

Abstract
Objective:
To analyze the oral motor, speech, and language phenotype in 20 children and adults with Dravet syndrome (DS) associated with mutations in SCN1A.
Methods:
Fifteen verbal and 5 minimally verbal DS patients with SCN1A mutations (aged 15 months-28 years) underwent a tailored assessment battery.
Results:
Speech was characterized by imprecise articulation, abnormal nasal resonance, voice, and pitch, and prosody errors. Half of verbal patients had moderate to severely impaired conversational speech intelligibility. Oral motor impairment, motor planning/programming difficulties, and poor postural control were typical. Nonverbal individuals had intentional communication. Cognitive skills varied markedly, with intellectual functioning ranging from the low average range to severe intellectual disability. Language impairment was congruent with cognition.
Conclusions:
We describe a distinctive speech, language, and oral motor phenotype in children and adults with DS associated with mutations in SCN1A. Recognizing this phenotype will guide therapeutic intervention in patients with DS.
Dravet syndrome (DS) is an infantile-onset developmental epileptic encephalopathy with poor outcome. Typically, a 6-month-old infant presents with febrile hemiclonic status epilepticus in the setting of reputedly normal development, and then develops multiple seizure types over the next 4 years, with developmental slowing from 1–2 years of age.1 More than 80% of cases have mutations of the sodium channel gene SCN1A. Intellectual disability (ID) is usual, with almost all patients having severe ID.
Speech and language function in adults and children with DS has not been specifically characterized. Three pediatric studies have examined language (understanding and use of words) in the context of a broader neuropsychological battery and include SCN1A positive and negative cases.2–4 The results are varied, ranging from cohorts with severe ID and severe language impairment to others with mild to moderate ID and borderline to average naming and comprehension.
In terms of speech (how speech sounds are produced or articulated), dysarthria and speech planning difficulties have been reported anecdotally.2,4–6 Oral motor skills have not been investigated.
We aimed to determine whether there was a characteristic developmental speech, language, and oral motor phenotype in children and adults with DS associated with mutations in SCN1A. Recognition of progressive patterns of dysfunction will inform diagnosis and guide therapeutic intervention.
METHODS
Patients attending a DS clinic were invited to participate; the entire cohort comprised 26 patients with the electroclinical features of DS and an SCN1A mutation. Three families refused and 3 were unavailable during the study. Diagnosis was confirmed by a pediatric neurologist with expertise in DS (I.E.S.). All SCN1A mutations were located in highly conserved regions or reported to alter protein expression or function.
Standard protocol approvals, registrations, and patient consents.
The study was approved by Austin Health Human Research Ethics Committee (Austin HREC H2011/04390). Written informed consent was obtained from all participants or their parents (for minors or those with ID). Consent covered use of video footage for publication.
Speech and language assessment.
Two batteries were administered depending on the patient's ability (table 1). Standardized assessments were used where possible, for comparison with typically developing children.
Table 1.
Comprehensive assessment battery
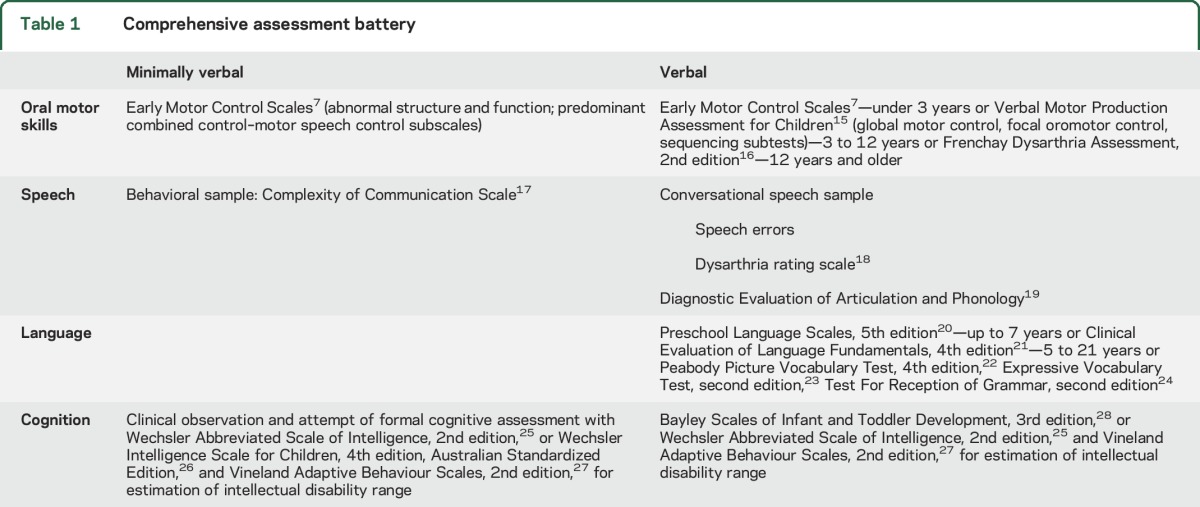
The minimally verbal patients (MV) had little or no speech and were unable to cooperate with standardized assessment, while the verbal group (V) had conversational speech. Testing focused on oral motor, speech, and language skills.
