

Abstract
In meiotic cells, the pachytene checkpoint or meiotic recombination checkpoint is a surveillance mechanism that monitors critical processes, such as recombination and chromosome synapsis, which are essential for proper distribution of chromosomes to the meiotic progeny. Failures in these processes lead to the formation of aneuploid gametes. Meiotic recombination occurs in the context of chromatin; in fact, the histone methyltransferase Dot1 and the histone deacetylase Sir2 are known regulators of the pachytene checkpoint in Saccharomyces cerevisiae. We report here that Sas2-mediated acetylation of histone H4 at lysine 16 (H4K16ac), one of the Sir2 targets, modulates meiotic checkpoint activity in response to synaptonemal complex defects. We show that, like sir2, the H4-K16Q mutation, mimicking constitutive acetylation of H4K16, eliminates the delay in meiotic cell cycle progression imposed by the checkpoint in the synapsis-defective zip1 mutant. We also demonstrate that, like in dot1, zip1-induced phosphorylation of the Hop1 checkpoint adaptor at threonine 318 and the ensuing Mek1 activation are impaired in H4-K16 mutants. However, in contrast to sir2 and dot1, the H4-K16R and H4-K16Q mutations have only a minor effect in checkpoint activation and localization of the nucleolar Pch2 checkpoint factor in ndt80-prophase-arrested cells. We also provide evidence for a cross-talk between Dot1-dependent H3K79 methylation and H4K16ac and show that Sir2 excludes H4K16ac from the rDNA region on meiotic chromosomes. Our results reveal that proper levels of H4K16ac orchestrate this meiotic quality control mechanism and that Sir2 impinges on additional targets to fully activate the checkpoint.
Keywords: meiosis, checkpoint, histone H4K16, chromatin modifications, Sir2, Pch2, Sas2
INTRODUCTION
Meiosis is a specialized type of cell division in which a single round of DNA replication is followed by two consecutive rounds of nuclear division (meiosis I and II), allowing the generation of haploid gametes from diploid progenitor cells 1,2. In the first meiotic division the segregation of homologous chromosomes (homologs) takes place, whereas during meiosis II sister chromatids separate one from each other.
Between DNA duplication and the first meiotic division, a complex series of events involving homologous chromosomes occur during the so-called meiotic prophase; namely, genetic recombination initiated by Spo11-induced DNA double-strand breaks (DSBs) 3, alignment of parental chromosomes (pairing) and tight association of homologs (synapsis) in the context of the synaptonemal complex (SC) 1,4. The SC is a highly conserved meiosis-specific tripartite structure that assembles along the lengths of paired homologous chromosomes. It consists of a central region, in which the S. cerevisiae Zip1 protein is the major component 5,6, and two lateral elements composed of the Hop1 and Red1 proteins. Problems in the recombinational repair of meiotic DSBs as well as defects in pairing and synapsis of homologs are situations that trigger the activation of a meiosis-specific surveillance mechanism, the so-called pachytene checkpoint or meiotic recombination checkpoint, that prevents meiotic nuclear division until those crucial processes have been completed 7,8,9. In the yeast Saccharomyces cerevisiae, the activation of this evolutionarily-conserved pathway by unrepaired meiotic DSBs relies on the same sensor proteins that the canonical DNA damage checkpoint operating in vegetative growing cells, specifically the Mec1 and Tel1 kinases (the yeast homologs of mammalian DNA damage sensor kinases ATR and ATM), Rad24 and the 9-1-1 complex 10,11,12,13,14. In addition, meiosis-specific proteins, present in the chromosomal axis, such as Red1 and Hop1 15,16,17, act as adaptors sustaining the activation and hyperphosphorylation of the meiosis-specific downstream effector kinase Mek1 18,19,20,21,22,23. The delay in the exit from meiotic prophase in S. cerevisiae is imposed predominantly by controlling the expression and localization of the meiosis-specific transcription factor Ndt80, which in turn promotes the activation of the majority of genes required for late meiotic development, including B-type cyclins and the polo-like kinase Cdc5 18,24,25,26,27, as well as by inhibiting the major cyclin-dependent kinase (CDK) Cdc28 through its Swe1-dependent phosphorylation 28,29. Budding yeast meiotic mutants such as zip1, defective in SC and crossover formation that leads to the accumulation of recombination intermediates 5,30,31, are invaluable genetic tools to activate and study the pachytene checkpoint.
Meiotic recombination and the checkpoint response occur in the context of chromatin, which is subject to a wide variety of histone post-translational modifications (PTMs). These histone PTMs include acetylation, methylation, phosphorylation or ubiquitylation and exert their functions either influencing the overall structure of chromatin or regulating the binding of effector molecules. Histone PTMs have important roles in transcription, replication, repair, establishment of euchromatin/heterochromatin and other aspects of eukaryotic chromosome dynamics. Various histone PTMs have been described to be involved in crucial meiotic processes, such as recombination and the pachytene checkpoint 8,9,32. In particular, it has been proposed that H3K4 trimethylation promotes the formation of Spo11-dependent meiotic DSBs in S. cerevisiae mediated by the tethering of the Ssp1 subunit of the Set1 complex to chromosome axes 33,34,35. Nevertheless, further mechanistic studies are required to confirm this model. In addition, previous reports have also revealed the requirement of Dot1 and Sir2 for the meiotic block triggered by the pachytene checkpoint in zip1 mutants lacking a component of the SC 21,36,37. Dot1 is the methyltransferase required for H3K79 methylation (H3K79me), whereas Sir2 is a histone deacetylase that establishes and maintains silencing within yeast heterochromatic-like regions at telomeres, ribosomal DNA (rDNA) and silenced mating-type loci, and whose preferred histone substrates are H3K56ac and H4K16ac 38,39,40,41,42. However, in some cases, the precise meiotic role of those epigenetic modifications is not well known yet.
In this work we have investigated the role of the acetylation of lysine 16 in histone H4 (H4K16ac) during meiosis and its regulation by Sas2 and Sir2. We demonstrate that global acetylation of H4K16 does not change in either unperturbed or challenged meiosis and found that proper H4K16ac is dispensable during normal meiotic divisions. However, it is required for meiotic checkpoint activity, as manifested by the effect of H4-K16R and H4-K16Q mutants on suppression of the checkpoint-induced meiotic delay of zip1. These mutants show a reduction in the activity of the Mek1 meiotic effector kinase, which is most probably due to impaired Hop1 phosphorylation at threonine 318. Our results also indicate that the effect of H4-K16R and H4-K16Q mutations on the meiotic checkpoint is exerted, at least in part, through a cross-talk between H4K16ac and H3K79me. We provide cytological evidence showing that Pch2 localization is slightly altered in the H4K16ac mutants and, finally, we unveil the meiotic chromosomal distribution of H4K16ac, which is excluded from the rDNA region in a Sir2-dependent manner.
RESULTS AND DISCUSSION
Global levels of H4K16ac do not change in either normal or challenged meiosis
In budding yeast, the lysine 16 of histone H4 (hereafter H4K16) is primarily acetylated by Sas2, a member of the MYST-type family of histone acetyltransferases (HATs) 43,44,45,46,47 and secondarily by the essential HAT Esa1 48,49. In turn, at least in vitro, H4K16ac is the preferred substrate, but not the only one, of the NAD+-dependent Sir2 deacetylase 40,44,50,51,52. Importantly, disruption of SIR2 leads to H4K16 hyperacetylation exclusively in heterochromatic-like regions, such as subtelomeric sequences, the rDNA locus and the silenced mating-type loci, but does not affect genome-wide H4K16ac 53. In fact, Sir2-dependent deacetylation of H4K16ac is a characteristic feature of silenced chromatin at those particular genomic domains 54. Since Sir2 has been shown to play a crucial role in the meiotic recombination checkpoint 36, we sought to explore the possible role of H4K16ac in this process.
To study the kinetics of H4K16ac accumulation during meiosis, we performed meiotic time courses as described in Materials and Methods and followed this histone mark by immunoblotting with an anti-H4K16ac antibody. A non-acetylatable H4-K16R mutant was used as a control for antibody specificity (Figure 1). In this preliminary approach to determine variations of this histone modification, we found that global levels of H4K16ac do not significantly change upon meiosis induction (compare time 0 with the remaining times) or during the whole length of the meiotic program (Figure 1, upper panels). Next, we wanted to determine if H4K16ac was affected by the activation of the meiotic recombination checkpoint; thus, we analyzed a zip1 mutant, which triggers the checkpoint. We found that H4K16ac levels were also unaltered during the meiotic time courses in the zip1 mutant (Fig. 1, lower panels), indicating that despite the role of Sir2 in the checkpoint, global levels of H4K16ac remain fairly constant when synapsis defects exist.
Figure 1. FIGURE 1: H4K16 acetylation remains unaltered during both normal and perturbed meiosis.
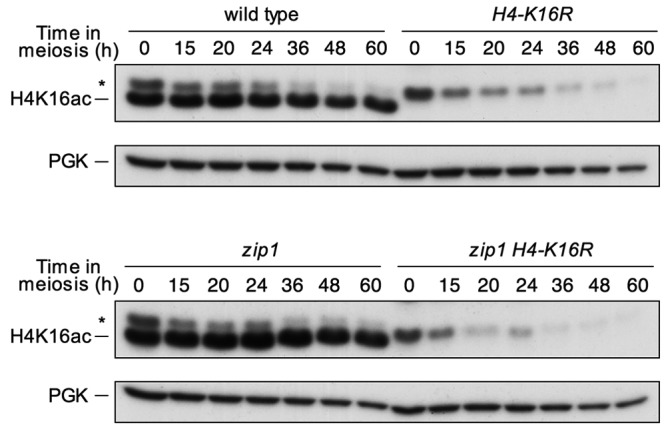
Western blot analysis of H4K16 acetylation throughout meiosis in wild-type (DP421) and zip1 (DP422) cells. The H4-K16R (DP994) and zip1 H4-K16R (DP995) mutant strains were used as controls for antibody specificity. PGK was used as a loading control. Asterisks mark a non-specific band.
