

Knockout of either of two Drosophila Troponin C genes that are expressed in either the flight muscle or the jump muscle resulted in expansion of transcription of its paralogue into the affected muscle. Although either isoform can support normal jumping, only the flight isoform can support flight.
Abstract
We investigated the functional overlap of two muscle Troponin C (TpnC) genes that are expressed in the adult fruit fly, Drosophila melanogaster: TpnC4 is predominantly expressed in the indirect flight muscles (IFMs), whereas TpnC41C is the main isoform in the tergal depressor of the trochanter muscle (TDT; jump muscle). Using CRISPR/Cas9, we created a transgenic line with a homozygous deletion of TpnC41C and compared its phenotype to a line lacking functional TpnC4. We found that the removal of either of these genes leads to expression of the other isoform in both muscle types. The switching between isoforms occurs at the transcriptional level and involves minimal enhancers located upstream of the transcription start points of each gene. Functionally, the two TpnC isoforms were not equal. Although ectopic TpnC4 in TDT muscles was able to maintain jumping ability, TpnC41C in IFMs could not effectively support flying. Simultaneous functional disruption of both TpnC genes resulted in jump-defective and flightless phenotypes of the survivors, as well as abnormal sarcomere organization. These results indicated that TpnC is required for myofibril assembly, and that there is functional specialization among TpnC isoforms in Drosophila.
INTRODUCTION
Numerous genes in higher eukaryotes exist as multigene families. In some instances, such as ribosomal DNA genes, the existence of multiple gene copies is believed to enable the production of an increased amount of gene product. By contrast, in many instances, the existence of gene duplicates has allowed for either subfunctionalization, by which individual gene family members acquire specialized functions, or neofunctionalization, by which a duplicate copy acquires a new or novel function (reviewed in Conant and Wolfe, 2008; Katju, 2012).
Although examples of neofunctionalization are relatively rare (see, e.g., Zhang et al., 2004), there are a number of examples of subfunctionalization. In mammals, several muscle structural genes are represented as duplicate copies, such as myosin heavy chain (MHC) genes. The mouse genome encodes 11 muscle-specific MHC genes that are expressed in different regions of the body and at different times of development; moreover, the encoded proteins have biochemical properties that differ from one another (reviewed in Weiss and Leinwand, 1996).
The existence of subfunctionalization raises questions of the degree to which the duplicate gene copies are functionally distinct and how the differences in patterns of gene expression for duplicate copies has arisen. For the myosin genes, as with other examples, there is evidence for functional nonredundancy in gene duplicates: loss of function of the murine Myh1 gene is partially compensated for by up-regulation of Myh2 expression. However, the mutant mice show a number of severe phenotypes, including muscle weakness and kyphosis, indicating that the two isoforms are functionally nonequivalent (Sartorius et al., 1998; Allen and Leinwand, 2001). Similarly, different muscle-specific actin genes in Drosophila were shown to be nonequivalent when tested for function in the adult indirect flight muscles (IFMs; Fyrberg et al, 1998). These studies, alongside many others, underline the adaptive potential of subfunctionalization.
Another muscle structural gene family in Drosophila encodes the Troponin C (TpnC) proteins. TpnC is the Ca2+-binding moiety of the troponin complex that regulates Ca2+-activated skeletal muscle contraction (reviewed in Lieber, 2010). There are five TpnC genes in the Drosophila genome, which show distinct patterns of gene expression. In particular, TpnC41C is expressed at high levels in some adult tubular muscles, including the tergal depressor of the trochanter (TDT; or jump) muscle, whereas TpnC4 is expressed almost exclusively in the adjacent IFMs (Qiu et al., 2003; Herranz et al., 2004).
Two recent studies evaluated the roles of TpnC in Drosophila adult muscles. Eldred et al. (2014) studied the roles of the two adult TpnC isoforms in adult muscle function. They detected low levels of the TDT-enriched TpnC, TpnC41C, in the flight muscles; knockdown of TpnC41C expression, however, did not affect flight muscle function. By contrast, knockdown of TpnC4 expression, encoding the predominant flight muscle isoform, resulted in up-regulation of TpnC41C in flight muscles and defects in muscle structure and function. In a similar study, Singh et al. (2014) demonstrated that flight muscle–specific knockdown of TpnC4 expression resulted in a reduction in flight ability and myofibrillar defects. These findings suggested that, at least in the flight muscles, TpnC41C and TpnC4 were not functionally equivalent.
