

Abstract
Myeloid differentiation factor (MD-2) is an extracellular protein, associated with the ectodomain of Toll-like receptor 4 (TLR4), that plays a critical role in the recognition of bacterial lipopolysaccharide (LPS). Despite high overall structural and functional similarity, human (h) and murine (m) MD-2 exhibit several species-related differences. hMD-2 is capable of binding LPS in the absence of TLR4, whereas mMD-2 supports LPS responsiveness only when mMD-2 and mTLR4 are coexpressed in the same cell. Previously, charged residues at the edge of LPS binding pocket have been attributed to this difference. In this study, site-directed mutagenesis was used to explore the hydrophobic residues within MD-2 binding pocket, as the source of functional differences between hMD-2 and mMD-2. While decreased hydrophobicity of residues 61 and 63 in hMD-2 binding pocket retained the characteristics of wild type hMD-2, a relatively minor change of valine to alanine at position 135 completely abolished the binding of LPS to the hMD-2 mutant. The mutant, however, retained the LPS binding in complex with TLR4 and also cell activation, resulting in a murine-like phenotype. These results were supported by the molecular dynamics simulation. We propose that the residue at position 135 of MD-2 governs the dynamics of the binding pocket and its ability to accommodate lipid A, which is allosterically affected by bound TLR4.
Introduction
Endotoxins (i.e. lipopolysaccharide, LPS)3, the main components of Gram-negative bacterial cell envelope, are able to activate the innate immune system at picomolar concentrations, leading to the release of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), IL-1, IL-6 and IL-8 (1, 2). Recognition of the LPS is a complex process where transmembrane protein Toll-like receptor 4 (TLR4), myeloid differentiation factor (MD-2) and CD14 play crucial roles (3). CD14 binds and accumulates LPS and presents it to the TLR4/MD-2 receptor complex which, by dimerization, delivers a signal through the plasma membrane. It is essential that glycoprotein MD-2 binds to both, LPS and the extracellular domain of TLR4 (TLR4ecd). MD-2 presents the exposed acyl chain of the hexa-acylated lipid A moiety of LPS to TLR4ecd, which triggers receptor dimerization. TLR4ecd adopts a horseshoe-like shape consisting of leucine-rich repeats (LRRs) (4), while MD-2 is characterized by a β-cup fold structure composed of two antiparallel β sheets forming a large hydrophobic pocket for ligand binding (5–7). Several crystal structures of MD-2 from different species and with different bound ligands, including agonists and antagonists, have been determined (6–8). In these structures, the four acyl chains of the lipid IVa fit into the MD-2 pocket. The sixth acyl chain of LPS remains at the surface of MD-2, partially exposed to the solvent, and forms a hydrophobic interface for the TLR4 from the neighboring TLR4/MD-2/LPS complex, triggering the receptor dimerization and activation. Despite the proposed global structural changes upon binding of agonist or antagonist, the crystal structure of TLR4/MD-2/LPS complex (9) and molecular studies of the MD-2 hydrophobic loop (10) demonstrated that the size of the MD-2 pocket remains unchanged and the structural changes are localized to the edge of the pocket.
Although there is a high degree of structural similarity, MD-2 orthologues exhibit some important functional differences, including the binding of certain endotoxin chemotypes (hypo-acylated lipid A) and mimetics (Taxol), or the ability of these ligands to act as TLR4 agonists (11, 12). Notably, murine (m) and human (h) MD-2 differ in their discrimination between lipid A (506) and lipid IVa (406) (13, 14). Mutagenesis studies revealed that several residues in the hydrophobic pocket of mMD2 (e.g. 42, 57, 61 and 69) and residues at the entrance of the hydrophobic pocket (e.g. 122, 125) influence the agonist activity of lipid IVa (13, 15). It has been further shown that residues 82 and 122 of MD-2 govern species-specific activation of TLR4 by tetra- and penta-acylated endotoxins (16). Finally, there is an apparent functional difference between the two species: hMD-2, but not mMD-2, is able to react with LPS when expressed and secreted in the absence of TLR4, to ultimately form a functional complex with TLR4 (17–20).
Our study aimed to identify the residues of hMD-2 required for the ligand binding to soluble MD-2 (sMD-2). Several residues in the hydrophobic pocket of hMD-2 have been replaced with the corresponding mMD-2 residues or with the residues with modified hydrophobicity. The majority of residues in the hydrophobic pocket of MD-2 are conserved. The residues at positions 61 and 63 that are occupied by hydrophobic residues did not cause any differences. Surprisingly however, the residue at position 135 exhibited a large effect, although it lies at the very bottom of the pocket. Replacement of Val-135 by Ala, as in mouse MD-2, inactivated MD-2 ability to bind LPS, while maintaining its ability to bind and activate cells if present in the complex with TLR4. This indicates that the residue at position 135 is important for the function of MD-2. We propose, based on the experimental results and a molecular dynamics simulation, that this residue plays a role in the dynamics of the MD-2 binding pocket, with an allosteric effect of the bound TLR4.
Materials and Methods
Cell culture and reagents
Human embryonic kidney (HEK) 293 cells were provided by Dr. J. Chow (Eisai Research Institute, Andover, MA, USA). HEK293 cells stably transfected with TLR4 (HEK293/TLR4 no. BF1) were provided by Dr. Douglas Golenbock (University of Massachusetts Medical Center, Worcester, MA, USA) and Dr. Andra Schromm (Research Center Borstel, Borstel, Germany). HEK293T cells, used for the analysis of complex formation between lipooligosaccharide (LOS) and MD-2 using gel filtration chromatography and in immunoblotting experiments, were provided by Dr. Fabio Re (University of Tennessee Health Sciences Center, Memphis, TN, USA). Expression plasmids containing the sequences of human TLR4 and MD-2 as well as the pELAM-1 firefly luciferase plasmid were a gift from Dr. C. Kirschning (University of Duisburg-Essen, Institute of Medical Microbiology, Germany). Expression plasmid containing the sequence of mouse TLR4 was purchased from InvivoGen (San Diego, CA, USA). Expression plasmid for mouse MD-2 was a gift from Dr. Y. Nagai (University of Tokyo, Japan). The Renilla luciferase phRL-TK plasmid was purchased from Promega (Fitchburg, WI, USA). The nucleotide sequences encoding MD-2 were cloned into pEF-BOS vector with Flag and His tags on the C-terminal. The nucleotide sequences encoding TLR4 were cloned into pUNO vector with C-terminal HA tag. Transfection reagent JetPEI was purchased from Polyplus-Transfection (Illkirch, France) and was used according to the manufacturer’s instructions. S-LPS (from Salmonella abortus equi HL83) was purchased from Sigma (St. Louis, MO, USA). Escherichia coli-type lipid A (compound 506) was obtained from the Peptide Institute (Osaka, Japan). Purified [3H]LOS (25,000 cpm/pmol) was isolated from an acetate auxotroph of Neisseria meningitidis serogroup B after metabolic labeling, as described (21). Sephacryl S500 and S200 HR size exclusion gel matrices were purchased from GE Healthcare (Buckinghamshire, UK). Human serum albumin (HSA) was obtained as an endotoxin-free, 25% stock solution (Baxter Health Care, Deerfield, IL, USA). Anti-Tetra-His antibodies and goat anti-mouse HRP-conjugated secondary Abs were from Qiagen (Valencia, CA, USA) and Jackson Immunologicals (West Grove, PA, USA), respectively. LPS-binding protein (LBP) and sCD14 were gifts from XOMA (Berkeley, CA, USA) and Amgen Corp. (Thousand Oaks, CA, USA), respectively. AlphaLISA IL-8 Immunoassay Research kit was acquired from Perkin Elmer (Waltham, MA, USA).
Site-directed Mutagenesis
All mutations were introduced into pEFBOS-h or -m MD-2-FLAG-His plasmid using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s instructions. All plasmids were sequenced to confirm the mutation. Primer sequences will be made available upon request.