Previous studies have shown that histone acetylation levels, including those of H4K16, dramatically increase during the induction of an HO-induced DSB lesion and decrease during the subsequent homologous recombinational repair, presumably due to the coordinated action of histone modifying enzymes, such as Esa1 and Sir2, that are recruited to the lesion 55. This ability to modify the levels of histone acetylation is essential to maintain cell viability after exposure to DNA damaging agents or during DNA repair by homologous recombination, either because changes in histone acetylation are necessary for the recruitment of DNA repair enzymes and/or chromatin remodelers, or because they are important in downstream signaling. In fact, different H3 and H4 lysines are found acetylated upon DNA damage in yeast 56,57. Meiosis involves the generation and subsequent repair of multiple DSBs across the genome and signal transduction in the meiotic checkpoint pathway shares many components with the mitotic DNA damage checkpoint 8. However, in this study, we show that global levels of H4K16ac do not change either with the induction of the meiotic program or when meiotic chromosome synapsis defects exist (Figure 1). Nevertheless, the precise meiotic errors (incomplete recombination, chromosome structural defects or both) triggering the checkpoint in the zip1 mutant remain to be established. In addition, in contrast to the situation in mitotic cells, meiotic DSB repair occurs in the special context of the SC with probably different chromatin modifications requirements. Moreover, in our study we have measured global levels of H4K16 acetylation and we cannot rule out the possibility that local modifications of H4K16 acetylation may occur at particular genomic regions.
H4K16 normal acetylation is required for efficient meiotic checkpoint regulation
To further investigate the role of H4K16ac in meiosis, several meiotic events were analyzed in H4-K16R (non-acetylatable) and H4-K16Q (mimicking constitutive acetylation) mutants, both in a wild-type (unperturbed meiosis) and a zip1 background (triggering meiotic checkpoint activation). The kinetics of meiotic nuclear divisions was monitored by DAPI staining of nuclei. Dityrosine fluorescence, a specific component of mature spores, was used as a semi-quantitative indicator for sporulation efficiency. Finally, spore viability that reflects the fidelity of meiotic chromosome segregation and the integrity of the spore genome was determined by tetrad dissection. In an otherwise wild-type background, the H4-K16R and H4-K16Q single mutants showed no or little meiotic defects (Figure 2). The progression through meiosis was normal (Figure 2B, S1A) and resulted in the formation of mature dityrosine-containing spores (Figure 2A) with a high viability similar to that of the wild type (Figure 2C). These observations suggest that normal regulation of H4K16ac is dispensable in unperturbed meiosis.
Figure 2. FIGURE 2: The meiotic recombination checkpoint is impaired in H4-K16R and H4-K16Q mutants.
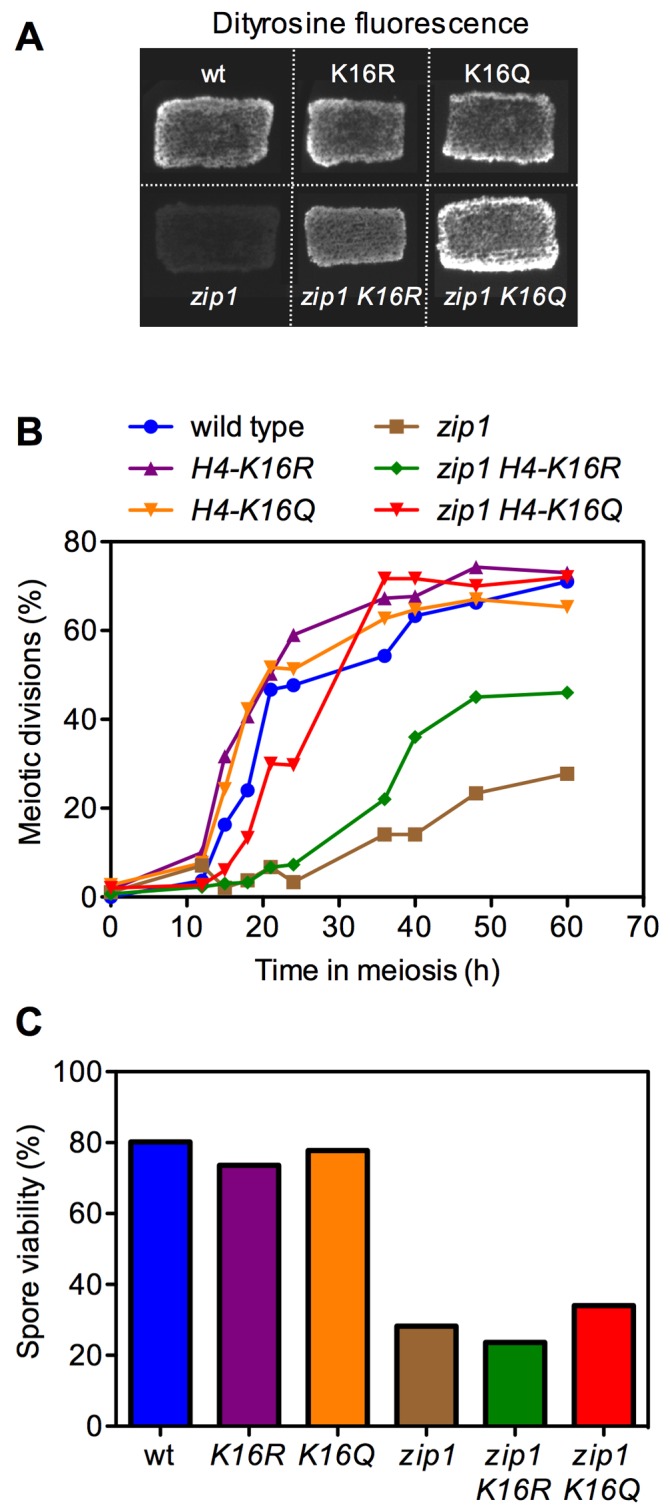
(A) Dityrosine fluorescence, as an indicator of sporulation, was examined after 3 days of sporulation on plates.
(B) Time course of meiotic nuclear divisions; the percentage of cells containing two or more nuclei is represented.
(C) Spore viability, as assessed by asci dissection, is presented. At least 144 spores were scored for each strain. Strains used in (A) are DP421 (wild type), DP994 (H4-K16R), DP1000 (H4-K16Q), DP422 (zip1), DP995 (zip1 H4-K16R) and DP1001 (zip1 H4-K16Q). Strains used in (B) and (C) are DP634 (wild type), DP635 (H4-K16R), DP636 (H4-K16Q), DP639 (zip1), DP640 (zip1 H4-K16R) and DP641 (zip1 H4-K16Q).
As previously described, the zip1 mutant, where the pachytene checkpoint is triggered, showed a strong delay in meiotic progression and the formation of mature spores was dramatically reduced (Figure 2A, 2B, S1A). Notably, the H4-K16R and H4-K16Q mutations were able to partially (K16R) or completely (K16Q) alleviate the checkpoint-dependent meiotic block: the zip1 H4-K16Q and zip1 H4-K16R double mutants progressed faster into meiosis (Figure 2B, S1A) and formed dityrosine-containing spores in a higher proportion than zip1 cells (Figure 2A); however, spore viability remained low (Figure 2C) indicating that although zip1 H4-K16Q and zip1 H4-K16R cells were able to progress into meiosis and to form mature spores, the problems caused by the lack of Zip1 persist. Thus, the status of H4K16ac modulates meiotic progression in the zip1 mutant. Interestingly, the H4-K16Q mutant mimicking constitutive acetylation shows a stronger checkpoint defect, similar to the lack of the Sir2 deacetylase 36 (see below).
H4-K16R and H4-K16Q mutants are defective in the maintenance, but not the establishment, of checkpoint-induced Mek1 activation
To investigate the meiotic checkpoint role of H4K16ac more directly at a molecular level, we followed the status of Mek1 activation throughout meiotic time courses in the zip1 H4-K16R and zip1 H4-K16Q mutants using high-resolution Phos-tag gels. The appearance of hyper-phosphorylated Mek1 isoforms is indicative of meiotic checkpoint activation 21. The threonine 11 of histone 3 has been identified as one of Mek1 downstream targets 58. Although the role of H3T11ph in meiosis, if any, is still unclear, it is a useful additional reporter for Mek1 kinase activity (Figure 3) 59. In wild-type cells, Mek1 levels rose transiently during meiotic prophase (peak at 20 hours) and then progressively declined as meiosis I and II and sporulation took place. Phosphorylated forms of Mek1 and H3T11ph remained at very low levels during the whole meiotic time course (Figure 3, upper panel). In contrast, robust Mek1 activation, as shown by the appearance of additional slow migrating and stronger phosphorylated Mek1 forms, and marked H3T11ph could be detected in the zip1 mutant (Figure 3, second panel), consistent with its pronounced meiotic delay triggered by the checkpoint (Figure 2B). We next examined the zip1 H4-K16R and zip1 H4-K16Q double mutants. Remarkably, according with the complete suppression of the meiotic delay (Figure 2B), Mek1 activation was severely impaired in the zip1 H4-K16Q double mutant, as manifested by the absence of the upper Mek1 phosphorylated forms and low levels of H3T11ph (Figure 3, third panel). The zip1 H4-K16R, which shows only a partial checkpoint defect (Figure 2B), showed a milder reduction in both the levels and the duration of Mek1 activation and H3T11ph (Figure 3, bottom panels).