Oral motor tasks and perceptual speech characteristics of conversational samples were independently rated by 2 speech pathologists (S.J.T., A.T.M.). The SCN1A mutation, psychological assessment results, medications, and seizure history were reviewed.
RESULTS
The cohort comprised 20 patients with DS (11 female), with 15 in the V and 5 in the MV group (table 2). Median age was 11½ years (mean 13 years, range 15 months–28 years). Fifteen had de novo SCN1A mutations and 3 inherited mutations. Inherited mutations were from an unaffected mother (patient 10), a father with genetic epilepsy with febrile seizures plus (GEFS+; patient 15), and a mother (patient 20 here) with DS and a de novo SCN1A mutation (patient 7). Inheritance for 2 individuals (6, 18) could not be confirmed as their fathers were not available for testing.
Table 2.
Patient details and results of oral motor and language assessment (n = 20)
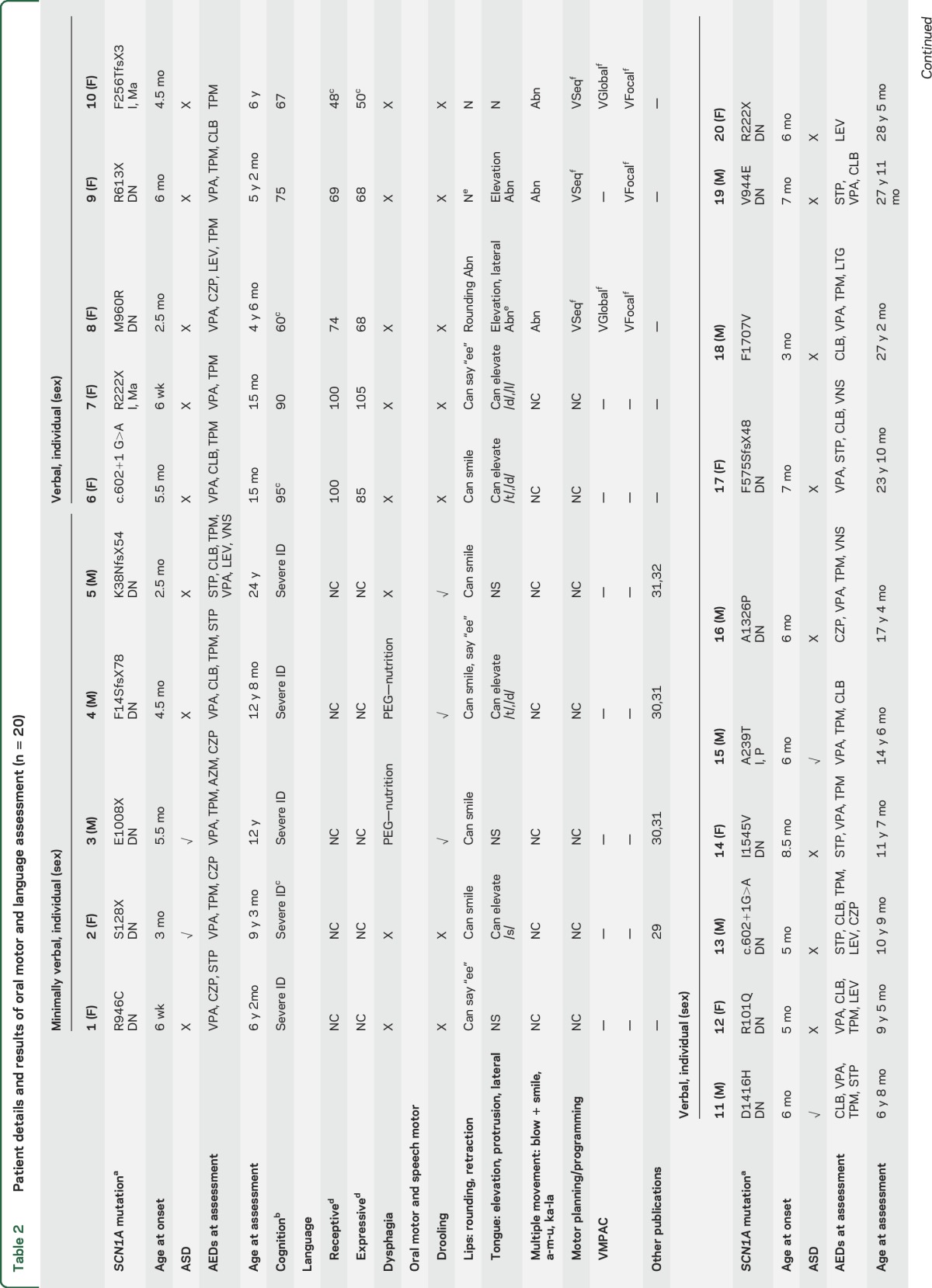
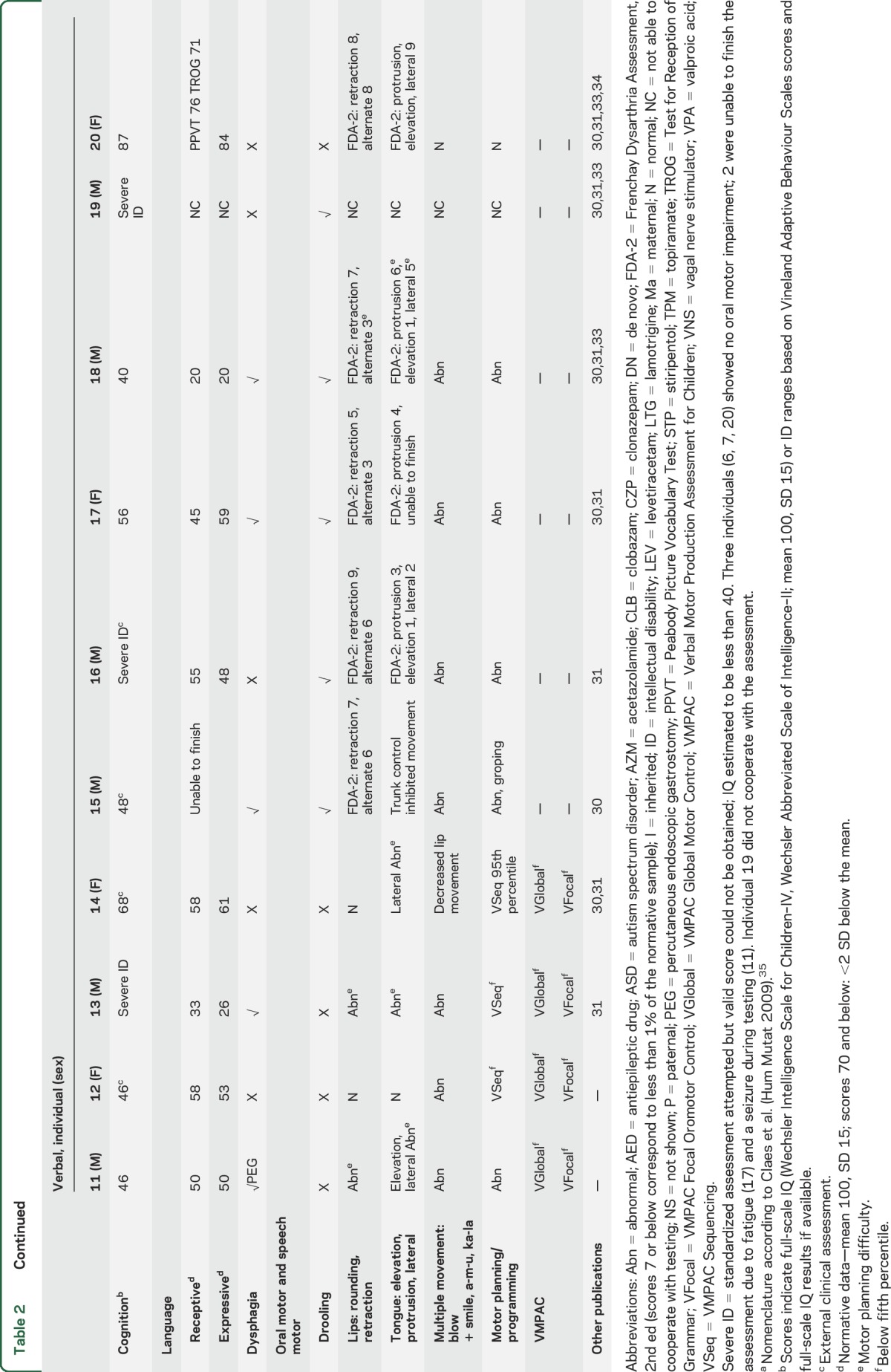