These two studies significantly enhanced our understanding of TpnC function and demonstrated that, at least in the context of the flight muscles, the two adult isoforms could not functionally replace one another. Nevertheless, several questions remain: are these results from RNA interference (RNAi) experiments supported by genetic loss-of-function experiments? Is there reciprocal functional nonredundancy between flight and jump muscle isoforms of TpnC in Drosophila? What is the phenotype of TpnC-null muscle? Finally, how is the up-regulation of expression of one protein in the absence of the other achieved? In this article, we demonstrate that TpnC4 and TpnC41C are expressed almost exclusively in the IFM and TDT muscles, respectively, and that they show reciprocal cross-regulation. Using knockdown and knockout combinations, we confirm that TpnC41C cannot compensate for the loss of TpnC4 in the IFMs. We additionally demonstrate that, in the converse situation, loss of TpnC41C function in the TDT results in an expansion of TpnC4 expression into the TDT, which enables normal jumping ability of adults. We also generate TpnC41C TpnC4 double-null mutants, which can neither fly nor jump and fail to form normal myofibrils during development. Finally, we demonstrate that the expansion of gene expression in each case occurs through transcriptional up-regulation via identified enhancer sequences. These studies complement and significantly extend earlier findings by demonstrating the functional significance of TpnC isoform diversity and providing insight into the mechanisms by which loss of expression of one gene results in a change in the expression of a paralogous gene.
RESULTS
Drosophila tubular and fibrillar muscle fiber types express different TpnC isoforms
Previous studies demonstrated that TpnC4 is the predominant IFM isoform, whereas TpnC41C is most strongly expressed in the tubular muscles, including the jump muscle. More detailed studies also indicated that the flight muscles, although predominantly expressing TpnC4, also express TpnC41C to some degree (Qiu et al., 2003; Herranz et al., 2004; Eldred et al., 2014). In our studies, we observed relatively little overlap in expression of the two genes: firstly, we assessed TpnC protein isoform composition in whole thoraces as well as dissected IFM and TDT muscles using Western blotting and probed with an antibody that recognizes both adult isoforms of TpnC. We found that TpnC4 protein was the only isoform detected in the IFMs, whereas TpnC41C was a specific TpnC isoform of TDT muscles (Figure 1A). Although TpnC4 and TpnC41C were reported earlier as major and minor isoforms found in the IFM, respectively (Eldred et al., 2014), we could not find any traces of TpnC41C expression on our Western blots when we loaded equal amounts of total protein of dissected muscles. Only when the flight muscle lane was overloaded to tenfold the amount of protein used in Figure 1A did we observe a faint band corresponding to TpnC41C.
FIGURE 1:

TpnC4 and TpnC41C are fiber-specific genes expressed in the muscles of the Drosophila thorax. (A) Western blot of protein samples prepared from Drosophila whole thoraces (THX, lane 1) and dissected IFM and TDT muscles (lanes 2 and 3, respectively) stained with TpnC antibody. Arrows indicate positions of TpnC4 and TpnC41C isoforms. Positions and molecular weights of protein standards are indicated on the left. (B, C) Top, diagrams of the D. melanogaster TpnC4 and TpnC41C genes with their exon composition and the reporter constructs generated, in which the indicated genomic regions were fused to lacZ. Protein-coding regions are shown in orange; untranslated regions are shown in gray. Numbers indicate starting and terminal nucleotides relative to the transcription start site for each tested cloned promoter region. Bottom, representative sections of Drosophila thoraces expressing either TpnC4-lacZ (B) or TpnC41C-lac-Z (C) and stained for β-Gal activity. IFMs stain blue in B, whereas TDTs stain blue in C. TDT muscles are outlined with dashed lines. Scale bar, 100 µm.