Figure 3. FIGURE 3: H4K16 acetylation is necessary for normal Mek1 and Hop1 phosphorylation.
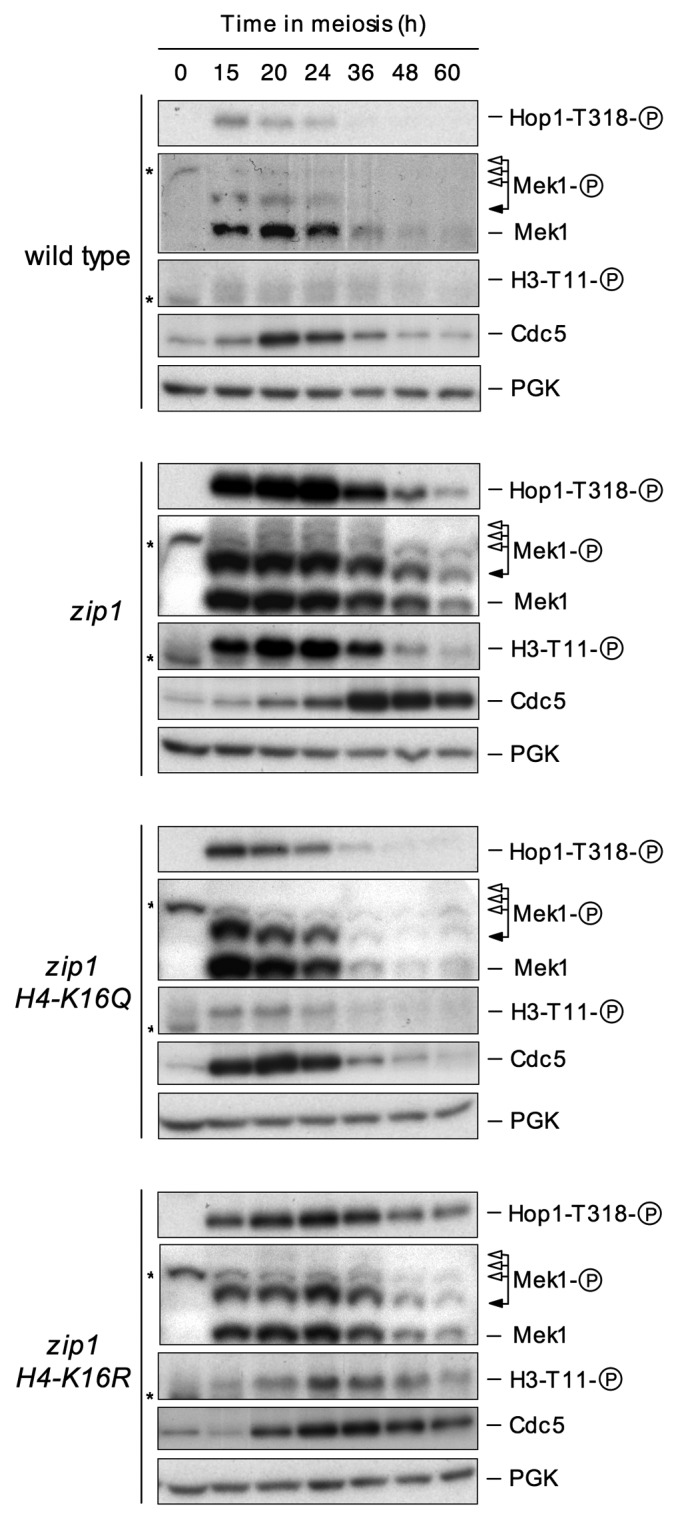
Western blot analysis of Mek1 and Hop1 activation in wild type (DP421), zip1 (DP422), zip1 H4-K16Q (DP1001) and zip1 H4-K16R (DP995) strains throughout meiosis. Black arrows point the Mec1/Tel1-dependent phosphorylated form of Mek1, whereas white arrows mark the bands resulting from Mek1 autophosphorylation 21. Asterisks mark non-specific bands. H3T11 phosphorylation and Cdc5 inhibition were used as additional molecular markers for checkpoint activation. PGK was used as a loading control.
Mec1/Tel1-dependent phosphorylation of Hop1 at defined S/T-Q sites is required for Mek1 hyperphosphorylation and activation, as well as for meiotic checkpoint activity 15. Among the several S/T-Q sites targeted by Mec1/Tel1 in Hop1, phosphorylation of threonine 318 together with phosphorylation of serine 298 are crucial events in the meiotic checkpoint network to coordinate recombination and meiotic progression 60. We examined the levels of Hop1-T318 phosphorylation throughout the meiotic time courses using a phospho-specific antibody as an upstream marker for zip1-induced checkpoint activation 59. During normal meiosis, only a very weak and transient Hop1-T318ph signal could be detected during the meiotic prophase, coinciding with the weak activation observed in Mek1 (Figure 3, upper panel). However, in zip1 mutant cells triggering the activation of the pachytene checkpoint, Hop1-T318ph dramatically increased (Figure 3, second panel). We next analyzed the zip1 H4-K16R and zip1 H4-K16Q double mutants and we found a reduction in Hop1-T318 phosphorylation, very similar to that observed in Mek1 activity (Figure 3, third and bottom panels). Again, the effect of H4-K16Q was much stronger.
To further support the results shown above, we also analyzed a downstream target of the meiotic recombination checkpoint, the Cdc5 polo-like kinase. Cdc5 is one of the most prominent members of a large set of genes under the control of the meiosis-specific Ndt80 transcription factor, with a number of functions in meiosis including the exit from pachytene and entry into the first meiotic division 18,24,61,62,63,64. In wild-type cells, low levels of Cdc5 were detected in vegetative cell cycle, prior to entering meiosis; those levels peaked during mid-meiosis and then declined. Meanwhile, in a zip1 mutant the production of Cdc5 was clearly delayed (Figure 3, top and second panels), according with the slower meiotic progression (Figure 2B). In contrast, earlier induction of Cdc5 production was completely or partially restored in the zip1 H4-K16Q and zip1 H4-K16R double mutants, respectively (Figure 3, third and bottom panels), which is again consistent with the meiotic progression of these mutants.
All together, these results confirm the effect of H4-K16Q and H4-K16R mutations in meiotic progression and indicate that the checkpoint defects observed most probably arise from the failure to efficiently phosphorylate Hop1 and Mek1. Thus, H4K16ac is required for both Hop1 phosphorylation and the ensuing Mek1 activation in the meiotic recombination checkpoint pathway.
Interestingly, the substitution of the lysine 16 of histone 4 with differently charged residues resulted in slightly different outcomes. Similar to the lack of the Sir2 deacetylase 36 the H4-K16Q substitution, mimicking the constitutively acetylated state of lysine, completely abolished the meiotic block imposed by ZIP1 disruption, as well as the phosphorylation of Mek1 and Hop1. Conversely, substitution of the lysine by arginine, a residue that cannot be acetylated, H4-K16R, only showed a partial effect on the meiotic progression as well as in the Hop1 and Mek1 phosphorylation (Figures 2 and 3). Curiously, similar consequences have been observed regarding the impact of H4K16ac mutants on other biological processes. For example, the H4-K16Q substitution significantly reduces lifespan whereas H4-K16R shows only a marginal effect 65. Likewise, the frequency of chromosome loss and the levels of rDNA recombination are also higher in H4-K16Q strains than in H4-K16R mutants 66,67. In line with these observations, our results raise the possibility that the dynamics of H4K16ac, more than only the exact state of such acetylation, is required to regulate the meiotic checkpoint, although the precise mechanism underlying such effect remains to be elucidated.
Figure 4. FIGURE 4: Analysis of Mek1 activation and localization in ndt80-arrested cells.
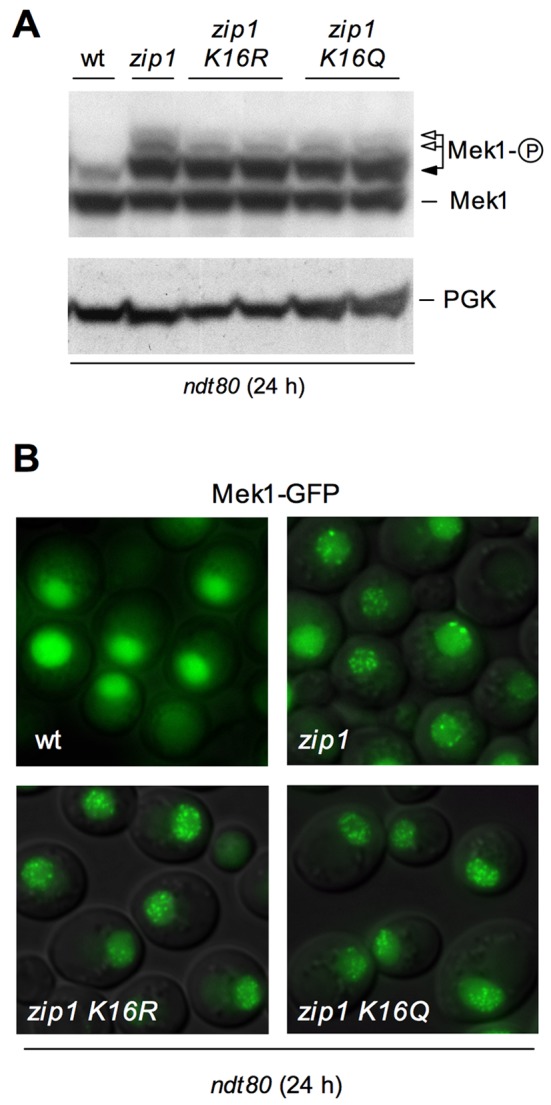
(A) Western blot analysis of different Mek1 phosphorylation forms in ndt80-arrested cells after 24 h in meiosis. PGK is shown as a loading control. Strains are DP424 (wild type), DP428 (zip1), DP996 (zip1 H4-K16R) and DP1002 (zip1 H4-K16Q). Two independent clones of DP966 and DP1002 were analyzed.