In children under 2 years of age, development was normal. In older patients, cognitive skills varied markedly, with intellectual functioning ranging from low average (1) to borderline (1) to mild (5), moderate (2), and severe (9) ID. No pattern of performance was seen on verbal vs perceptual reasoning tasks. All patients were on antiepileptic drugs, with 17/20 taking 3 or more. Three individuals had a vagal nerve stimulator (VNS) (table 2).
Oral motor skills.
See table 2 and video 1 (at Neurology.org). All 5 MV individuals showed impaired oral motor control apart from individual 2 who had independent jaw and tongue movement.7
Lip and tongue movement was reduced, asymmetrical, or poorly coordinated in 12/15 V individuals. Notably, 2/3 without impairment were the youngest patients in the cohort aged under 2 years. Lip retraction (say “ee”) was generally within normal limits, while lip rounding (say “oo”) was weak or asymmetrical. Two individuals (17 and 18) could not overcome an open mouth posture at rest to round the lips. Tongue protrusion, elevation, and lateral movement were also impaired, with 6 (8, 9, 11, 15, 16, 18) unable to elevate the tongue and 3 (16, 17, 18) showing involuntary tongue movement.
Overriding impaired motor programming and planning issues affected performance of speech motor and nonspeech oral motor tasks (table 2). Poor postural control of the trunk and head also affected lip and tongue movement.
Saliva control issues were prominent in 8/20 individuals, likely compounded by benzodiazepine therapy in 7 cases. Saliva control management included medication in 3 (3, 5, 18) and salivary duct surgery in 1 that was not beneficial (18). Mild dysphagia was reported in 5/20 individuals, including 1 with a VNS (17). Three had percutaneous endoscopic gastrostomy for nutrition (3, 4, 11).
Speech.
See table 3 and video 2. All MV individuals had intentional communication. Three had potentially communicative behavior (Complexity of Communication Scale [CCS] score 7b) such as eye contact, gesture, vocalization (1, 5), or using an adult's hand as a tool (3) regarded as nonsymbolic communication. Two (2, 4) had symbolic communication (CCS score 10), using single words recognized by an unfamiliar observer.
Table 3.