Preparation of [3H]LOSagg and [3H]LOS:sCD14 Complex
[3H]LOSagg and [3H]LOS:sCD14 complex were prepared as previously described (22–24). Briefly, [3H]LOSagg (Mr > 20 × 106) were obtained after hot phenol extraction of [3H]LOS followed by ethanol precipitation of [3H]LOSagg and ultracentrifugation. Monomeric [3H]LOS:CD14 complexes (Mr ~ 60,000) were prepared by treatment of [3H]LOSagg for 30 min at 37°C with a substoichiometric LBP (molar ratio LOS:LBP = 100:1) and 1–1.5 x molar excess sCD14 followed by gel exclusion chromatography (Sephacryl S200, 1.6 × 70 cm column) in PBS pH 7.4, 0.03% HSA to isolate monomeric [3H]LOS:sCD14 complex. Radiochemical purity of [3H]LOSagg and [3H]LOS:sCD14 was confirmed by Sephacryl S500 (LOSagg) or S200 ([3H]LOS:sCD14) chromatography (21, 24).
Production and Reaction of Secreted (s)MD-2 and sMD-2/TLR4 ectodomain (TLR4ecd) with [3H]LOS:sCD14
HEK293T cells were plated in a 6-well plate with 10% FBS in DMEM. Cells were transfected the following day with an expression plasmid encoding MD-2 alone (wild-type (wt) or mutant) or cotransfected with expression plasmids encoding MD-2 and TLR4ecd using PolyFect reagent (Qiagen), as previously described (25). After 12–16 h, the medium was replaced with 1.5 ml of serum-free medium (DMEM) + 0.1% HSA. The medium was spiked with [3H]LOS:sCD14 (1 nM) at the time of medium replacement to permit the reaction of [3H]LOS:sCD14 with the newly secreted MD-2 ± wt TLR4ecd. The reaction products were analyzed by Sephacryl HR S200 (1.6 × 30 cm) chromatography in PBS. Fractions (0.5 ml) were collected at a flow rate of 0.5 ml/min at room temperature using AKTA Purifier or Explorer 100 fast protein liquid chromatography (GE Healthcare). Radioactivity in collected fractions was analyzed by liquid scintillation spectroscopy (Beckman LS liquid scintillation counter). In all cases, the recovery rate of [3H]LOS was ≥ 70%. All the solutions used were pyrogen-free and sterile-filtered.
HEK293 cell activation assays: Dual Luciferase Reporter Assay and AlphaLISA Assay
HEK293 cells were seeded into 96-well Costar plates (Corning, NY, USA) with 10% FBS in DMEM, at 3 × 104 cells/well, and incubated overnight in a humidified atmosphere (5% CO2) at 37°C. The next morning, cells were co-transfected for 4 h with pEFBOS- (wt or mutant, h or m) MD-2-FLAG-His and pUNO- (h or m) TLR4-HA together with NF-κB-dependent luciferase and constitutive Renilla reporter plasmids using Lipofectamine 2000 (Invitrogen, Life Technologies Waltham, MA, USA). After 4 h, medium was removed and replaced with DMEM + 10% FBS. The following day, cells were incubated with S-LPS for 16 h, as indicated. In selected experiments, HEK293 cells were seeded into 96-well plates (3 × 104 cells/well) and separately transfected either with mMD-2 (wt or mutant) or with TLR4 (h or m) together with NF-κB-dependent luciferase and constitutive Renilla reporter plasmids. After 16 h, aliquots of the conditioned medium of HEK293 cells containing sMD-2 were added to HEK293TLR4 cells where the supernatants have been removed. The cells were then activated with lipid A for 16h. In a different approach, HEK293 cells were seeded into 12-well plates (3 × 105 cells/well) and transfected either with mMD-2 (wt or mutant) or with TLR4 (h or m) together with NF-κB-dependent luciferase and constitutive Renilla reporter plasmids. After 16 h, the cells were resuspended in fresh medium containing 10% serum, joined in 1:1 ratio and re-seeded together in 96-well plates to yield co-cultures of cells separately expressing MD-2 or TLR4. The following day, the cells were incubated in the presence of S-LPS or lipid A for 16 h. After the activation with S-LPS or lipid A, the supernatants were harvested and the cells were lysed in 1 x reporter assay lysis buffer (Promega, Fitchburg, WI, USA) and analyzed for reporter gene activities using a dual-luciferase reporter assay system on a Mithras LB940 luminometer. Relative luciferase activity (RLA) was calculated by normalizing each sample’s luciferase activity for constitutive Renilla activity measured within the same sample. When plotting data the value of the unstimulated sample with wt hMD-2 was set to 1 and other values were adjusted accordingly. In the supernatants of HEK293 cells, IL-8 concentrations were determined with AlphaLISA IL-8 Immunoassay Research kit (Perkin Elmer), according to the manufacturer’s instructions.
Immunoblotting
To detect polyhistidine labeled wt and mutant MD-2, an anti-polyhistidine Ab (Tetra-His Ab, Qiagen) was used. We expressed wt MD-2 and MD-2 mutants in HEK293T cells, which do not express MD-2 without transfection with expression plasmids encoding MD-2 (11, 18). HEK293T cells were transiently transfected with wt or mutant MD-2 using PolyFect (Qiagen) as a transfection reagent. The medium was changed 12 h posttransfection and replaced with serum-free medium. Aliquots of conditioned medium from transfected and mock-transfected HEK293T cells were harvested after 24 h. Equal volumes of the medium and Laemmli sample buffer containing dithiothreitol were combined, and each sample was electrophoresed (Bio-Rad mini gel system) through a 4–15% gradient acrylamide gel (Tris/HEPES/SDS buffer) and transferred to nitrocellulose membrane. The membrane was washed with Tris-buffered saline, pH 7.5, containing 0.05% Tween 20 and 0.2% Triton X-100 (TBSTT), blocked to reduce nonspecific background with 5% dried nonfat milk in TBSTT for 1 h at 25 °C, and incubated with the anti-His4 antibody in the blocking solution overnight. After washing with TBSTT, the blot was incubated with goat antimouse IgG conjugated to HRP for 1 h at 25°C in the blocking solution and washed extensively with TBSTT. Blots were developed using the Pierce SuperSignal substrate system. By reducing immunoblot samples, each MD-2 species was converted to the monomeric form, migrating as a triplet (hMD-2) or quadruplet (mMD-2) due to the differences in glycosylation (26). Recovered extracellular medium (supernatants) were immunoblotted to confirm that levels of expression of wt and mutant MD-2 were comparable.
Computational Methods
Preparation of the macromolecules and myristic acids
The 3D coordinates of human and mouse MD-2 proteins were obtained from the corresponding X-Ray crystallographic structure. Mutation of Val-135 to Ala was done with PyMOL mutation wizard (www.pymol.org) in all studied systems. Caps to the N-Terminal and C-Terminal residues were added, ligands (if present) and crystallographic water molecules were deleted, missing hydrogens were added and protonation state of ionisable groups was computed by using Maestro Protein Preparation Wizard (Maestro, version 9.3, Schrödinger, LLC, New York, NY, 2012). Atom types and charges were assigned according to AMBER ff10 force field (27). Each resulting system was immersed in a rectangular box of explicit TIP3P water molecules (28) extending 10 Å away from any protein atom for simulating the aqueous environment with the help of AmberTools 13 (University of California, San Francisco, CA, USA). Coordinates for myristic acids were extracted from PDB 2e56, and parameters from gaff BCC force field were used. Myristate ionization state was considered for the calculations.