(B) Representative images of checkpoint-induced Mek1-GFP foci in wild type (DP584), zip1 (DP582), zip1 H4-K16R (DP1089) and zip1 H4-K16Q (DP1090) ndt80-arrested cells after 24 h in meiosis.
In principle, the differences observed in Mek1 phosphorylation between zip1 H4-K16R and zip1 H4-K16Q double mutants and the zip1 single mutant could be a consequence of their different kinetics in meiotic progression (zip1 exhibits a profound delay that is bypassed in zip1 H4-K16R and zip1 H4-K16Q; Figure 2B) or could arise from a direct effect of H4K16 acetylation on Mek1 activation. To distinguish between these two possibilities, we monitored Mek1 phosphorylation in pachytene-arrested ndt80 cells. Ndt80 is a meiosis-specific transcription factor required for induction of meiotic middle genes 25,68,69 promoting exit from prophase 70; thus, ndt80 cells arrest in pachytene independently of the meiotic checkpoint allowing us to analyze the status of checkpoint activation independent of meiotic progression. If H4K16 acetylation were not involved in the establishment of checkpoint-induced Mek1 activation but only in its maintenance, we will expect Mek1 phosphorylation to be similar in zip1 and in zip1 H4K16 acetylation mutants, in a ndt80 background. As shown in Figure 4A, in an ndt80 background, zip1 H4-K16R and zip1 H4-K16Q double mutants are only slightly impaired in Mek1 activation. Previous studies have shown that zip1-induced checkpoint activation results in different Mek1 phosphorylated forms 21. In Figure 4A we can observe that H4-K16R and H4-K16Q mutants slightly affected only the upper phosphorylated bands, corresponding to Mek1 autophosphorylation (Figure 4A; white arrows), while the band immediately above the basal form, which depends on Mec1/Tel1 21, remained intact (Figure 4A; black arrow). Moreover, when we analyzed the phosphorylation of H3T11 and Hop1-T318 as additional markers of checkpoint activation in ndt80 cells, we observed little if any reduction in their phosphorylation levels in the zip1 H4-K16R and zip1 H4-K16Q mutants when compared to zip1 (Figure 5). This is in clear contrast with the results of a zip1 dot1 double mutant in which H3T11ph and Hop1-T318ph were practically abolished (Figure 5), consistent with Dot1 being absolutely required both for checkpoint activation and maintenance 21. These results suggest that the correct acetylation of H4K16 is not required for the establishment of checkpoint-induced Mek1 and Hop1 phosphorylation, but more probably only for its maintenance. If the meiotic prophase block is artificially imposed by means of the ndt80 mutation, then H4K16ac becomes dispensable to sustain Hop1 and Mek1 activation.
Figure 5. FIGURE 5: The sir2 mutant, but not H4-K16Q, H4-K16R or sas2, is defective in establishing early markers of checkpoint activation.
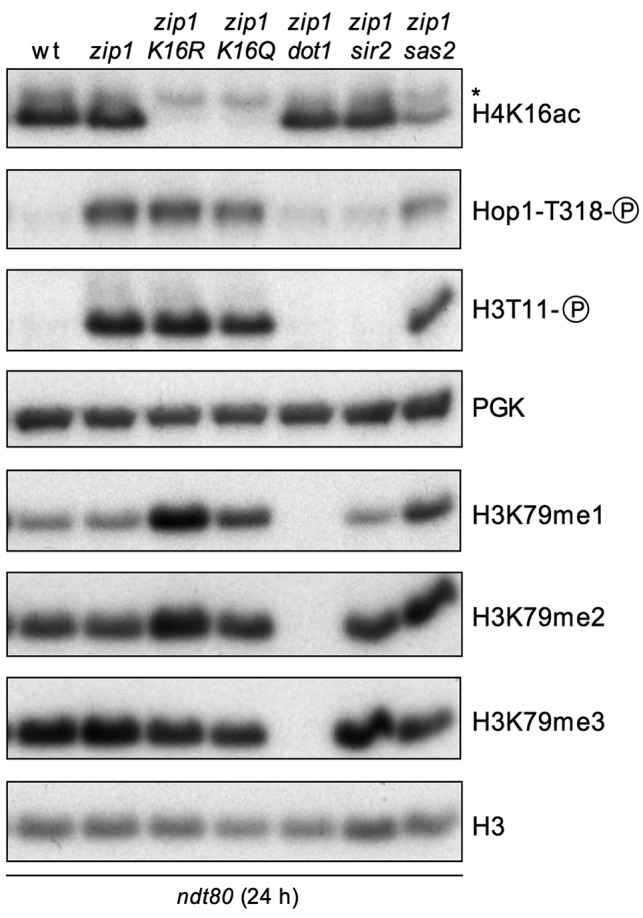
Western blot analysis of zip1-induced Hop1-T318 and H3-T11 phosphorylation, as well as H3K79 methylation, 24 h after meiosis induction in ndt80-arrested cells. PGK and total H3 were used as loading controls. Strains are: DP424 (wild type), DP428 (zip1), DP996 (zip1 H4-K16R), DP1002 (zip1 H4-K16Q), DP655 (zip1 dot1), DP1086 (zip1 sir2) and DP1073 (zip1 sas2).
It has been previously demonstrated that, upon meiotic checkpoint activation, the Mek1 effector kinase localizes to discrete nuclear foci that can be detected both on chromosome spreads and in live meiotic cells 12,21. To investigate in more detail the role of H4K16ac in the meiotic checkpoint, we assessed the localization of Mek1-GFP in wild-type, zip1, zip1 H4-K16R and zip1 H4-K16Q cells, always in an ndt80 background. As expected, zip1 mutant cells accumulated multiple discrete Mek1-GFP foci during meiotic prophase (Figure 4B) and most zip1 H4-K16R and zip1 H4-K16Q cells displayed a similar pattern of Mek1 localization (Figure 4B), indicating that formation of zip1-induced Mek1 foci is not defective in the absence of normal H4K16ac. This observation suggests that, although H4K16ac is required for sustained meiotic checkpoint activity, it is not necessary for the checkpoint-induced association of Mek1 to meiotic chromosomes.
The Sir2 and Sas2 proteins are required for proper meiotic checkpoint response
To further investigate the role of H4K16ac in the meiotic recombination checkpoint we studied mutants affecting the metabolism of this residue, such as sir2 (deficient in a H4K16ac deacetylase), and sas2 (lacking the main H4K16 acetyltransferase). The relationship of Sir2 with the meiotic checkpoint has been previously reported 36, but a detailed analysis of meiotic progression and checkpoint activity was not described.
We found that deletion of SIR2 completely suppressed the meiotic delay imposed by the checkpoint in the zip1 mutant; that is, the zip1 sir2 double mutant showed similar kinetics of meiotic progression than the wild type (Figure 6A, S1B) and displayed high levels of sporulation (Figure 6B). Hop1T318 phosphorylation and H3T11 phosphorylation (as a marker of Mek1 activity) were drastically reduced in zip1 sir2 compared to zip1 and, according with the meiotic progression, Cdc5 production was restored to wild-type kinetics in zip1 sir2 (Figure 6C). Disruption of SAS2 also alleviated the zip1 meiotic block, but to a lesser extent than zip1 sir2 did (Figure 6A, 6B, S1B). Consistent with this intermediate effect, Hop1T318 and H3T11 phosphorylation showed a moderate reduction, and Cdc5 dynamics was only partially restored in zip1 sas2 (Figure 6C). Thus, in NDT80+ cells competent for meiotic progression, the checkpoint phenotype resulting from the lack of the Sir2 deacetylase is similar to that produced by the H4-K16Q mutation mimicking constitutive acetylation, and the effect produced by the absence of the Sas2 acetyltransferase parallels that of the H4-K16R mutation preventing acetylation of this residue (Figures 2 and 3).
Figure 6. FIGURE 6: The meiotic recombination checkpoint response is impaired in the absence of Sir2 or Sas2.

(A) Time course of meiotic nuclear divisions; the percentage of cells containing two or more nuclei is represented.
(B) Dityrosine fluorescence, as a visual indicator of sporulation, and sporulation efficiency, quantified by microscopic examination of at least 300 cells, were examined after 3 days of sporulation on plates.
(C) Western blot analysis of the indicated proteins during meiosis. Strains are DP421 (wild type), DP422 (zip1), DP1401 (zip1 sir2) and DP1410 (zip1 sas2).