Perceptual speech assessment in verbal patients with conversational speech (n = 13a)
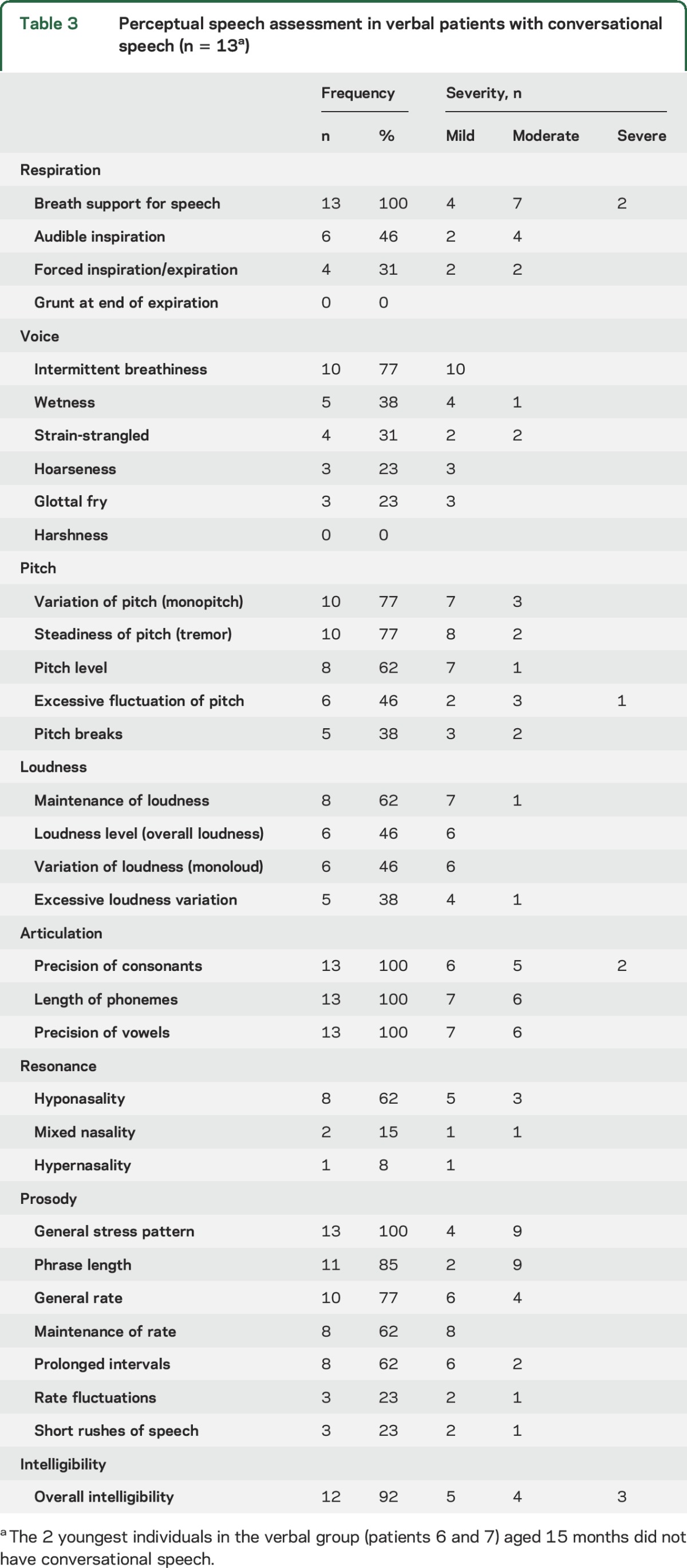
In the V group, conversational speech intelligibility was severely impaired in 3 (16, 18, 19), moderately impaired in 4 (11, 13, 15, 17), mildly reduced in 5 (8, 9, 10, 12, 14), and normal in 1 (20). All had inadequate breath support for speech. Speech was typified by imprecise articulation of consonants and vowels, abnormal nasal resonance, breathy or strain-strangled voice, low pitch, and prosodic errors (e.g., excess stress on unstressed parts of speech, slow rate, short phrases). Sound errors included voicing errors, distortion of fricatives /s, z, ∫/, affricates /t∫, dʒ/ and /l/, delayed phonological processes (final consonant deletion, gliding, fronting, stopping, cluster reduction, /f/ for /θ/, /d/ for /ð/) and atypical phonological processes (backing, replacing sounds with /j/ or /h/, insertion of schwa vowel). The vocal quality of individuals 16 and 17 may be attributed to VNS functioning; however, their voice was similar to patients without a VNS.