Second, to complement this analysis, we tested the activities in the adult muscles of β-galactosidase (β-Gal) reporter constructs in which the regulatory sequences of either TpnC4 or TpnC41C were fused to a lacZ reporter (Figure 1, B and C, diagrams). To locate the TpnC4 regulatory sequences, we tested the enhancer activities of 5′- and 3′-adjacent sequences of the gene, as well as its introns, and the only construct that had reporter expression was −730/+579 TpnC4-lacZ. The TpnC41C-lacZ reporter was described in Chechenova et al. (2015).
In the thoraces of transgenic flies, the cloned TpnC4 enhancer selectively directed reporter expression in the fibrillar IFMs, whereas reporter expression was excluded from tubular TDT muscles (Figure 1B). By contrast, the TpnC41C enhancer was active in TDT muscles but not in the IFMs (Figure 1C). These findings underline the published observations that different thoracic muscle fibers use different isoforms of TpnC protein, which probably reflect the differences in functional demands for different types of highly specialized adult muscles. Our data differ to a small extent from those of Eldred et al. (2014), in that we show that the contribution of TpnC41C in the flight muscles is very minor, which might simply reflect line-specific differences in gene expression.
The TpnC4 isoform functionally substitutes for TpnC41C
To analyze the functional importance of TpnC41C in TDT muscles, we sought to inactivate its expression. We achieved gene inactivation by two methods: expressing a short hairpin RNA (shRNA) targeting TpnC41C and generating a TpnC41C-null allele.
The TpnC41C-null allele was created using the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome-editing approach. We generated two plasmids containing sequences to express single guide RNAs targeting 5′ and 3′ regions of the gene (Figure 2A). The plasmids were injected into embryos expressing Cas9 in their germline, and potential mutant lines were identified as homozygotes in the second generation. Western blotting identified one line, TpnC41C∂#2, that did not accumulate TpnC41C in the thorax, and we determined that this line had arisen through the designed deletion of the genomic sequence between the two target sites based on PCR and sequencing genomic DNA from this line (Figure 2A, bottom).
FIGURE 2:

Loss of TpnC41C is functionally compensated by expansion of TpnC4 expression. (A) Design and analysis of a TpnC41C genomic deletion. Top, GBrowse image from flybase.org shows the TpnC41C locus and its annotated transcripts. Bottom, locations of CRISPR target sites (X marks), primers used to confirm the deletion (arrows), and deletion boundaries in the TpnC41C∂#2 allele (T-bars). (B) TpnC isoform composition in TpnC41C-knockdown and -knockout mutants. Proteins from whole thoraces and dissected IFM and TDT muscles were separated by SDS–PAGE, followed by Western blotting and staining with TpnC antibody (top) and α-actinin antibody to serve as a loading control (bottom). Protein samples from w1118 control flies (lines 1, 2, and 6) were analyzed alongside lysates obtained from either Act79B>TpnC41C RNAi flies (C41-KD, lines 3–5) or TpnC41C∂#2 homozygotes (C41-KO, lines 7 and 8). Vertical gray line between lanes 6 and 7 indicates that these samples were analyzed on the same blot but that intervening lanes were cropped out. (C) Jump test results. The offspring of Act79B-Gal4 crossed to the RNAi control line 36303 (BDSC) (79B>WT, dark gray bar) were used as a control for the line with TpnC41C knockdown in jump muscles. Act79B>UAS-TpnC41C RNAi (79B>C41-KD, light gray bar) are the knockdown animals. Heterozygous flies from the cross of w1118 x TpnC41C∂#2 (w1118 xC41-KO, dark green bar) were used as controls for TpnC41C∂#2 homozygous mutants (C41-KO, light green bar). Error bars show SD for average jumping distance in millimeters. TpnC41C-knockout and -knockdown flies showed reduced jumping ability compared with controls (*p < 0.01). (D) Flight test results. The offspring of 1151-Gal4 crossed with the RNAi control line 36303 (1151>WT, dark gray bars) were used as a control for flies with the TpnC41C knockdown in both flight and jump muscles (n = 96). 1151>UAS-TpnC41C RNAi (1151>C41-KD, light grey bars) represent knockdown animals (n = 129). Heterozygous offspring from the cross of w1118 with TpnC41C∂#2 (w1118 x C41-KO, dark green bars) was used as a control for the TpnC41C∂#2 homozygous mutants (C41-KO, light green bars; n = 72 and 96, respectively). Letters on the top show direction of flight: D, downward; H, horizontally; N, no flight at all; U, upward. TpnC41C-knockdown and -knockout flies showed only slight reductions in flight ability compared with their controls.