The checkpoint impact of SIR2 and SAS2 deletions was also analyzed in ndt80 mutant cells by monitoring the levels of zip1-induced Hop1T318 and H3T11 phosphorylation. In the case of sir2, we found a complete abrogation of both phosphorylation events (Figure 5), indicating that, in contrast to H4-K16Q, the sir2 mutant is defective both in the establishment and maintenance of the meiotic checkpoint, in a similar way to dot1. The fact that the lack of the H4K16ac Sir2 deacetylase does not cause exactly the same effect as the mimicked constitutive acetylation of the H4-K16Q mutant in ndt80 strains suggests that Sir2 has additional checkpoint functions. On the other hand, in ndt80 cells, SAS2 disruption only showed a marginal effect on both H3T11 and Hop1 phosphorylation, similar to what we observed with the acetylation-defective H4-K16R mutant (Figure 5), indicating that Sas2 is primarily involved in checkpoint maintenance.
We also monitored the state of H4K16ac and, as we showed above (Figure 1), it was also unaffected when the checkpoint was triggered by zip1 in ndt80-arrested cells (Figure 5). Strikingly, we found that the disruption of SIR2 did not significantly increase global levels of H4K16ac in either NDT80 or ndt80 cells (Figures 5 and 6C), consistent with the notion that Sir2 is not the main genome-wide H4K16ac deacetylase and its action may be specifically restricted to precise heterochromatic domains 53,54. On the other hand, SAS2 deletion clearly, but not completely, reduced H4K16ac (Figures 5 and 6C), suggesting that Sas2 is the main, but not the only, H4K16 acetyltransferase acting in the meiotic cell cycle.
Cross-talk between H4K16 acetylation and H3K79 methylation
Previous studies have shown that some histone PTMs positively or negatively affect other histone marks in what has been described as histone cross-talk, adding an extra layer of complexity to the control of different chromatin processes 71,72,73. One example is the tri-methylation of H3K79 by Dot1, which is completely dependent upon the prior ubiquitylation of H2BK123 by Rad6/Bre1 74. It has also been described that H4K16ac modulates Dot1-dependent H3K79 methylation by promoting Dot1 binding to a short basic patch in the histone H4 tail in competition with Sir3 75. Since Dot1-dependent H3K79 methylation is required for the meiotic recombination checkpoint 21,37 it was possible that the impact of H4K16ac on the checkpoint (Figures 2 and 3) was exerted via regulation of H3K79me. To explore this possibility, we first analyzed the effect of H4-K16R and H4-K16Q mutations on H3K79 mono-, di- and tri-methylation in zip1 ndt80 checkpoint-activated and pachytene-arrested cells (Figures 5 and 7A, 7B). Given the distributive mode of action of the Dot1 methyltransferase 76, an impaired Dot1 catalytic activity is manifested as a reduction in H3K79me3 concomitant with an increase in H3K79me1 and H3K79me2 21,76. Indeed, we observed higher levels of H3K79me1 and H3K79me2 in both H4K16ac-defective mutants, as well as a reduction in those of H3K79me3 (Figures 5 and 7A, 7B), which is the most relevant form to sustain the meiotic checkpoint response 21. Thus, these findings suggest that H4K16ac mutants affect the activity of Dot1. We also observed that, like H4-K16R, the absence of the H4K16 acetyltransferase Sas2 also increased H3K79me1 and H3K79me2 and reduced H3K79me3 (Figure 5). Curiously, disruption of SIR2, did not have any effect on global H3K79me levels (Figure 5), again consistent with the notion that Sir2 meiotic checkpoint function can be exerted, at least in part, in a way that is independent from a global activity on H4K16ac.
Since Dot1 catalytic activity appears to be compromised in H4K16ac mutants, we explored whether DOT1 overexpression would restore normal H3K79me3 levels and meiotic checkpoint function in zip1 H4-K16R or zip1 H4-K16Q double mutants. DOT1 was overexpressed from a high-copy plasmid (Figure S2) and the pattern of H3K79me was analyzed at 0 h and 20 h after meiotic induction (Figure 7A, 7B). We found that the increased H3K79me1 and H3K79me2 levels observed in the zip1 H4-K16R and zip1 H4-K16Q mutants were reduced upon DOT1 overexpression (Figure 7A, 7B). On the contrary, high doses of Dot1 increased the amount of H3K79me3 in zip1 H4-K16R and zip1 H4-K16Q, although it did not reach normal wild-type levels (Figure 7A, 7B). These observations confirm that overexpression of DOT1 can partially compensate for the crippled Dot1 methyltransferase activity when H4K16ac metabolism is altered; therefore, we analyzed the effect on the meiotic checkpoint by monitoring the kinetics of meiotic divisions.
Figure 7. FIGURE 7: DOT1 overexpression partially restores the meiotic checkpoint in H4K16ac-deficient mutants.

(A) Western blot analysis of H3K79 methylation species in vegetative (T=0h) and meiotic cells (T=20h). Total H3 and PGK were used as loading controls.
(B) Quantification of relative levels of the H3K79 methylation forms at T=0 from the blots shown in (A). Total H3 signal was used for normalization.
(C) Time course of meiotic nuclear divisions; the percentage of cells with two or more nuclei is presented. Strains are DP422 (zip1), DP995 (zip1 H4-K16R) and DP1001 (zip1 H4-K16Q), transformed either with an empty vector or with the high-copy pSS63 DOT1 overexpression plasmid (DOT1-OE).
We have shown before that H4-K16R releases the checkpoint-dependent zip1 meiotic delay to some extent and H4-K16Q completely alleviates the zip1 block (Figure 2B). Interestingly, DOT1 overexpression resulted in less efficient meiotic progression in zip1 H4-K16R and zip1 H4-K16Q cells compared to the controls transformed with empty vector (Figure 7C, S1C), consistent with a partial restoration of the checkpoint. Altogether, these results suggest that the effect of H4-K16R and H4-K16Q mutations on the meiotic checkpoint triggered by a zip1 mutant is exerted, at least in part, through their effect on modulating proper H3K79 methylation pattern.
Relationship between Sir2, H4K16ac and Pch2 nucleolar localization
In Saccharomyces cerevisiae, the Pch2 protein is a negative regulator of Hop1 chromosomal abundance in synapsed chromosomes 77,78, but it is required for the zip1-induced meiotic checkpoint promoting Hop1 phosphorylation at T318 36,59. The majority of Pch2 localizes to the unsynapsed nucleolar region of chromosome XII that contains the ribosomal RNA genes (rDNA), where it is required to exclude the meiosis-specific Hop1 protein from the nucleolus. This nucleolar localization of Pch2 is completely dependent on the Sir2 deacetylase, which is also located in the rDNA 36, and deletion of SIR2 impairs the meiotic checkpoint (Figures 5 and 6). Moreover, the Dot1 meiotic checkpoint factor regulates both Sir2 and Pch2 nucleolar localization 21,37. This scenario points to a pivotal role for the nucleolar Pch2 in the pachytene checkpoint 59 and prompted us to investigate if H4-K16R and/or H4-K16Q mutations affected the nucleolar localization of Pch2 on meiotic chromosome spreads.
In zip1 cells, when the meiotic checkpoint is activated, Pch2 localization is limited to the nucleolar (rDNA) region (Figure 8). As it has been previously shown 36, in zip1 sir2 cells the nucleolar concentration of Pch2 was lost and the protein appeared in form of foci dispersed throughout the meiotic chromosomes (Figure 8). Then, we analyzed Pch2 distribution in the zip1 H4-K16Q and zip1 H4-K16R mutants. We found that the Pch2 signal was still located in a restricted chromosomal area, presumably the rDNA region, but it was somehow more diffused although to a lesser extent than in zip1 sir2 (Figure 8). Thus, like Dot1 and Sir2, these findings point to a role for H4K16ac in delimiting the nucleolar confinement of Pch2 and its exclusion from the rest of the chromatin, although the action of Sir2 must not be exerted only on H4K16ac because the effect of SIR2 deletion on Pch2 localization is stronger than that of H4K16 mutations.
Figure 8. FIGURE 8: Analysis of Pch2 localization in H4K16ac-deficient mutants.

Immunofluorescence of meiotic chromosome spreads from zip1 (DP1123), zip1 sir2 (DP1124), zip1 H4-K16R (DP1121) and zip1 H4-K16Q (DP1139) stained with DAPI (blue) as well as with anti-HA to detect Pch2-HA (red) and anti-H4K16ac (green) antibodies. The arrows point to the rDNA region where Pch2 accumulates and is devoid of H4K16ac. Representative nuclei are shown. Spreads were prepared after 24 h of meiotic induction in ndt80 strains.
In addition, we also examined the distribution of H4K16ac on meiotic chromosomes. We used an antibody that recognizes the nucleolar Nsr1 protein involved in ribosome biogenesis 79 to unambiguously identify the rDNA region, which often appears as a chromatin loop on preparations of spread meiotic chromosomes. Strikingly, the H4K16ac histone mark was completely excluded from the rDNA in both wild-type and zip1 nuclei (Figure 9), also displaying an exclusive localization pattern with that of nucleolar Pch2 (Figure 8, arrow). However, in the absence of Sir2, H4K16ac was distributed all along the chromatin, showing a complete co-localization with the DAPI staining, including the rDNA region marked by the nucleolar Nsr1 protein (Figure 9). Consistent with microarray studies in vegetative cells 53 and with our western blot analysis of global meiotic levels of H4K16ac (Figures 5 and 6), the H4K16ac signal on the bulk of the genome was not significantly altered in sir2 mutants (Figure 9). These results indicate that Sir2 is the major deacetylase specifically responsible for preventing H4K16 acetylation in the rDNA during meiosis.
