doi: 10.1002/nbm.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu H, Xi ZX, Gitajn L, Rea W, Yang Y, Stein EA. Cocaine-induced brain activation detected by dynamic manganese-enhanced magnetic resonance imaging (MEMRI) Proc Natl Acad Sci U S A. 2007;104:2489–2494. doi: 10.1073/pnas.0606983104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu YH, Chen CC, Zechariah A, Yen CC, Yang LC, Chang C. Neuronal dysfunction of a long projecting multisynaptic pathway in response to methamphetamine using manganese-enhanced MRI. Psychopharmacology (Berl) 2008;196:543–553. doi: 10.1007/s00213-007-0990-x. [DOI] [PubMed] [Google Scholar]
- 26.den Hollander B, Dudek M, Ojanpera I, Kankuri E, Hyytia P, Korpi ER. Manganese-enhanced magnetic resonance imaging reveals differential long-term neuroadaptation after methamphetamine and the substituted cathinone 4-methylmethcathinone (mephedrone) Int J Neuropsychopharmacol. 2014;18 doi: 10.1093/ijnp/pyu106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jandova K, Maresova D, Pokorny J. Fast and delayed locomotor response to acute high-dose nicotine administration in adult male rats. Physiol Res. 2013;62(Suppl 1):S81–8. doi: 10.33549/physiolres.932610. [DOI] [PubMed] [Google Scholar]
- 28.Rowell PP, Li M. Dose-response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors. J Neurochem. 1997;68:1982–9. doi: 10.1046/j.1471-4159.1997.68051982.x. [DOI] [PubMed] [Google Scholar]
- 29.Yang K, Buhlman L, Khan GM, Nichols RA, Jin G, McIntosh JM, Whiteaker P, Lukas RJ, Wu J. Functional nicotinic acetylcholine receptors containing alpha6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J Neurosci. 2011;31:2537–48. doi: 10.1523/JNEUROSCI.3003-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 31.Uberti MG, Boska MD, Liu Y. A semi-automatic image segmentation method for extraction of brain volume from in vivo mouse head magnetic resonance imaging using Constraint Level Sets. J Neurosci Methods. 2009;179:338–344. doi: 10.1016/j.jneumeth.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bade AN, Gorantla S, Dash PK, Makarov E, Sajja BR, Poluektova LY, Luo J, Gendelman HE, Boska MD, Liu Y. Manganese-enhanced magnetic resonance imaging reflects brain pathology during progressive HIV-1 infection of humanized mice. Mol Neurobiol. 2016;53:3286–3297. doi: 10.1007/s12035-015-9258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malin DH. Nicotine dependence: studies with a laboratory model. Pharmacol Biochem Behav. 2001;70:551–9. doi: 10.1016/s0091-3057(01)00699-2. [DOI] [PubMed] [Google Scholar]
- 34.Malin DH, Lake JR, Newlin-Maultsby P, Roberts LK, Lanier JG, Carter VA, Cunningham JS, Wilson OB. Rodent model of nicotine abstinence syndrome. Pharmacol Biochem Behav. 1992;43:779–84. doi: 10.1016/0091-3057(92)90408-8. [DOI] [PubMed] [Google Scholar]
- 35.Subramaniyan M, Dani JA. Dopaminergic and cholinergic learning mechanisms in nicotine addiction. Ann N Y Acad Sci. 2015;1349:46–63. doi: 10.1111/nyas.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jasinska AJ, Zorick T, Brody AL, Stein EA. Dual role of nicotine in addiction and cognition: a review of neuroimaging studies in humans. Neuropharmacology. 2014;84:111–122. doi: 10.1016/j.neuropharm.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nestler EJ. Cellular basis of memory for addiction. Dialogues Clin Neurosci. 2013;15:431–443. doi: 10.31887/DCNS.2013.15.4/enestler. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–669. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry CJ, Zbukvic I, Kim JH, Lawrence AJ. Role of cues and contexts on drug-seeking behaviour. Br J Pharmacol. 2014;171:4636–4672. doi: 10.1111/bph.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaznick N, Tranel D, McNutt A, Bechara A. Basal ganglia plus insula damage yields stronger disruption of smoking addiction than basal ganglia damage alone. Nicotine Tob Res. 2014;16:445–453. doi: 10.1093/ntr/ntt172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brody AL, Mandelkern MA, Jarvik ME, Lee GS, Smith EC, Huang JC, Bota RG, Bartzokis G, London ED. Differences between smokers and nonsmokers in regional gray matter volumes and densities. Biol Psychiatry. 2004;55:77–84. doi: 10.1016/s0006-3223(03)00610-3. [DOI] [PubMed] [Google Scholar]
- 42.Domino EF, Minoshima S, Guthrie S, Ohl L, Ni L, Koeppe RA, Zubieta JK. Nicotine effects on regional cerebral blood flow in awake, resting tobacco smokers. Synapse. 2000;38:313–321. doi: 10.1002/1098-2396(20001201)38:3<313::AID-SYN10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 43.Domino EF, Tsukada H. Nicotine sensitization of monkey striatal dopamine release. Eur J Pharmacol. 2009;607:91–95. doi: 10.1016/j.ejphar.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Gallinat J, Meisenzahl E, Jacobsen LK, Kalus P, Bierbrauer J, Kienast T, Witthaus H, Leopold K, Seifert F, Schubert F, Staedtgen M. Smoking and structural brain deficits: a volumetric MR investigation. Eur J Neurosci. 2006;24:1744–1750. doi: 10.1111/j.1460-9568.2006.05050.x. [DOI] [PubMed] [Google Scholar]