Language.
Thirteen patients cooperated with language testing. Severely impaired receptive and expressive language (>2 SD below the mean) was seen in 9/13, a severe expressive deficit in patient 8 (receptive moderate), and moderately impaired receptive language in patient 20. The 2 youngest patients scored in the average range at age 15 months (table 2).
DISCUSSION
A distinctive speech and language phenotype was found in 20 patients with DS associated with SCN1A mutations. Oral motor impairment was common, compounded by poor postural control of the trunk, neck, and head. Motor planning and programming difficulties were striking. Speech was characterized by imprecise articulation of consonants and vowels, abnormal nasal resonance, breathy or strain-strangled voice, and errors in pitch and prosody. Language impairment involving receptive and expressive language was seen in all bar the 2 youngest children. Nonverbal individuals had intentional communication.
Our language findings were more severe than previously reported in 2 earlier studies, which found borderline to average comprehension (9/12 and 9/9 children) and naming (8/12 and 4/9 children).3,4 This disparity is likely due to past studies including children of a younger age range (up to 13 years of age) and with better cognitive profile. Further, around half of previously reported patients had SCN1A mutations. Our findings are comparable to a cohort aged up to 16 years, in which 3/20 children had preserved language.2
We found a trend towards a more severe speech phenotype in adults than children, with 3/4 of verbal adults being moderately to severely unintelligible. Current therapeutic regimens for DS are more targeted than in the past, which may lead to amelioration of speech impairment. The oldest adult had very mild impairments in respiration, articulation, phonation, and prosody; however, her phenotype is distinct from the rest of the cohort, as she had normal speech intelligibility, no oral motor impairment, and normal intellect, which is rare in DS. The youngest girls, aged 15 months, presented with age-appropriate language and oral motor skills, which likely reflects the typical developmental trajectory of DS, with normal development slowing in the second year of life.
Interestingly, 5 patients (aged 6–27 years) reported mild dysphagia, similar to the frequency reported in older adults from their fourth decade (5/22 patients).5 Larger numbers of patients are needed to determine whether there is a correlation between the severity of the speech phenotype and features such as age, type and inheritance of SCN1A mutation, seizure types, and medication. Looking at cognitive outcome more broadly, previous studies have shown no correlation of SCN1A mutation class, age at seizure onset, type, and number, and MRI abnormalities.8,9
The voltage-gated sodium channel NaV1.1, encoded by SCN1A, is found in brain regions important for speech and language function including the cerebellum, sensory motor cortex, basal ganglia, hippocampus, middle temporal gyrus, and middle frontal gyrus.10 Hyperexcitability due to loss of function of GABAergic inhibitory interneurons expressing NaV1.1 underlies seizures in DS.11 Abnormal inhibition may also be important for speech and language function in DS. Interestingly, abnormal excitation due to mutations in the excitatory glutamate receptor subunit gene GRIN2A is associated with motor speech impairment in epilepsy-aphasia syndromes.12 Studies in milder SCN1A phenotypes such as GEFS+ may clarify the role of sodium channels in speech and language impairment.
Moreover, structural changes in speech and language brain regions have been reported and include precentral gyrus, cerebellum, brainstem, corpus callosum, corticospinal tracts, and association fibers (left inferior fronto-occipital fasciculus, left uncinate fasciculus),13 and influence phenotypic heterogeneity.
Understanding the speech and language phenotype in DS is crucial to planning early intervention. Targeted dysarthria therapy has been successful in other pediatric populations with mild to severe dysarthria14 and could potentially also improve speech intelligibility of verbal patients with DS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the families for their participation in this study; Associate Professor Nancy Brady for use of the Complexity of Communication Scale; Deborah Hayden and Brookes Publishing for permission to use the prepublication version of the Early Motor Control Scales; and Natalie Bryant and Annie Roten for assistance with filming assessments.