Figure 9. FIGURE 9: Sir2 excludes H4K16ac from the rDNA region.

Immunofluorescence of meiotic chromosome spreads from wild type (BR2495), sir2 (DP262), zip1 (DP1123) and zip1 sir2 (DP1124), stained with DAPI (red) as well as with anti-H4K16ac (green) and anti-Nsr1 (blue) antibodies. The arrows point to the rDNA region identified by Nsr1 staining. Representative nuclei are shown. Spreads were prepared after 16 h of meiotic induction for BR2495 and DP262 and 24 h for DP1123 and DP1124.
Besides the impact on the meiotic recombination checkpoint, it has been shown that SIR2 disruption significantly alters the genomic distribution of Spo11-induced DSBs; with some genes displaying increased levels of DSBs whereas others experience reduced levels of DSBs in the absence of Sir2 80. Two defined genomic domains, such as subtelomeric regions and the rDNA array, show elevated recombination in the sir2 mutant 80. Pch2 also prevents recombination at the rDNA by excluding Hop1 from the nucleolar region 36,59. Moreover, Sir2 and Pch2 modulate the protection of DSB-induced meiotic instability at the rDNA borders 81. It has been proposed that the effect of Sir2 on recombination at subtelomeric regions is exerted through the regulation of H4K16ac; however, the heterogeneous effect of Sir2 on the global meiotic DSB landscape implies that multiple factors and targets must be involved in addition to H4K16ac 80.
Concluding remarks
In this work we have explored the functional contribution of H4K16ac, the Sir2 deacetylase and the Sas2 acetyltransferase in the meiotic recombination checkpoint triggered by synaptonemal complex defects. In line with previous observations, our results indicate that an intricate network of histone PTMs fine-tune this meiotic quality control mechanism (Figure 10). We propose that reduced levels of Dot1-mediated H3K79me3 at the rDNA enable the enrichment of Sir2 in the nucleolus. The presence of Sir2 at the rDNA region is responsible for the low level of H4K16ac in this area and, together with an additional unknown Sir2 target, confines Pch2 in the nucleolus. The Pch2 ATPase is critical to orchestrate the proper balance between the amount of Hop1 bound to chromosome axes and phosphorylated Hop1, which in turn sustains Mek1 activation 21,59. Nevertheless, the precise mechanism by which nucleolar Pch2 regulates the phosphorylation status of the Hop1 checkpoint adaptor located at the axes and excluded from the rDNA remains to be determined.
Figure 10. FIGURE 10: A model for the regulation of the meiotic checkpoint by histone post-translational modifications. See text for details.
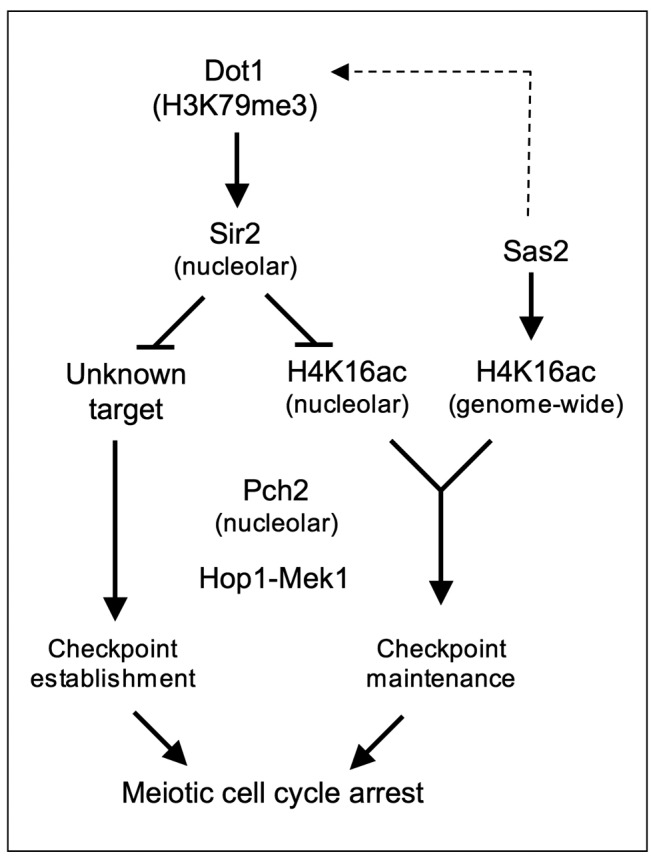
Curiously, when meiotic progression is prevented by the ndt80 mutation, we have observed different checkpoint activity phenotypes resulting from deletion of SIR2 compared with H4-K16 or sas2 mutants. Whereas Sir2 is required for Mek1 activation in any condition, Sas2/H4K16ac only affect the maintenance of Mek1 activation in NDT80-proficient cells, thus supporting the notion that Sir2 acts on additional targets.
We hypothesize that the general status of H4K16ac modulates DNA repair pathways involved in the resolution of recombination intermediates accumulated in zip1 triggering the checkpoint arrest. Alteration of H4K16ac dynamics by SAS2 deletion or H4-K16 mutations, would allow the Ndt80-dependent repair of those intermediates thus allowing meiotic progression in zip1. Further experimental work will be required to explore this possibility.
Materials and Methods
Yeast strains
Yeast strains genotypes are listed in Table S1. All the strains are in the BR1919 or BR2495 genetic background 82. Gene deletion and tagging were performed using a PCR-based approach or by genetic crosses always in an isogenic background. The dot1::URA3, zip1::LYS2, zip1::LEU2, sir2::URA3 and ndt80::LEU2 deletions were previously described 5,26,31,37,83. In the plasmid-borne H4-K16R and H4-K16Q mutants, both genomic copies of the histone H3-H4 encoding genes (HHT1-HHF1 and HHT2-HHF2) were deleted and the wild-type HHT2-HHF2 genes or the modified HHT2-hhf2(K16R) or HHT2-hhf2(K16Q) versions were expressed from the centromeric plasmids pRM204, pWD23 and pWD25, respectively, as the only source of H3-H4 histones 65. Alternatively, both copies of the histone H4-encoding genes HHF1 and HHF2 were mutated in their genomic loci to K16R and K16Q following the delitto perfetto approach 84. N-terminal tagging of Pch2 with three copies of the HA epitope and the MEK1-GFP construct were previously described 21,36. DOT1-HA was overexpressed from the pSS63 plasmid 37.
Meiotic time courses
Strains were grown on 2xSC (3,5 ml) for 20-24 h and then transferred to 2,5 ml of YPDA where they were incubated to saturation for an additional 8 h. Cells were then harvested, washed with 2% potassium acetate (KAc), resuspended into 10 ml of KAc and incubated at 30°C with vigorous shaking (235 rpm) to induce meiosis and sporulation. 20 mM adenine and 10 mM uracil was added to both YPDA and KAc media. Culture volumes were scaled up when needed. Aliquots of cells were removed at different time points for analysis. To analyze meiotic divisions, cells were fixed in 70% ethanol, washed in phosphate-buffered saline (PBS) and stained with 1 mg/ml DAPI for 15 min at room temperature. Nuclei were observed by fluorescence microscopy and at least 300 cells were scored for each strain at each time point in every experiment. Meiotic kinetics experiments were repeated several times and representative experiments are shown. Dityrosine fluorescence was analyzed as previously described 37 and spore viability was determined by tetrad dissection.
Western blotting
TCA yeast whole cell extracts from 5-10 ml aliquots of meiotic cultures were prepared as described previously 18 and proteins were resolved by SDS-PAGE and then transferred to PVDF membranes. To resolve the phosphorylated forms of Mek1, 10% SDS-PAGE gels with a 29:1 ratio of acrylamide:bisacrylamide containing 37,5 μM Phos-tag reagent (Wako) and 75 μM MnCl2 were prepared as described 18,21, whereas normal 15% or 10% gels (acrylamide:bisacrylamide 37,5:1) were used for detection of H4K16ac, H3T11ph and H3K79me or Mek1, Hop1-T318ph, Cdc5 and Dot1-HA, respectively. Blots were probed with the following primary antibodies: rabbit polyclonal antibodies raised against Mek1 (1:1000) 13, Hop1-T318 (1:1000; kindly provided by J. Carballo), H3T11ph (1:2000; Abcam 5168), H4K16ac (1:2000; Millipore 07-329), H3K79-me1 (1:1000; Abcam ab2886), H3K79-me2 (1:2000; Abcam ab3594) and H3K79-me3 (1:2000; Abcam ab2621); goat polyclonal antibody against Cdc5 (1:1000; Santa Cruz Biotechnology sc-6733); mouse monoclonal antibody against the HA epitope (1:2000; Roche 12CA5). A mouse monoclonal antibody directed against 3-phosphoglycerate kinase (PGK) (1:10000; Molecular Probes A-6457) or a rabbit polyclonal antibody against histone H3 (1:5000; Abcam ab1791) were used as loading controls. HRP-conjugated secondary antibodies were from GE Healthcare (NA934 and NA931) or Santa Cruz Biotechnology (sc-2033). The Pierce ECL or ECL-2 reagents (Thermo Scientific) were used for detection and the signal was captured on film (Amersham Hyperfilm ECL; GE Healthcare) and/or with a ChemiDoc XRS (BioRad) system, using the Quantity One software (Bio-Rad). The same software was used to quantify protein levels.