MD Simulations
Molecular dynamics (MD) simulations were run with Amber 12 (University of California, San Francisco, CA, USA). Before the MD simulations, all hydrated systems were equilibrated under the following protocol: initial 8000 steps of steepest descent minimization, followed by heating of the system with position restrain (force constant of 20 kcal mol−1 Å−2) for all protein atoms with during 10 ps of MD simulation increasing the temperature from 100°C to 300°C plus additional 15 ps at constant 300 °C. Position restrain was gradually decreased during 100 ps at constant 300 °C, until the full system was under no restrains with constant temperature and pressure. After equilibration, 50 ns of MD simulation were run for all systems at constant temperature (300 °C) and pressure (1 atm). Short and long range forces were calculated every one and two time steps, respectively (each time step = 2.0 fs), constraining the covalent bonds involving hydrogen atoms to their equilibrium values. Long range electrostatic interactions were accounted for using the particle mesh Ewald approach (29), applying periodic boundary conditions. The root mean square deviation (RMSD) as a function of time with respect to the starting structure for the α-C atoms was computed for all the studied systems showing main changes in the first 5 ns and complete convergence beyond 10 ns till the end of simulation.
Average structures
With the help of the ptraj module of AmberTools 13, for each MD simulation of the eight systems, five average structures were extracted corresponding to the following time slots: from 0 to 2.5 ns (AVG-1), from 2.5 to 5 ns (AVG-2), from 5 to 10 ns (AVG-3), from 10 to 20 ns (AVG-4) and from 20 to 50 ns (AVG-5). All average structures were minimized with 5000 steps of steepest descent minimization with position restrain (force constant of 10 kcal mol−1 Å−2) for all non-hydrogen atoms, plus 5000 steps of steepest descent minimization with no restrains.
SASA and volume calculations
Solvent accessible surface area (SASA) and solvent accesible volume were calculated on hydrophobic pockets of MD-2 by using CASTp server (30). CASTp server identifies and computes the molecular area and volume for cavities and pockets of a given protein. In our case, we computed SASA and volumes of the hydrophobic pocket of MD-2 for the 40 structures corresponding to the five average structures derived from each studied molecular system.
Results
Mutations in the hydrophobic pocket of hMD-2 do not affect hMD-2 expression and TLR4 activation
MD-2 has a narrow and deep binding pocket with hydrophobic residues lining the internal surface and positively charged residues located at the opening rim of the cavity (6, 7). This pocket accommodates acyl chains of the lipid A. In order to study the role of hydrophobic amino acids in the binding pocket of hMD-2, we prepared point mutations at positions within the hydrophobic binding pocket where human and mouse MD-2 differ, namely residues Leu-61, Ile-63, and Val-135 (Fig. 1A). Hydrophobic residues are conserved at these positions, however, the size and hydrophobicity of the residues might underlie the species-specific differences. To test this hypothesis, residues 61, 63, and 135 of hMD-2 were replaced with the corresponding mMD-2 residues, each smaller and less hydrophobic than their hMD-2 counterparts. Next, additional mutants of hMD-2 with more hydrophobic residues at positions Cys-133 and Val-135 were tested. Finally, two mMD-2 mutants with corresponding hMD-2 residues were prepared and evaluated (A135V and A135V E122K).
FIGURE 1. The hydrophobic pocket of human MD-2.

A, Left, an electrostatic surface representation of hMD-2 (PDB ID 2E56) (Coulombic coloring), showing the hydrophobic pocket (white surface). Right, a closeup of the hydrophobic pocket with amino acid residues L61, I63, C133 and V135 shown in purple, pink, yellow and orange, respectively. B, Amino acid alignment of human, murine and equine MD-2.
We first examined the effects of these mutations on the expression and secretion of MD-2. The cell lysates and extracellular media from transiently transfected HEK293T cells expressing either wt hMD-2 or hMD-2 mutants were analyzed by immunoblotting. Figure 2A shows that all hMD-2 mutants were expressed and secreted at a similar level as wt hMD-2. This facilitated the interspecies comparison of the hMD-2 ability to promote cellular response to endotoxin. Figures 2B, C and D demonstrate that none of the mutations affected hMD-2 coreceptor functional activity, as assessed by the ability of hMD-2 to support LPS-induced TLR4 activation when coexpressed in cells with h or mTLR4.
FIGURE 2. Effect of mutations at residues 61, 63, 133 and 135 of hMD-2.

A, Secretion of mutants into the medium from HEK293T cells transfected with expression plasmids encoding wt hMD-2 or mutant hMD-2. Multiple bands reflect differences in MD-2 glycosylation. B, C and D, NFkB-dependent reporter activity or IL-8 production of lipid A stimulated HEK293 cells transfected with of hMD-2 with replaced residues at positions 61, 63, 133 and 135; with residues with increased (C133F, C133L, C133F V135L, C133L V135L) or decreased (V135A, L61V, L61V I63V, L61V I63V V135A) hydrophobicity. HEK293 cells were transiently transfected with expression plasmids encoding MD-2 together with h or m TLR4 and reporter luciferase plasmids. Results are representative of two or more experiments.
Effect of mutations in the hydrophobic pocket of hMD-2 on LPS binding
To investigate the effect of mutations in the hydrophobic pocket of hMD-2 on endotoxin binding, we initially assayed the transfer of added radiolabeled monomeric [3H]LOS:sCD14 to soluble wt or mutant MD-2 that were expressed and secreted by transiently transfected HEK293T cells. MD-2 requires presentation of LOS/LPS as part of a monomeric LOS(LPS):protein complex with CD14 for high affinity (pM) binding of endotoxin. Transfer of [3H]LOS from CD14 (Mr ~ 60,000) to sMD-2 (Mr ~ 25,000) was monitored by the size exclusion chromatography, as previously described (24). Although all the mutants preserved the competence of wt hMD-2 to support LPS-triggered TLR4 activation (Fig. 2B–D), not all the mutant proteins retained the ability to bind [3H]LOS:sCD14 and form monomeric [3H]LOS:MD-2 complex (Fig. 3). Mutants with increased hydrophobicity at residues Cys-133 (C133F) and Val-135 (V125L) were found to bind LOS and form the LOS:MD-2 monomer at the same elution volume as the wt hMD-2 (Fig. 3A, B). Furthermore, less hydrophobic single or double mutants at Leu-61 and Ile-63 of hMD-2 (L61V, L61V I63V) also exhibited LPS binding similar to wt hMD-2, as shown by reactivity with LOS:sCD14 (Fig. 3C) and supported normal (wt hMD-2) TLR4-dependent cell activation by endotoxin (Fig. 2C, D). In contrast, mutation of a single residue, Val-135 to Ala, completely abolished the transfer of [3H]LOS from [3H]LOS:sCD14 to the MD-2 mutant (Fig. 3D). Yet there were no changes in the functional activities of hMD-2 V135A and L61V I63V V135A variants, as measured by luciferase reporter assay or AlphaLISA assay performed in HEK293/TLR4 cells (Fig. 2C, D).
FIGURE 3. The effect of changes in hydrophobicity of amino acid residues at 61, 63, 133 and 135 of hMD-2 on the ability of sMD-2 to react with [3H]LOS:sCD14 and form monomeric [3H]LOS:MD-2 complex.