GLOSSARY
- CCS
Complexity of Communication Scale
- DS
Dravet syndrome
- GEFS+
genetic epilepsy with febrile seizures plus
- ID
intellectual disability
- MV
minimally verbal
- V
verbal
- VNS
vagal nerve stimulator
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
S.J.T., A.T.M., and I.E.S. designed the study and wrote the manuscript. A.T.M., I.E.S., and V.A. supervised the study. S.J.T. and A.T.M. performed phenotypic analysis. A.B. and M.A. performed most standardized developmental/intellectual functioning assessments.
STUDY FUNDING
S.J.T. is supported by National Health and Medical Research Council (NHMRC) Postgraduate Scholarship (101777), Australian National University Gowrie Scholarship, and Speech Pathology Australia Nadia Verrall Memorial Research Grant. V.A. is supported by NHMRC Senior Practitioner Fellowship (2010–2019). A.T.M. is supported by NHMRC Career Development Award (607315, 2010–2015) and Practitioner Fellowship (1105008, 2016–2020). I.E.S. is supported by NHMRC Program Grant (628952, 2011–2015; 1091593, 2016–2020) and Senior Practitioner Fellowship (1006110, 2011–2015; 1104831, 2016–2020). The project is also supported by Australian Research Council Discovery Project (DP120100285) to A.T.M. and I.E.S.
DISCLOSURE
S. Turner, A. Brown, M. Arpone, V. Anderson, and A. Morgan report no disclosures relevant to the manuscript. I. Scheffer serves on the editorial boards of Neurology® and Epileptic Disorders; may accrue future revenue on a pending patent re: Therapeutic compound; has received speaker honoraria from Athena Diagnostics, UCB, GSK, Eisai, and Transgenomics; has received funding for travel from Athena Diagnostics, UCB, and GSK; and receives/has received research support from the NHMRC, ARC, NIH, Health Research Council of New Zealand, March of Dimes, the Weizmann Institute, CURE, US Department of Defense, and the Perpetual Charitable Trustees. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Dravet C, Bureau M, Oguni H, Cokar O, Guerrini R. Dravet syndrome (severe myoclonic epilepsy in infancy). In: Bureau M, Genton P, Dravet C, et al., eds. Epileptic Syndromes in Infancy, Childhood and Adolescence, 5th ed. Montrouge, France: John Libbey Eurotext Ltd.; 2012:125–156. [Google Scholar]
- 2.Casse-Perrot C, Wolff M, Dravet C. Neuropsychological aspects of severe myoclonic epilepsy in infancy. In: Jambaque I, Lassonde M, Dulac O, eds. The Neuropsychology of Childhood Epilepsy. New York: Plenum Press/Kluwer Academic; 2001:131–140. [Google Scholar]
- 3.Battaglia D, Chieffo D, Siracusano R, et al. Cognitive decline in Dravet syndrome: is there a cerebellar role? Epilepsy Res 2013;106:211–221. [DOI] [PubMed] [Google Scholar]
- 4.Chieffo D, Battaglia D, Lettori D, et al. Neuropsychological development in children with Dravet syndrome. Epilepsy Res 2011;95:86–93. [DOI] [PubMed] [Google Scholar]
- 5.Catarino CB, Liu JY, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain 2011;134:2982–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia 2011;52(suppl 2):44–49. [DOI] [PubMed] [Google Scholar]
- 7.Hayden D, Wetherby AM, Cleary JE, Prizant BM. Early Motor Control Scales: Prepublication Version. Baltimore: Paul H. Brookes Publishing Co.; 2011. [Google Scholar]
- 8.Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012;135:2329–2336. [DOI] [PubMed] [Google Scholar]
- 9.Villeneuve N, Laguitton V, Viellard M, et al. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav 2014;31:143–148. [DOI] [PubMed] [Google Scholar]
- 10.Whitaker WR, Faull RL, Waldvogel HJ, Plumpton CJ, Emson PC, Clare JJ. Comparative distribution of voltage-gated sodium channel proteins in human brain. Brain Res Mol Brain Res 2001;88:37–53. [DOI] [PubMed] [Google Scholar]