Cytology
Whole cell images were captured with a Nikon Eclipse 90i fluorescence microscope controlled with the MetaMorph software (Molecular Devices) and equipped with an Orca-AG (Hamamatsu) CCD camera and a PlanApo VC 100X 1.4 NA objective. To analyze Mek1-GFP foci in live meiotic cells, exposure time was 1 second and stacks of 11 planes at 0,4 μm were captured. Maximum intensity projections were generated with the NIH ImageJ software (http://rsb.info.nih.gov/ij ). To outline the contour of the cells in the representative whole-cell images presented, an overlay of the DIC image with 15-20% transparency over the GFP signal is shown. Immunofluorescence of meiotic chromosome spreads was performed as previously described 36. To detect the HA-tagged Pch2 and H4K16ac, a mouse monoclonal anti-HA antibody (12CA5, Roche) or a rabbit polyclonal anti H4K16ac (Millipore 07-329) were used at 1:200 dilution. Nsr1 was detected with a mouse monoclonal antibody (clone 31C4, ThermoFisher MA1-10030) used at 1:200 dilution. Alexa-Fluor-488 and Alexa-Fluor-594-conjugated secondary antibodies from Molecular Probes were used at 1:200 dilution. Images were captured with the same equipment as indicated above.
SUPPLEMENTAL MATERIAL
All supplemental data for this article are also available online at http://microbialcell.com/researcharticles/impact-of-histone-h4k16-acetylation-on-the-meiotic-recombination-checkpoint-in-saccharomyces-cerevisiae/.
Funding Statement
We are grateful to Shelly Berger, Félix Prado and Jesús Carballo for plasmids and antibodies and to Natalia Calixto for help with strain construction. SC was partially supported by a postdoctoral JAE-Doc contract and DO by a predoctoral JAE-Predoc contract from the Consejo Superior de Investigaciones Científicas (CSIC) of Spain. This work was funded by grants BFU2012-35748 and BFU2015-65417-R, from the Ministry of Economy and Competitiveness of Spain (MINECO), and grant CSI084U16 from Junta de Castilla y León (Spain), to PSS.
References
- 1.Petronczki M, Siomos MF, Nasmyth K. Un menage a quatre: the molecular biology of chromosome segregation in meiosis. Cell. 2003;112(4):423–440. doi: 10.1016/S0092-8674(03)00083-7. [DOI] [PubMed] [Google Scholar]
- 2.Kleckner N. Meiosis: how could it work? Proc Natl Acad Sci USA. 1996;93(16):8167–8174. doi: 10.1073/pnas.93.16.8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/S0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- 4.Roeder GS. Meiotic chromosomes: it takes two to tango. Genes Dev. 1997;11(20):2600–2621. doi: 10.1101/gad.11.20.2600. [DOI] [PubMed] [Google Scholar]
- 5.Sym M, Engebrecht JA, Roeder GS. ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell. 1993;72(3):365–378. doi: 10.1016/0092-8674(93)90114-6. [DOI] [PubMed] [Google Scholar]
- 6.Dong H, Roeder GS. Organization of the yeast Zip1 protein within the central region of the synaptonemal complex. J Cell Biol. 2000;148(3):417–426. doi: 10.1083/jcb.148.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roeder GS, Bailis JM. The pachytene checkpoint. Trends Genet. 2000;16(9):395–403. doi: 10.1016/S0168-9525(00)02080-1. [DOI] [PubMed] [Google Scholar]
- 8.Subramanian VV, Hochwagen A. The meiotic checkpoint network: step-by-step through meiotic prophase. Cold Spring Harb Perspect Biol. 2014;6(10):a016675. doi: 10.1101/cshperspect.a016675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacQueen AJ, Hochwagen A. Checkpoint mechanisms: the puppet masters of meiotic prophase. Trends Cell Biol. 2011;21(7):393–400. doi: 10.1016/j.tcb.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 11.Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst) 2009;8(9):1004–1008. doi: 10.1016/j.dnarep.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong EJ, Roeder GS. A role for Ddc1 in signaling meiotic double-strand breaks at the pachytene checkpoint. Genes Dev. 2002;16(3):363–376. doi: 10.1101/gad.938102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Refolio E, Cavero S, Marcon E, Freire R, San-Segundo PA. The Ddc2/ATRIP checkpoint protein monitors meiotic recombination intermediates. J Cell Sci. 2011;124(Pt 14):2488–2500. doi: 10.1242/jcs.081711. [DOI] [PubMed] [Google Scholar]
- 14.Lydall D, Nikolsky Y, Bishop DK, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383(6603):840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- 15.Carballo JA, Johnson AL, Sedgwick SG, Cha RS. Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell. 2008;132(5):758–770. doi: 10.1016/j.cell.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 16.Woltering D, Baumgartner B, Bagchi S, Larkin B, Loidl J, de los Santos T, Hollingsworth NM. Meiotic segregation, synapsis, and recombination checkpoint functions require physical interaction between the chromosomal proteins Red1p and Hop1p. Mol Cell Biol. 2000;20(18):6646–6658. doi: 10.1128/MCB.20.18.6646-6658.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eichinger CS, Jentsch S. Synaptonemal complex formation and meiotic checkpoint signaling are linked to the lateral element protein Red1. Proc Natl Acad Sci USA. 2010;107(25):11370–11375. doi: 10.1073/pnas.1004248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acosta I, Ontoso D, San-Segundo PA. The budding yeast polo-like kinase Cdc5 regulates the Ndt80 branch of the meiotic recombination checkpoint pathway. Mol Biol Cell. 2011;22(18):3478–3490. doi: 10.1091/mbc.E11-06-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailis JM, Roeder GS. Pachytene exit controlled by reversal of Mek1-dependent phosphorylation. Cell. 2000;101(2):211–221. doi: 10.1016/S0092-8674(00)80831-4. [DOI] [PubMed] [Google Scholar]
- 20.Niu H, Li X, Job E, Park C, Moazed D, Gygi SP, Hollingsworth NM. Mek1 kinase is regulated to suppress double-strand break repair between sister chromatids during budding yeast meiosis. Mol Cell Biol. 2007;27(15):5456–5467. doi: 10.1128/MCB.00416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ontoso D, Acosta I, van Leeuwen F, Freire R, San-Segundo PA. Dot1-dependent histone H3K79 methylation promotes activation of the Mek1 meiotic checkpoint effector kinase by regulating the Hop1 adaptor. PLoS Genet. 2013;9(1):e1003262. doi: 10.1371/journal.pgen.1003262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rockmill B, Roeder GS. A meiosis-specific protein kinase homolog required for chromosome synapsis and recombination. Genes Dev. 1991;5(12B):2392–2404. doi: 10.1101/gad.5.12b.2392. [DOI] [PubMed] [Google Scholar]
- 23.Wan L, de los Santos T, Zhang C, Shokat K, Hollingsworth NM. Mek1 kinase activity functions downstream of RED1 in the regulation of meiotic double strand break repair in budding yeast. Mol Biol Cell. 2004;15(1):11–23. doi: 10.1091/mbc.E03-07-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sourirajan A, Lichten M. Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev. 2008;22(19):2627–2632. doi: 10.1101/gad.1711408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu S, Herskowitz I. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell. 1998;1(5):685–696. doi: 10.1016/S1097-2765(00)80068-4. [DOI] [PubMed] [Google Scholar]
- 26.Tung KS, Hong EJ, Roeder GS. The pachytene checkpoint prevents accumulation and phosphorylation of the meiosis-specific transcription factor Ndt80. Proc Natl Acad Sci USA. 2000;97(22):12187–12192. doi: 10.1073/pnas.220464597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Chang CY, Wu JF, Tung KS. Nuclear localization of the meiosis-specific transcription factor Ndt80 is regulated by the pachytene checkpoint. Mol Biol Cell. 2011;22(11):1878–1886. doi: 10.1091/mbc.E10-12-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leu JY, Roeder GS. The pachytene checkpoint in S.cerevisiae depends on Swe1-mediated phosphorylation of the cyclin-dependent kinase Cdc28. . Mol Cell. 1999;4(5):805–814. doi: 10.1016/S1097-2765(00)80390-1. [DOI] [PubMed] [Google Scholar]
- 29.Pak J, Segall J. Role of Ndt80, Sum1, and Swe1 as targets of the meiotic recombination checkpoint that control exit from pachytene and spore formation in Saccharomyces cerevisiae. Mol Cell Biol. 2002;22(18):6430–6440. doi: 10.1128/MCB.22.18.6430-6440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storlazzi A, Xu L, Schwacha A, Kleckner N. Synaptonemal complex (SC) component Zip1 plays a role in meiotic recombination independent of SC polymerization along the chromosomes. Proc Natl Acad Sci USA. 1996;93(17):9043–9048. doi: 10.1073/pnas.93.17.9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sym M, Roeder GS. Crossover interference is abolished in the absence of a synaptonemal complex protein. Cell. 1994;79(2):283–292. doi: 10.1016/0092-8674(94)90197-X. [DOI] [PubMed] [Google Scholar]
- 32.Brachet E, Sommermeyer V, Borde V. Interplay between modifications of chromatin and meiotic recombination hotspots. Biol Cell. 2012;104(2):51–69. doi: 10.1111/boc.201100113. [DOI] [PubMed] [Google Scholar]
- 33.Borde V, Robine N, Lin W, Bonfils S, Geli V, Nicolas A. Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J. 2009;28(2):99–111. doi: 10.1038/emboj.2008.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acquaviva L, Drogat J, Dehe PM, de La Roche Saint-Andre C, Geli V. Spp1 at the crossroads of H3K4me3 regulation and meiotic recombination. Epigenetics. 2013;8(4):355–360. doi: 10.4161/epi.24295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sommermeyer V, Beneut C, Chaplais E, Serrentino ME, Borde V. Spp1, a member of the Set1 Complex, promotes meiotic DSB formation in promoters by tethering histone H3K4 methylation sites to chromosome axes. Mol Cell. 2013;49(1):43–54. doi: 10.1016/j.molcel.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 36.San-Segundo PA, Roeder GS. Pch2 links chromatin silencing to meiotic checkpoint control. Cell. 1999;97(3):313–324. doi: 10.1016/S0092-8674(00)80741-2. [DOI] [PubMed] [Google Scholar]