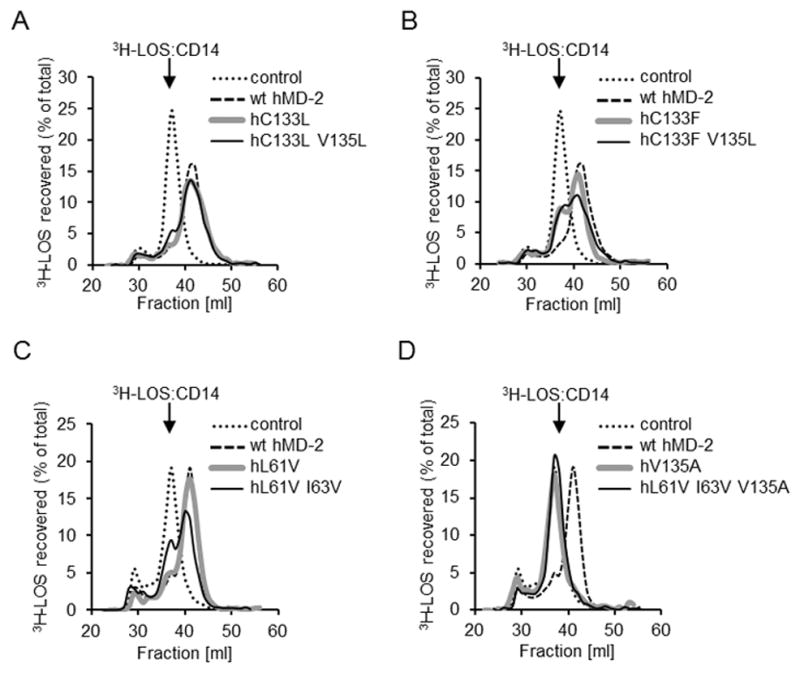
Soluble MD-2 (wt or mutant) was produced using transiently transfected HEK293T cells and tested for the ability to bind LOS (i.e. transfer LOS from the [3H]LOS:CD14 complex) using Sephacryl S200 chromatography as described under “Materials and Methods.” Note that the peaks of elution of [3H]LOS:sCD14 and [3H]LOS:MD-2 were at 37 and 42 ml, respectively. The results shown are from one experiment, representative of at least two independent determinations.
Activity of soluble V135A hMD-2 is strongly reduced in comparison to soluble wt hMD-2
LPS can activate human cells through binding to hMD-2 that is already associated with TLR4ecd or through binding of the hMD-2/LPS complex to the ectodomain of TLR4 (22, 24, 31). Murine MD-2 differs from its human orthologue as it does not form a detectable mMD-2/LPS complex (17, 20). The inability of secreted V135A hMD-2 to react with [3H]LOS:sCD14 suggested that this mutant may have impaired ability to support LPS-induced activation in the cells expressing TLR4 without MD-2 and whose LPS sensing ability depends on the addition of soluble MD-2. To test this hypothesis, we compared the ability of secreted (soluble) wt hMD-2 and V135A hMD-2 to confer activation of HEK293/TLR4 cells by LPS. As shown in figures 4A and B, cells transfected with wither h- or mTLR4 were unable to respond to LPS in the presence of a medium containing soluble V135A hMD-2. This coincided with the differences observed in the reaction of V135A hMD-2 with [3H]LOS:sCD14 (Fig. 3D). It has been reported that MD-2 is secreted as a labile molecule that, over a relatively short period of time, may lose its biological activity at physiological temperature in a serum-free medium (32). If, however, freshly synthesized soluble hMD-2 is exposed to LPS and CD14, it converts to a stable MD-2/LPS complex that is capable of activating TLR4 (24, 33–35). Since the presence of LPS significantly stabilized MD-2, we therefore performed the similar experiment by spiking the cell medium with LPS so that all secreted MD-2 could immediately bind LPS and increase its stability. However, relative to the wt hMD-2, the biological activity of V135A mutant was reduced to the same extent as when LPS was added subsequently (Fig. 4A, B). Most MD-2 orthologues have a valine residue at position 135, with notable exceptions of murine and horse (e) MD-2 that have alanine at this position. Consequently, we predicted that isolated eMD-2, like mMD-2, would not be able to bind LPS in solution. Indeed, soluble eMD-2 did not confer activation of HEK293/mTLR4 cells by LPS (Fig. 4B), however, co-transfection of h- or mTLR4 with eMD-2 resulted in LPS-triggered activation (16). On the other hand, co-culture of cells secreting MD-2 variants and cells expressing TLR4 and luciferase reporter demonstrated only slightly lower response by the mutated V135A hMD-2 (Figs. 4C, D and 4E, F for hTLR4 and mTLR4, respectively). This suggests that the secreted V135A MD-2 mutant is functional, yet it loses the competence to mediate LPS signaling unless it is rapidly recruited to TLR4.
FIGURE 4. Cell activation by LPS, mediated by MD-2 variants via soluble proteins or cell co-culture.

A, and B, HEK293 cells were transiently transfected with plasmids encoding MD-2. After 24 h, harvested medium containing sMD-2 was incubated with lipid A (50, 200 or 500 ng/ml) and added to HEK293 cells that had been transfected with a plasmid encoding h or mTLR4 together with NF-κB-dependent luciferase reporter plasmid. C, D, E, and F, Co-culture of HEK293 cells transfected with plasmids encoding wt or V135A hMD-2 and cells transfected with TLR4 and NF-κB-dependent luciferase reporter plasmids. Co-cultured cells were stimulated with increasing concentration of S-LPS or lipid A for 16 h. G, Inhibition of TLR4 signaling with soluble V135A hMD-2. Culture medium of HEK293 cells transfected with plasmids encoding wt hMD-2 together with V135A hMD-2, in ratios from 1:0 to 1:10, was incubated with lipid A (10, 500 or 1000 ng/ml) and then added to HEK293 cells that had been transfected with a plasmid encoding hTLR4 together with NF-κB-dependent luciferase reporter plasmid. All the results shown are means ± SEM RLA from three independent experiments.
Next, we assessed whether soluble V135A hMD-2 mutant, apart from failing to bind endotoxin, also lacks the ability to bind TLR4, which would be expected if the mutation affects the global protein fold. For this purpose, TLR4 expressing HEK293 cells were incubated with a constant amount of soluble wt hMD-2 and increasing amounts of soluble V135A hMD-2. As indicated in figure 4G, soluble biologically inactive V135A hMD-2 caused a dose-dependent inhibition of TLR4 activation that was otherwise induced by the added wt hMD-2 + lipid A. This result is most compatible with a selective deficiency in LPS (but not TLR4) binding, resulting at higher doses in a reduced fraction of TLR4 available to bind wt hMD-2 and respond to the added lipid A.
LPS binding to soluble hMD-2 coexpressed with TLR4ecd
Previous research has shown that TLR4 ectodomain can rescue the functional activity (i.e. LPS binding) of MD-2 variants prone to aggregation by stabilizing the functional, monomeric state of MD-2 (10, 36–38). In our study, the retained functional responsiveness to LPS of the cells coexpressing TLR4 and V135A hMD-2 (Fig. 2D) indicated that pre-association of V135A hMD-2 with TLR4 ectodomain might sustain the interaction of V135A hMD-2 with [3H]LOS:sCD14, as compared to secreted V135A hMD-2 in the absence of TLR4. To test this hypothesis, experiments were repeated with cells coexpressing wt hMD-2 or V135A hMD-2 with hTLR4ecd. As seen from figure 5, the addition of [3H]LOS:sCD14 to the culture medium of the cells expressing wt hMD-2 and hTLR4ecd resulted in a nearly complete conversion of [3H]LOS:sCD14 to the earlier eluting (Mr ~ 190,000) and later eluting (Mr ~ 25,000) [3H]LOS-containing complexes representing ([3H]LOS:MD-2:TLR4ecd)2 (38) and [3H]LOS:MD-2 (35), respectively. As predicted, coexpression of V135A hMD-2 with hTLR4ecd resulted in a transition of [3H]LOS:sCD14 to ([3H]LOS: V135A MD-2:TLR4ecd)2 but not to [3H]LOS: V135A MD-2 (Fig. 5). This demonstrates the association of V135A hMD-2 with TLR4ecd is prerequisite for the reaction of V135A hMD-2 with [3H]LOS:sCD14. V135A hMD-2 fails to bind LPS in the absence of TLR4 (Figs. 3 and 5) and TLR4 activation is achieved only when V135A hMD-2 and TLR4 are expressed in the same cell (Fig. 2C, D) or by the neighboring cells (Fig. 4C–F).