- 11.Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006;9:1142–1149. [DOI] [PubMed] [Google Scholar]
- 12.Turner SJ, Mayes AK, Verhoeven A, Mandelstam SA, Morgan AT, Scheffer IE. GRIN2A: an aptly named gene for speech dysfunction. Neurology 2015;84:586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez A, Garcia-Penton L, Canales-Rodriguez EJ, et al. Brain morphometry of Dravet syndrome. Epilepsy Res 2014;108:1326–1334. [DOI] [PubMed] [Google Scholar]
- 14.Pennington L, Parker NK, Kelly H, Miller N. Speech therapy for children with dysarthria acquired before three years of age. Cochrane Database Syst Rev 2016;7:CD006937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayden D, Square P. The Verbal Motor Production Assessment for Children. San Antonio, TX: The Psychological Corporation; 1999. [Google Scholar]
- 16.Enderby P, Palmer R. Frenchay Dysarthria Assessment, 2nd ed. Austin, TX: PRO-ED; 2008. [Google Scholar]
- 17.Brady NC, Fleming K, Thiemann-Bourque K, et al. Development of the communication complexity scale. Am J Speech-Language Pathol 2012;21:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murdoch BE. Dysarthria: A Physiological Approach to Assessment and Treatment. Cheltenham, UK: Stanley Thornes; 1998. [Google Scholar]
- 19.Dodd B, Hua Z, Crosbie S, Holm A, Ozanne A. Diagnostic Evaluation of Articulation and Phonology. London: Pearson Assessment; 2002. [Google Scholar]
- 20.Zimmerman I, Steiner V, Pond R. Preschool Language Scales, Australian and New Zealand Language Adapted Edition, 5th ed. Sydney: Pearson; 2011. [Google Scholar]
- 21.Semel E, Wiig E, Secord W. Clinical evaluation of language fundamentals. In: Australian Standardised Edition, 4th ed. Marrickville, Australia: Harcourt Assessment; 2006. [Google Scholar]
- 22.Dunn LM, Dunn DM. The Peabody Picture Vocabulary Test, 4th ed. Minneapolis, MN: NCS Pearson Inc.; 2007. [Google Scholar]
- 23.Williams KT. Expressive Vocabulary Test, 2nd ed. London: Pearson Assessment; 2007. [Google Scholar]
- 24.Bishop DJ. Test for Reception of Grammar, 2nd ed. London: Pearson Assessment; 2003. [Google Scholar]
- 25.Wechsler D. The Wechsler Abbreviated Scale of Intelligence, 2nd ed. London: Pearson; 2011. [Google Scholar]
- 26.Wechsler D. Wechsler Intelligence Scale for Children, Australian Standardised Edition, 4th ed. Sydney: Pearson Clinical and Talent Assessment; 2005. [Google Scholar]
- 27.Sparrow SS, Cicchetti DV, Balla DA. Vineland Adaptive Behavior Scales, 2nd ed. London: Pearson; 2005. [Google Scholar]
- 28.Bayley N. Bayley Scales of Infant and Toddler Development, 3rd ed. London: Pearson; 2005. [Google Scholar]
- 29.Carvill GL, Weckhuysen S, McMahon JM, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 2014;82:1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007;130:843–852. [DOI] [PubMed] [Google Scholar]
- 31.Rodda JM, Scheffer IE, McMahon JM, Berkovic SF, Graham HK. Progressive gait deterioration in adolescents with Dravet syndrome. Arch Neurol 2012;69:873–878. [DOI] [PubMed] [Google Scholar]
- 32.Singh R, Andermann E, Whitehouse WP, et al. Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia 2001;42:837–844. [DOI] [PubMed] [Google Scholar]
- 33.Jansen FE, Sadleir LG, Harkin LA, et al. Severe myoclonic epilepsy of infancy (Dravet syndrome): recognition and diagnosis in adults. Neurology 2006;67:2224–2226. [DOI] [PubMed] [Google Scholar]
- 34.Vadlamudi L, Dibbens LM, Lawrence KM, et al. Timing of de novo mutagenesis: a twin study of sodium-channel mutations. New Engl J Med 2010;363:1335–1340. [DOI] [PubMed] [Google Scholar]
- 35.Claes LRF, Deprez L, Suls A, et al. The SCN1A variant database: a novel research and diagnostic tool. Hum Mutat 2009;30:E904–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.