- 37.San-Segundo PA, Roeder GS. Role for the silencing protein Dot1 in meiotic checkpoint control. Mol Biol Cell. 2000;11(10):3601–3615. doi: 10.1091/mbc.11.10.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052–1058. doi: 10.1016/S0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 39.Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16(12):1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 41.Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 42.Xu F, Zhang Q, Zhang K, Xie W, Grunstein M. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol Cell. 2007;27(6):890–900. doi: 10.1016/j.molcel.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meijsing SH, Ehrenhofer-Murray AE. The silencing complex SAS-I links histone acetylation to the assembly of repressed chromatin by CAF-I and Asf1 in Saccharomyces cerevisiae. Genes Dev. 2001;15(23):3169–3182. doi: 10.1101/gad.929001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet. 2002;32(3):378–383. doi: 10.1038/ng1017. [DOI] [PubMed] [Google Scholar]
- 45.Kimura A, Umehara T, Horikoshi M. Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet. 2002;32(3):370–377. doi: 10.1038/ng993. [DOI] [PubMed] [Google Scholar]
- 46.Sutton A, Shia WJ, Band D, Kaufman PD, Osada S, Workman JL, Sternglanz R. Sas4 and Sas5 are required for the histone acetyltransferase activity of Sas2 in the SAS complex. J Biol Chem. 2003;278(19):16887–16892. doi: 10.1074/jbc.M210709200. [DOI] [PubMed] [Google Scholar]
- 47.Shia WJ, Osada S, Florens L, Swanson SK, Washburn MP, Workman JL. Characterization of the yeast trimeric-SAS acetyltransferase complex. J Biol Chem. 2005;280(12):11987–11994. doi: 10.1074/jbc.M500276200. [DOI] [PubMed] [Google Scholar]
- 48.Chang CS, Pillus L. Collaboration between the essential Esa1 acetyltransferase and the Rpd3 deacetylase is mediated by H4K12 histone acetylation in Saccharomyces cerevisiae. Genetics. 2009;183(1):149–160. doi: 10.1534/genetics.109.103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suka N, Suka Y, Carmen AA, Wu J, Grunstein M. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol Cell. 2001;8(2):473–479. doi: 10.1016/S1097-2765(01)00301-X. [DOI] [PubMed] [Google Scholar]
- 50.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43(30):9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 51.Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, Boeke JD. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97(12):6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braunstein M, Sobel RE, Allis CD, Turner BM, Broach JR. Efficient transcriptional silencing in Saccharomyces cerevisiae requires a heterochromatin histone acetylation pattern. Mol Cell Biol. 1996;16(8):4349–4356. doi: 10.1128/MCB.16.8.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robyr D, Suka Y, Xenarios I, Kurdistani SK, Wang A, Suka N, Grunstein M. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell. 2002;109(4):437–446. doi: 10.1016/S0092-8674(02)00746-8. [DOI] [PubMed] [Google Scholar]
- 54.Gartenberg MR, Smith JS. The Nuts and Bolts of Transcriptionally Silent Chromatin in Saccharomyces cerevisiae. Genetics. 2016;203(4):1563–1599. doi: 10.1534/genetics.112.145243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamburini BA, Tyler JK. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol. 2005;25(12):4903–4913. doi: 10.1128/MCB.25.12.4903-4913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436(7048):294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 57.Yu Y, Teng Y, Liu H, Reed SH, Waters R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc Natl Acad Sci USA. 2005;102(24):8650–8655. doi: 10.1073/pnas.0501458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Govin J, Dorsey J, Gaucher J, Rousseaux S, Khochbin S, Berger SL. Systematic screen reveals new functional dynamics of histones H3 and H4 during gametogenesis. Genes Dev. 2010;24(16):1772–1786. doi: 10.1101/gad.1954910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herruzo E, Ontoso D, Gonzalez-Arranz S, Cavero S, Lechuga A, San-Segundo PA. The Pch2 AAA+ ATPase promotes phosphorylation of the Hop1 meiotic checkpoint adaptor in response to synaptonemal complex defects. Nucleic Acids Res. 2016;44(16):7722–7741. doi: 10.1093/nar/gkw506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Penedos A, Johnson AL, Strong E, Goldman AS, Carballo JA, Cha RS. Essential and Checkpoint Functions of Budding Yeast ATM and ATR during Meiotic Prophase Are Facilitated by Differential Phosphorylation of a Meiotic Adaptor Protein, Hop1. PLoS One. 2015;10(7):e0134297. doi: 10.1371/journal.pone.0134297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hollingsworth NM. Deconstructing meiosis one kinase at a time: polo pushes past pachytene. Genes Dev. 2008;22(19):2596–2600. doi: 10.1101/gad.1727508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee BH, Amon A. Polo kinase--meiotic cell cycle coordinator. Cell Cycle. 2003;2(5):400–402. doi: 10.4161/cc.2.5.475. [DOI] [PubMed] [Google Scholar]
- 63.Lee BH, Amon A. Role of Polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science. 2003;300(5618):482–486. doi: 10.1126/science.1081846. [DOI] [PubMed] [Google Scholar]
- 64.Clyne RK, Katis VL, Jessop L, Benjamin KR, Herskowitz I, Lichten M, Nasmyth K. Polo-like kinase Cdc5 promotes chiasmata formation and cosegregation of sister centromeres at meiosis I. Nat Cell Biol. 2003;5(5):480–485. doi: 10.1038/ncb977. [DOI] [PubMed] [Google Scholar]
- 65.Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459(7248):802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choy JS, Acuna R, Au WC, Basrai MA. A role for histone H4K16 hypoacetylation in Saccharomyces cerevisiae kinetochore function. Genetics. 2011;189(1):11–21. doi: 10.1534/genetics.111.130781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cesarini E, D'Alfonso A, Camilloni G. H4K16 acetylation affects recombination and ncRNA transcription at rDNA in Saccharomyces cerevisiae. Mol Biol Cell. 2012;23(14):2770–2781. doi: 10.1091/mbc.E12-02-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hepworth SR, Friesen H, Segall J. NDT80 and the meiotic recombination checkpoint regulate expression of middle sporulation-specific genes in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18(10):5750–5761. doi: 10.1128/MCB.18.10.5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282(5389):699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 70.Xu L, Ajimura M, Padmore R, Klein C, Kleckner N. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15(12):6572–6581. doi: 10.1128/MCB.15.12.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 73.Millar CB, Grunstein M. Genome-wide patterns of histone modifications in yeast. Nat Rev Mol Cell Biol. 2006;7(9):657–666. doi: 10.1038/nrm1986. [DOI] [PubMed] [Google Scholar]
- 74.Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131(6):1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 75.Altaf M, Utley RT, Lacoste N, Tan S, Briggs SD, Cote J. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol Cell. 2007;28(6):1002–1014. doi: 10.1016/j.molcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frederiks F, Tzouros M, Oudgenoeg G, van Welsem T, Fornerod M, Krijgsveld J, van Leeuwen F. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol. 2008;15(6):550–557. doi: 10.1038/nsmb.1432. [DOI] [PubMed] [Google Scholar]
- 77.Borner GV, Barot A, Kleckner N. Yeast Pch2 promotes domainal axis organization, timely recombination progression, and arrest of defective recombinosomes during meiosis. Proc Natl Acad Sci USA. 2008;105(9):3327–3332. doi: 10.1073/pnas.0711864105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Joshi N, Barot A, Jamison C, Borner GV. Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet. 2009;5(7):e1000557. doi: 10.1371/journal.pgen.1000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan C, Melese T. Multiple regions of NSR1 are sufficient for accumulation of a fusion protein within the nucleolus. J Cell Biol. 1993;123(5):1081–1091. doi: 10.1083/jcb.123.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mieczkowski PA, Dominska M, Buck MJ, Lieb JD, Petes TD. Loss of a histone deacetylase dramatically alters the genomic distribution of Spo11p-catalyzed DNA breaks in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2007;104(10):3955–3960. doi: 10.1073/pnas.0700412104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vader G, Blitzblau HG, Tame MA, Falk JE, Curtin L, Hochwagen A. Protection of repetitive DNA borders from self-induced meiotic instability. Nature. 2011;477(7362):115–119. doi: 10.1038/nature10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rockmill B, Roeder GS. Meiosis in asynaptic yeast. Genetics. 1990;126(3):563–574. doi: 10.1093/genetics/126.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chien CT, Buck S, Sternglanz R, Shore D. Targeting of SIR1 protein establishes transcriptional silencing at HM loci and telomeres in yeast. Cell. 1993;75(3):531–541. doi: 10.1016/0092-8674(93)90387-6. [DOI] [PubMed] [Google Scholar]
- 84.Storici F, Resnick MA. The Delitto Perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 2006;409:329–345. doi: 10.1016/S0076-6879(05)09019-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.