FIGURE 5. Binding of [3H]LOS:sCD14 to V135A hMD-2 in the presence of TLR4ecd.
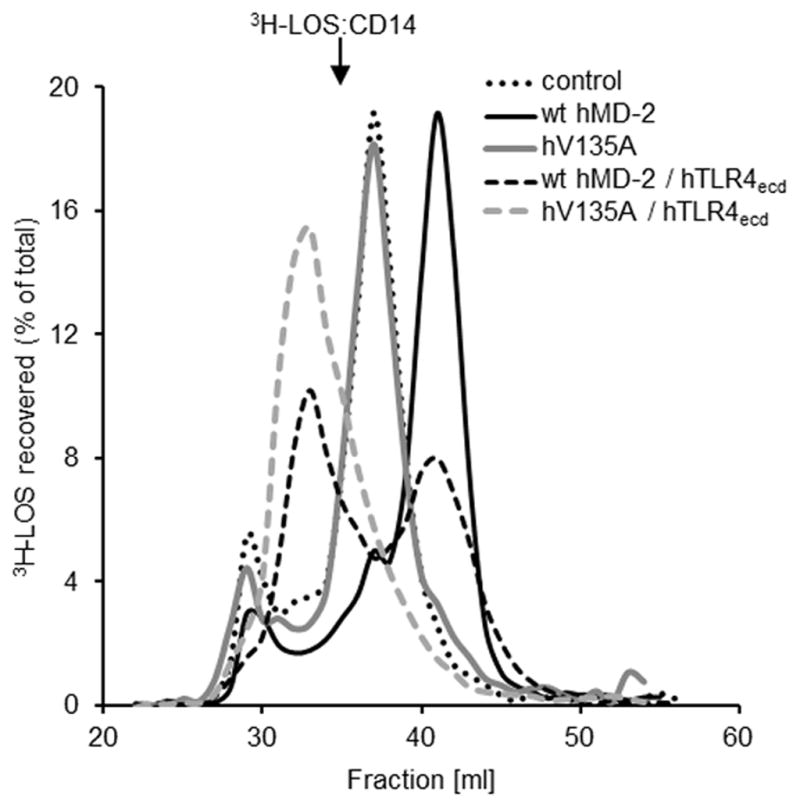
HEK293T cells were cotransfected with plasmids encoding hTLR4ecd and wt or mutant hMD-2, as indicated. After 24 h, the transfection medium was changed with serum-free medium and spiked with 1 nM [3H]LOS:sCD14. Medium was harvested at 24 h and analyzed by Sephacryl S500 size exclusion chromatography. Resolved reactants and products were monitored by liquid scintillation spectroscopy. Note that the reaction of [3H]LOS:sCD14 with MD-2:TLR4ecd yields ([3H]LOS:MD-2/TLR4ecd)2 complex, whereas the interaction of [3H]LOS:sCD14 with sMD-2 leads to the formation of [3H]LOS:MD-2. The results shown are representative of two or more experiments.
The reverse mMD-2 A135V mutant confirms the role of Val-135 in soluble hMD-2 to bind endotoxin
The above findings indicate that the ability of wt hMD-2 to form bioactive soluble MD-2 can be completely abolished by a single substitution of valine 135 with alanine that is present in wt mMD-2. To test the role of Ala-135 for the inability of mMD-2 to form bioactive sMD-2 when expressed in the absence of TLR4, residue 135 in mMD-2 was replaced with valine, as the corresponding amino acid present in wt hMD-2 (A135V). wt mMD-2 is secreted in a much lower amount in comparison to wt hMD-2 (20) (Fig 6A). Although recovery of secreted mMD-2 was not measurably improved by the single A135V mutation (Fig. 6A), increased TLR4 activation by lipid A was observed, especially in the cells expressing mTLR4 and co-cultured with cells expressing and secreting A135V mMD-2 (Fig. 6B–E). Combination of the A135V mutation with a mutation (E122K) that alone markedly increases secretion of mMD-2 without conferring functional reactivity and responses to LPS (20) yielded a much greater recovery of the secreted MD-2 (Fig. 6A) and greater TLR4 responsiveness to lipid A (Fig. 6B–E). These findings demonstrate that the Ala-135 residue of mMD-2 contributes to the restriction of its biological activity in the soluble form.
FIGURE 6. The effect of mMD-2 mutations on the ability to activate cells by lipid A.

A, wt mMD-2 exhibits drastically lower extracellular accumulation compared to wt hMD-2. E122K substitution induces a substantial increase in secretion and recovery of extracellular mMD-2. B, and C, Activity of secreted soluble mMD-2 is increased with A135V/E122K double mutation. Medium of HEK293 cells transfected with plasmids encoding MD-2 was added to HEK293 cells transfected with a plasmid encoding h or mTLR4 together with NF-κB-dependent luciferase reporter plasmid. Cells were stimulated with lipid A (10, 100, 1000 ng/ml) and assayed for luciferase activity. D, and E, Co-culture of HEK293 cells transiently transfected with plasmids encoding wt or mutant mMD-2 and HEK293 cells transfected with h or mTLR4 and NF-κB-dependent luciferase reporter plasmids. Co-cultured cells were stimulated with lipid A (10, 100, 1000 ng/ml) for 16 h and assayed for luciferase activity. All the results shown are means ± SEM RLA from three independent experiments.
MD simulations of WT and V135A mutant TLR4/MD-2 systems: computational studies of the binding pocket collapse
In order to obtain a molecular insight of the putative conformational changes in the ligand-free MD-2 prior to LPS binding, MD simulations were performed with MD-2 or TLR4/MD-2 systems in aqueous solvent without lipids, as these structures represent an obligatory intermediate step before the binding of LPS, and also in complex with three myristic acids, as observed in some X-ray crystallographic structures. Ten molecular systems were built corresponding to hMD-2 protein alone (systems 1–4, wt and mutant, agonist and antagonist conformation), hMD-2 protein in complex with TLR4 (systems 5 and 6, wt and mutant), mMD-2 (systems 7 and 8, agonist and antagonist conformation) and hMD-2 protein in complex with three myristic acids (systems 9 and 10, wt and mutant, antagonist conformation). Systems 1–8, lacking any lipid inside the MD-2 pocket, were submitted to MD simulations in explicit solvent (water) during 50 ns. Changes of the solvent accessible surface area (SASA) and solvent accessible volume of the hydrophobic pocket of MD-2 were monitored along the simulation time, as well as conformational changes in different regions of the protein. Wild type hMD-2 structures were observed to suffer a hydrophobic collapse, accordingly to a quick decrease of the pocket volume and SASA, leading to closed pocket structures (Fig. 7). In the case of mutant hMD-2, and similarly mMD-2, the pocket volume and SASA also decreased at the first stages of the simulation, but stuck at some point in a half-close pocket, and the full collapse was not observed. This could point to a decreased flexibility of V135A hMD-2 and wt mMD-2. This loss of plasticity could account for a poorer ability to bind LPS in accordance with the biological assays. In the case of hTLR4/MD2 complexes, both systems 5 and 6 (wt and mutant) exhibited similar dynamics, showing high values of pocket volume and SASA along the simulation, with no meaningful differences (Fig. 7B). We observed that the pocket dynamics of MD-2 in complex with TLR4 ectodomain was significantly reduced.
FIGURE 7. Molecular dynamics simulation analysis of free MD-2 and MD-2 bound to TLR4 ectodomain.