- 45.Kuhn S, Schubert F, Gallinat J. Reduced thickness of medial orbitofrontal cortex in smokers. Biol Psychiatry. 2010;68:1061–1065. doi: 10.1016/j.biopsych.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Marenco S, Carson RE, Berman KF, Herscovitch P, Weinberger DR. Nicotine-induced dopamine release in primates measured with [11C] raclopride PET. Neuropsychopharmacology. 2004;29:259–268. doi: 10.1038/sj.npp.1300287. [DOI] [PubMed] [Google Scholar]
- 47.Rose EJ, Ross TJ, Salmeron BJ, Lee M, Shakleya DM, Huestis M, Stein EA. Chronic exposure to nicotine is associated with reduced reward-related activity in the striatum but not the midbrain. Biol Psychiatry. 2012;71:206–213. doi: 10.1016/j.biopsych.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rose EJ, Ross TJ, Salmeron BJ, Lee M, Shakleya DM, Huestis MA, Stein EA. Acute nicotine differentially impacts anticipatory valence- and magnitude-related striatal activity. Biol Psychiatry. 2013;73:280–288. doi: 10.1016/j.biopsych.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson SJ, Smyth JM, MacLean RR. Integrating ecological momentary assessment and functional brain imaging methods: new avenues for studying and treating tobacco dependence. Nicotine Tob Res. 2014;16(Suppl 2):S102–110. doi: 10.1093/ntr/ntt129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menossi HS, Goudriaan AE, de Azevedo-Marques Perico C, Nicastri S, de Andrade AG, D’Elia G, Li CS, Castaldelli-Maia JM. Neural bases of pharmacological treatment of nicotine dependence - insights from functional brain imaging: a systematic review. CNS Drugs. 2013;27:921–941. doi: 10.1007/s40263-013-0092-8. [DOI] [PubMed] [Google Scholar]
- 51.Jasinska AJ, Stein EA, Kaiser J, Naumer MJ, Yalachkov Y. Factors modulating neural reactivity to drug cues in addiction: a survey of human neuroimaging studies. Neurosci Biobehav Rev. 2014;38:1–16. doi: 10.1016/j.neubiorev.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartwell KJ, Prisciandaro JJ, Borckardt J, Li X, George MS, Brady KT. Real-time fMRI in the treatment of nicotine dependence: a conceptual review and pilot studies. Psychol Addict Behav. 2013;27:501–509. doi: 10.1037/a0028215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Courtney KE, Schacht JP, Hutchison K, Roche DJ, Ray LA. Neural substrates of cue reactivity: association with treatment outcomes and relapse. Addict Biol. 2016;21:3–22. doi: 10.1111/adb.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huettel SA, Song AW, McCarthy G. Functional magnetic resonance imaging. Sunderland, Mass: Sinauer Associates; 2009. [Google Scholar]
- 55.Buxton RB. Introduction to functional magnetic resonance imaging: principles and techniques. Cambridge, New York: Cambridge University Press; 2009. [Google Scholar]
- 56.Zubieta JK, Heitzeg MM, Xu Y, Koeppe RA, Ni L, Guthrie S, Domino EF. Regional cerebral blood flow responses to smoking in tobacco smokers after overnight abstinence. Am J Psychiatry. 2005;162:567–577. doi: 10.1176/appi.ajp.162.3.567. [DOI] [PubMed] [Google Scholar]
- 57.Vafaee MS, Gjedde A, Imamirad N, Vang K, Chakravarty MM, Lerch JP, Cumming P. Smoking normalizes cerebral blood flow and oxygen consumption after 12-hour abstention. J Cereb Blood Flow Metab. 2015;35:699–705. doi: 10.1038/jcbfm.2014.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanabe J, Crowley T, Hutchison K, Miller D, Johnson G, Du YP, Zerbe G, Freedman R. Ventral striatal blood flow is altered by acute nicotine but not withdrawal from nicotine. Neuropsychopharmacology. 2008;33:627–633. doi: 10.1038/sj.npp.1301428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shinohara T, Nagata K, Yokoyama E, Sato M, Matsuoka S, Kanno I, Hatazawa J, Domino EF. Acute effects of cigarette smoking on global cerebral blood flow in overnight abstinent tobacco smokers. Nicotine Tob Res. 2006;8:113–121. doi: 10.1080/14622200500431759. [DOI] [PubMed] [Google Scholar]
- 60.Peebles KC, Horsman H, Tzeng YC. The influence of tobacco smoking on the relationship between pressure and flow in the middle cerebral artery in humans. PLoS One. 2013;8:e72624. doi: 10.1371/journal.pone.0072624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malheiros JM, Paiva FF, Longo BM, Hamani C, Covolan L. Manganese-enhanced MRI: biological applications in neuroscience. Front Neurol. 2015;6:161. doi: 10.3389/fneur.2015.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cacace AT, Brozoski T, Berkowitz B, Bauer C, Odintsov B, Bergkvist M, Castracane J, Zhang J, Holt AG. Manganese enhanced magnetic resonance imaging (MEMRI): a powerful new imaging method to study tinnitus. Hear Res. 2014;311:49–62. doi: 10.1016/j.heares.2014.02.003. [DOI] [PubMed] [Google Scholar]