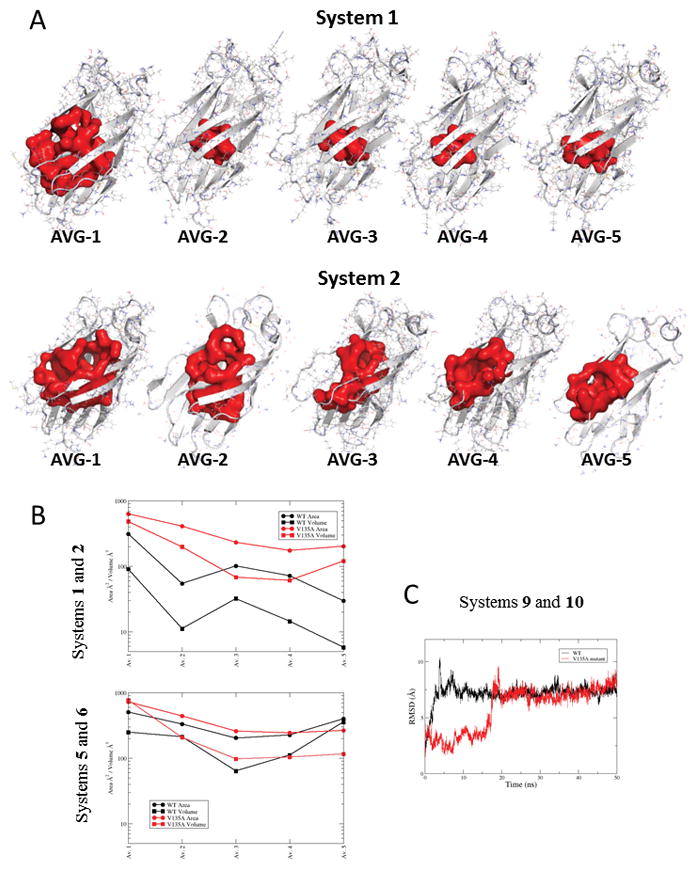
A, CASTp-calculated volumes of the MD-2 pocket for the molecular systems 1 (wt) and 2 (V135A mutant) corresponding to hMD-2 in the agonist conformation from PDB-ID 3FXI. B, CASTp-calculated SASA and volumes of the MD-2 pocket for the molecular systems 1 and 2 (corresponding to hMD-2 in the agonist conformation from PDB-ID 3FXI), 5 and 6 (corresponding to hMD-2 in the antagonist conformation from PDB-ID 2E59). C, Variation of the RMSD of the heavy atoms of the three myristic acids with respect to the starting geometry along the 50ns MD simulation for the complex of wt MD-2 with three myristic acids (system 9, in black) and for the complex of mutant V135A hMD-2 with three myristic acids (system 10, in red). The average structures come from the MD simulations (50 ns, explicit water). Ordinate values are expressed in the logarithm scale.
In wt hMD-2, Val-135 is surrounded by three phenyl rings from the corresponding Phe76, Phe147, and Phe151 residues, and stability of this association can be supported through CH-π interactions between the Val-135 side chain and the aromatic ring from Phe76. In mMD-2, naturally occurring Ala-135 is also surrounded by three Phe side chains, with similar spatial disposition. Scrutiny of the conformational change events revealed that, in wt hMD-2, the Phe76 and Phe147 side chains remarkably change their initial position during the simulations. Phe76 orients the phenyl ring toward the Phe147 away from Val-135. This movement pushes the rotation of Phe147 side chain, while Phe151 side chain almost maintains its initial crystallographic position. In the mutant and, interestingly, in the mMD-2, the three Phe side chains do not move significantly from the starting crystal-derived geometry. This observation seems to point to a lower plasticity of the V135A variant, both the mutant hMD-2 and wt mMD-2. Analysis of the structural changes in the protein-protein interfaces (dimerization and primary interfaces) does not allow us to conclude any consequence for the dimerization event.
MD simulations of the hMD-2 protein in complex with three myristic acids (systems 9 and 10, wt and mutant) led to the observation that the wt type complex (system 9) gains stability much faster along the simulation time, while in the case of the complex of the V135A mutant hMD-2 (system 10), the myristic acids are stabilized only after 20 ns of simulation (Fig. 7C). This slower adaptation to the lipids supports the hypothesis of a lower plasticity for the V135A hMD-2 mutant and a decreased ability to accommodate lipid-based ligands.
Discussion
The MD-2 protein is profoundly involved in cellular response to endotoxins from Gram-negative bacteria. It is also required for TLR4 signaling in nearly all investigated cases of endogenous sterile inflammation agonists investigated so far (39). Functional differences between human and murine MD-2 have been extensively investigated by the modeling, docking and point mutations (12, 20, 40, 41). In this study, we focused on the hydrophobic interactions between the acyl chains of LPS and the amino acid residues in the hydrophobic cavity of MD-2. Increasing the hydrophobicity of hMD-2 binding pocket preserved both, cellular responsiveness to endotoxin (Fig. 2B) and transfer of endotoxin from CD14 to the MD-2 (Fig. 3A, B). Moreover, decreased hydrophobicity of the pocket at Leu-61 and Ile-63 retained the characteristics of wt hMD-2 (Fig. 2C). Surprisingly, a relatively minute change of valine to alanine at position 135 significantly impaired the binding of LPS to soluble MD-2 (Fig. 3D). This led to a hypothesis that Ala-135 impairs the ability of mMD-2 to bind LPS in the absence of TLR4. The striking effect of a single amino acid residue at this position could play a critical role in the cellular response to endotoxins, contributing to the interspecies differences.
An important clue on the ability of alanine residue at position 135 to disable the bioactivity of soluble mMD-2 is provided by the fact that coexpression of mMD-2 with mTLR4 eliminates the need for a valine at this site (Fig. 2D). This finding, along with the location of Val-135 at the bottom of the pocket, indicates that the valine residue is not required for direct interactions of mMD-2 with mTLR4 but rather to preserve the bioactivity of soluble MD-2 secreted in the absence of TLR4. Subsequently, as a soluble extracellular protein, MD-2 can interact with endotoxin, presented by CD14, and/or with TLR4. The increased activity of soluble mMD-2 A135V in comparison to wt mMD-2 (Fig. 6) confirmed our hypothesis that valine 135, either directly or indirectly, preserves the bioactivity of soluble MD-2. In contrast to wt hMD-2, recombinant hMD-2 V135A was unable to bind LPS presented by CD14 or activate TLR4-transfected HEK293 cells. Accordingly, we propose that, despite the small difference in chemical properties between valine in human vs alanine in murine MD-2, valine increases the stability of bioactive MD-2 monomers in the absence of TLR4. Of note, equine MD-2 has alanine at position 135 and it phenocopies murine MD-2 by being biologically inactive when secreted without TLR4 but active when co-expressed with TLR4, further supporting our conclusions/hypothesis.
Both LPS binding (i.e., transfer of LPS monomer from CD14 to MD-2) and TLR4 binding require and stabilize MD-2 monomers (35). Thus, conceivably the effect of the alanine for valine substitution at residue 135 could be on the stability of MD-2 monomers. However, the ability of secreted V135A hMD-2 to competitively inhibit activation of TLR4 by lipid A when added in excess of wt hMD-2 suggests a much more discrete structural effect on MD-2 monomers that selectively affects LPS/lipid A binding. The finding that secreted V135A hMD-2 can partially reconstitute functional MD-2/TLR4 receptors on the neighboring cells expressing TLR4 alone (Fig. 4C–F) indicates that TLR4 binding soon after secretion can rescue the LPS binding function of V135A hMD-2. The inability of later exposure of secreted V135A hMD-2 to TLR4 to reconstitute functional MD-2/TLR4 receptors (Fig. 4A, B and G) indicates a time-dependent selective loss of LPS binding by MD-2(/TLR4) that cannot be reversed by subsequent association with TLR4 and, hence, not mediated by a more direct role of TLR4 in the binding of LPS to V135A hMD-2/TLR4 heterodimers.
Secreted wt mMD-2 has no detectable functional activity, as assessed either by reactivity with [3H]LOS:sCD14 spiked into the culture medium (20) or via the LPS-triggered activation of HEK293/mTLR4 cells (Fig. 6C). Although single residue substitution E122K in mMD-2 yielded detectable amounts of soluble MD-2 (20), it had no functional activity. However, the substitutions A135V ± E122K led to a significant increase in the bioactivity of soluble mMD-2 (Fig. 6). Taken together, these findings suggest the contribution of Lys-122 in enhanced secretion of MD-2 and Val-135 in maintaining the LPS-binding competence of soluble mMD-2. Molecular mechanism underlying the rescue of the functionality of soluble mMD-2 by association with TLR4 is in part the result of prevented aggregation due to its low solubility. Comparison of the surface charge distribution of MD-2 from different species shows significant differences in the electrostatic potential around the ligand–binding cavity (40). On the other hand, even the soluble mMD-2 mutants with increased net charge were unable to bind LPS, suggesting an additional factor in maintaining the competence for LPS binding. Position of Ala-135 deep inside the binding pocket could not affect its solubility but rather the binding pocket dynamics. We speculate that the structure of soluble V135A hMD-2 has a higher propensity to collapse in the absence of TLR4 and blunt the cellular signaling. In fact, MD simulations point out the possibility of a collapse of the binding pocket in the absence of bound residential lipids. It is likely that different lipid ligands may bind to MD-2 in the absence of LPS as the myristates were observed in the crystal structure of human MD-2 (6). Although the simulation of the whole process of the residential lipid dissociation from MD-2 and the kinetics of LPS binding to MD-2 is beyond the current computational ability of the MD simulations, the simulations presented here provide a plausible explanation accounting for a decreased plasticity of the V135A hMD-2 protein, thus leading to a diminished ability to bind host lipids such as myristic acid. We propose as a plausible scenario that the closing of the hydrophobic cavity occurs rapidly upon mMD-2 or V135A hMD-2 mutant secretion from the cell in the absence of TLR4. This collapsed conformation resembles the structure of the MD-2 homolog Der p 2 (42). The structure of Der p 2 has been investigated by both NMR (43) and X-ray crystallography (42). An overlay of the two Der p 2 structures indicated significant flexibility of the protein structure. The beta sheets in the Der p 2 X-ray structure are significantly further apart than in the NMR model and create an internal cavity, which is occupied by hydrophobic ligand, while the NMR structure displays a collapsed binding pocket. Hence we propose the existence of two MD-2 modes, closed and opened, depending on the cellular environment (i.e. presence of TLR4, LPS, other lipids). We suggest that soluble mMD-2 has a propensity for a closed, biologically inactive mode in the absence of bound TLR4 ectodomain and lower concentrations of surrounding host lipids, whereas soluble hMD-2, due to the Val-135, has a higher population of an opened mode, ready to bind the lipid-like ligands regardless of the presence of TLR4. This hypothesis might be confirmed by a high resolution structure of soluble mMD-2, since only the TLR4-bound (stabilized) structure of mMD-2 has been determined so far (44).
In conclusion, results of this study provide first evidence for the role of specific hydrophobic residues that are responsible for the competence of the circulating MD-2 for binding of LPS in the absence of TLR4, allowing LPS responsiveness of cells that do not express MD-2. The different MD-2 orthologues presented here (hMD-2, mMD-2, eqMD-2) suggest that subtle differences in molecular structure can have significant influence on the signaling process. Excessive production of inflammatory mediators may be harmful to host tissue, hence MD-2 represents an attractive therapeutic target of inflammatory and immune diseases. Our findings thus provide important information on the hydrophobic LPS binding site that may support development of potential therapeutic agents.
Acknowledgments
The authors have no financial conflicts of interest.
Footnotes
The project was supported by grants from the Slovenian research agency (J1-9795, J1-2271, P4-0176, Z1-3673), a bilateral Slovenian-USA collaborative grant, grants from the National Institute of Allergy and Infectious Disease (AI05732; to J.W.) and the Veterans’ Administration (to T.L.G.). Grants from Spanish MINECO (CTQ2011-22724 and CTQ2014-57141-R) and European Commission H2020-MSC-ETN-642157 TOLLerant project are gratefully acknowledged.
Abbreviations used in this paper: HEK, human embryonic kidney; h, human; LOS, lipooligosaccharide; LPS, lipopolysaccharide; LBP, LPS-binding protein; m, murine; MD, molecular dynamics; MD-2, Myeloid differentiation factor; RLA, relative luciferase activity; s, soluble; SASA, solvent accessible surface area; TLR4, Toll-like receptor 4; TLR4ecd, TLR4 ectodomain; wt, wild type.
References
- 1.Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ, Hauschildt S. LPS-induced Cytokine Production in Human Monocytes and Macrophages. Crit Rev Immunol. 2011;31:379–446. doi: 10.1615/critrevimmunol.v31.i5.20. [DOI] [PubMed] [Google Scholar]
- 2.Verhasselt V, Buelens C, Willems F, De Groote D, Haeffner-Cavaillon N, Goldman M. Bacterial lipopolysaccharide stimulates the production of cytokines and the expression of costimulatory molecules by human peripheral blood dendritic cells: evidence for a soluble CD14-dependent pathway. J Immunol. 1997;158:2919–25. [PubMed] [Google Scholar]
- 3.Jerala R. Structural biology of the LPS recognition. Int J Med Microbiol. 2007;297:353–63. doi: 10.1016/j.ijmm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Bella J, Hindle KL, McEwan PA, Lovell SC. The leucine-rich repeat structure. Cell Mol Life Sci. 2008;65:2307–33. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gruber A, Mancek M, Wagner H, Kirschning CJ, Jerala R. Structural model of MD-2 and functional role of its basic amino acid clusters involved in cellular lipopolysaccharide recognition. J Biol Chem. 2004;279:28475–28482. doi: 10.1074/jbc.M400993200. [DOI] [PubMed] [Google Scholar]
- 6.Ohto U, Fukase K, Miyake K, Satow Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science. 2007;316:1632–4. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- 7.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–17. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Jin MS, Lee JO. Structures of the toll-like receptor family and its ligand complexes. Immunity. 2008;29:182–91. doi: 10.1016/j.immuni.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 10.Resman N, Vasl J, Oblak A, Pristovsek P, Gioannini TL, Weiss JP, Jerala R. Essential roles of hydrophobic residues in both MD-2 and toll-like receptor 4 in activation by endotoxin. J Biol Chem. 2009;284:15052–60. doi: 10.1074/jbc.M901429200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawasaki K, Akashi S, Shimazu R, Yoshida T, Miyake K, Nishijima M. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J Biol Chem. 2000;275:2251–4. doi: 10.1074/jbc.275.4.2251. [DOI] [PubMed] [Google Scholar]
- 12.Resman N, Gradisar H, Vasl J, Keber MM, Pristovsek P, Jerala R. Taxanes inhibit human TLR4 signaling by binding to MD-2. FEBS Lett. 2008;582:3929–34. doi: 10.1016/j.febslet.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 13.Muroi M, Tanamoto K. Structural regions of MD-2 that determine the agonist-antagonist activity of lipid IVa. J Biol Chem. 2006;281:5484–91. doi: 10.1074/jbc.M509193200. [DOI] [PubMed] [Google Scholar]
- 14.Meng J, Lien E, Golenbock DT. MD-2-mediated ionic interactions between lipid A and TLR4 are essential for receptor activation. J Biol Chem. 2010;285:8695–702. doi: 10.1074/jbc.M109.075127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng J, Drolet JR, Monks BG, Golenbock DT. MD-2 residues tyrosine 42, arginine 69, aspartic acid 122, and leucine 125 provide species specificity for lipid IVA. J Biol Chem. 2010;285:27935–43. doi: 10.1074/jbc.M110.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oblak A, Jerala R. Species-specific activation of TLR4 by hypoacylated endotoxins governed by residues 82 and 122 of MD-2. PLoS One. 2014;9:e107520. doi: 10.1371/journal.pone.0107520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visintin A, Halmen Ka, Khan N, Monks BG, Golenbock DT, Lien E. MD-2 expression is not required for cell surface targeting of Toll-like receptor 4 (TLR4) J Leukoc Biol. 2006;80:1584–92. doi: 10.1189/jlb.0606388. [DOI] [PubMed] [Google Scholar]
- 18.Hajjar AM, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat Immunol. 2002;3:354–9. doi: 10.1038/ni777. [DOI] [PubMed] [Google Scholar]
- 19.Fujimoto T, Yamazaki S, Eto-Kimura A, Takeshige K, Muta T. The amino-terminal region of toll-like receptor 4 is essential for binding to MD-2 and receptor translocation to the cell surface. J Biol Chem. 2004;279:47431–7. doi: 10.1074/jbc.M408724200. [DOI] [PubMed] [Google Scholar]
- 20.Vasl J, Oblak A, Gioannini TL, Weiss JP, Jerala R. Novel roles of lysines 122, 125, and 58 in functional differences between human and murine MD-2. J Immunol. 2009;183:5138–45. doi: 10.4049/jimmunol.0901544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giardina PC, Gioannini T, Buscher BA, Zaleski A, Zheng DS, Stoll L, Teghanemt A, Apicella MA, Weiss J. Construction of acetate auxotrophs of Neisseria meningitidis to study host-meningococcal endotoxin interactions. J Biol Chem. 2001;276:5883–5891. doi: 10.1074/jbc.M009273200. [DOI] [PubMed] [Google Scholar]
- 22.Gioannini TL, Teghanemt A, Zhang D, Levis EN, Weiss JP. Monomeric endotoxin:protein complexes are essential for TLR4-dependent cell activation. J Endotoxin Res. 2005;11:117–123. doi: 10.1179/096805105X35198. [DOI] [PubMed] [Google Scholar]
- 23.Gioannini TL, Zhang D, Teghanemt A, Weiss JP. An essential role for albumin in the interaction of endotoxin with lipopolysaccharide-binding protein and sCD14 and resultant cell activation. J Biol Chem. 2002;277:47818–25. doi: 10.1074/jbc.M206404200. [DOI] [PubMed] [Google Scholar]
- 24.Gioannini TL, Teghanemt A, Zhang DS, Coussens NP, Dockstader W, Ramaswamy S, Weiss JP. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc Natl Acad Sci U S A. 2004;101:4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasl J, Prohinar P, Gioannini TL, Weiss JP, Jerala R. Functional Activity of MD-2 Polymorphic Variant Is Significantly Different in Soluble and TLR4-Bound Forms: Decreased Endotoxin Binding by G56R MD-2 and Its Rescue by TLR4 Ectodomain. J Immunol. 2008;180:6107–6115. doi: 10.4049/jimmunol.180.9.6107. [DOI] [PubMed] [Google Scholar]
- 26.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–3. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Kollman PA. Automatic parameterization of force field by systematic search and genetic algorithms. J Comput Chem. 2001;22:1219–1228. [Google Scholar]
- 28.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- 29.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089. [Google Scholar]
- 30.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–97. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gioannini TL, Teghanemt A, Zhang D, Esparza G, Yu L, Weiss J. Purified monomeric ligand.MD-2 complexes reveal molecular and structural requirements for activation and antagonism of TLR4 by Gram-negative bacterial endotoxins. Immunol Res. 2014;59:3–11. doi: 10.1007/s12026-014-8543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy MN, Mullen GED, Leifer CA, Lee C, Mazzoni A, Dileepan KN, Segal DM. A complex of soluble MD-2 and lipopolysaccharide serves as an activating ligand for Toll-like receptor 4. J Biol Chem. 2004;279:34698–34704. doi: 10.1074/jbc.M405444200. [DOI] [PubMed] [Google Scholar]
- 33.Schromm AB, Lien E, Henneke P, Chow JC, Yoshimura A, Heine H, Latz E, Monks BG, Schwartz DA, Miyake K, Golenbock DT. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: a point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J Exp Med. 2001;194:79–88. doi: 10.1084/jem.194.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teghanemt A, Prohinar P, Gioannini TL, Weiss JP. Transfer of monomeric endotoxin from MD-2 to CD14: characterization and functional consequences. J Biol Chem. 2007;282:36250–6. doi: 10.1074/jbc.M705995200. [DOI] [PubMed] [Google Scholar]
- 35.Teghanemt A, Re F, Prohinar P, Widstrom R, Gioannini TL, Weiss JP. Novel roles in human MD-2 of phenylalanines 121 and 126 and tyrosine 131 in activation of Toll-like receptor 4 by endotoxin. J Biol Chem. 2008;283:1257–66. doi: 10.1074/jbc.M705994200. [DOI] [PubMed] [Google Scholar]
- 36.Vasl J, Prohinar P, Gioannini TL, Weiss JP, Jerala R. Functional activity of MD-2 polymorphic variant is significantly different in soluble and TLR4-bound forms: decreased endotoxin binding by G56R MD-2 and its rescue by TLR4 ectodomain. J Immunol. 2008;180:6107–15. doi: 10.4049/jimmunol.180.9.6107. [DOI] [PubMed] [Google Scholar]
- 37.Mitsuzawa H, Nishitani C, Hyakushima N, Shimizu T, Sano H, Matsushima N, Fukase K, Kuroki Y. Recombinant soluble forms of extracellular TLR4 domain and MD-2 inhibit lipopolysaccharide binding on cell surface and dampen lipopolysaccharide-induced pulmonary inflammation in mice. J Immunol. 2006;177:8133–9. doi: 10.4049/jimmunol.177.11.8133. [DOI] [PubMed] [Google Scholar]
- 38.Prohinar P, Re F, Widstrom R, Zhang DS, Teghanemt A, Weiss JP, Gioannini TL. Specific high affinity interactions of monomeric endotoxin center dot protein complexes with Toll-like receptor 4 ectodomain. J Biol Chem. 2007;282:1010–1017. doi: 10.1074/jbc.M609400200. [DOI] [PubMed] [Google Scholar]
- 39.Manček-Keber M, Jerala R. Postulates for validating TLR4 agonists. Eur J Immunol. 2015;45:356–70. doi: 10.1002/eji.201444462. [DOI] [PubMed] [Google Scholar]
- 40.Walsh C, Gangloff M, Monie T, Smyth T, Wei B, Mckinley TJ, Maskell D, Gay N, Bryant C. Elucidation of the MD-2/TLR4 Interface Required for Signaling by Lipid IVa. J Immunol. 2008;181:1245–54. doi: 10.4049/jimmunol.181.2.1245. [DOI] [PubMed] [Google Scholar]
- 41.Muroi M, Tanamoto K. Structural regions of MD-2 that determine the agonist-antagonist activity of lipid IVa. J Biol Chem. 2006 doi: 10.1074/jbc.M509193200. [DOI] [PubMed] [Google Scholar]
- 42.Derewenda U, Li J, Derewenda Z, Dauter Z, Mueller GA, Rule GS, Benjamin DC. The crystal structure of a major dust mite allergen Der p 2, and its biological implications. J Mol Biol. 2002;318:189–197. doi: 10.1016/S0022-2836(02)00027-X. [DOI] [PubMed] [Google Scholar]
- 43.Mueller GA, Benjamin DC, Rule GS. Tertiary structure of the major house dust mite allergen Der p 2: sequential and structural homologies. Biochemistry. 1998;37:12707–14. doi: 10.1021/bi980578+. [DOI] [PubMed] [Google Scholar]
- 44.Ohto U, Fukase K, Miyake K, Shimizu T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc Natl Acad Sci U S A. 2012;109:7421–6. doi: 10.1073/pnas.1201193109. [DOI] [PMC free article] [PubMed] [Google Scholar]