We next compared the effects of RNAi-mediated knockdown (KD) of TpnC41C with the TpnC41C-null allele. For the KD, we observed a loss of TpnC41C protein from whole thoraces (Figure 2B, lane 3), yet when we analyzed the IFM and TDT muscles separately, we observed that the jump muscles accumulated a TpnC with the same electrophoretic mobility as the flight muscle TpnC4 isoform (Figure 2B, lanes 4 and 5). This result suggested that loss of TpnC41C from the jump muscles caused an expansion of TpnC4 expression into the TDT. To confirm that this result was not an artifact of the RNAi system being used, we studied TpnC accumulation in the muscles of homozygous TpnC41C∂#2-null mutants. Again, we observed a loss of the TpnC41C-specific band in TDT samples but the appearance of a band corresponding to TpnC4 (Figure 2B, lane 8). Together these data indicate that loss of TpnC41C from the jump muscles caused an isoform switch in the TDT.
To define the functional consequences of this isoform switch, we jump tested the TpnC41C KD and null homozygotes because the TDT is the sole muscle responsible for jumping in the adult (Elliott et al., 2007). Of interest, although the KD and mutant animals showed a small but significant reduction in jumping distance compared with controls, they maintained jumping ability (Figure 2C), indicating that there is a functional overlap between TpnC4 and TpnC41C in the context of jump muscle function.
As mentioned earlier, some publications reported a low expression of TpnC41C in IFMs (Eldred et al., 2014). To determine whether such minor TpnC41C presence had an effect on flight ability, we performed flight testing on flies with TpnC41C deficiency. We found that 90% of homozygotes for the TpnC41C deletion were able to fly (Figure 2D, C41-KO), which was comparable to a heterozygous line lacking one copy of the TpnC41C gene (99% able to fly; Figure 2D, w1118xC41KD). To down-regulate TpnC41C expression in flight muscles, we crossed the 1151-Gal4 driver to a UAS-TpnC41C RNAi line. This resulted in a complete loss of TpnC41C from TDT muscles (unpublished data) and therefore presumably also the loss of any small amount of TpnC41C present in the flight muscles. Among the offspring of this cross, 18% were flight impaired and flew downward, although the majority of the flies were able to fly normally (67%; Figure 2D). These results confirmed the observations of Eldred et al. (2014) that the TpnC41C isoform does not play a noticeable role in the function of the IFMs. Overall our studies demonstrate that, in the absence of TpnC41C in the jump muscles, there is an expansion of TpnC4 expression into the jump muscles that rescues jump muscle function.
The TpnC41C isoform does not functionally substitute for TpnC4
The finding that the TpnC4 gene was activated in TpnC41C-deficient TDTs encouraged us to examine the possibility of a reciprocal effect. We asked whether the tubular muscle TpnC isoform can substitute for its fibrillar-muscle paralogue in flight muscles of TpnC4-deficient flies. To achieve this, we analyzed a TpnC4-knockdown mutant created by crossing the 1151-Gal4 driver with the UAS-TpnC4 RNAi line. We also analyzed the TpnC4-null mutant, termed TpnC4F2–3, described in Adame et al. (2016). Thoracic muscles of both lines were dissected and assessed for TpnC expression by Western blotting.
In our Western blots, we observed that knockdown of TpnC4 in the flight muscles resulted in a loss of the corresponding protein in whole-thorax samples (Figure 3A, lane 3). Moreover, we observed that flight muscles of these knockdown animals accumulated a TpnC isoform with mobility matching that of TpnC41C (Figure 3A, lane 4). A similar observation was made in the TpnC4-null animals, in which loss of TpnC4 in the IFMs resulted in accumulation of TpnC41C in those muscles (Figure 3A, lanes 7–9). Of interest, in the TpnC4-knockout mutants, the accumulation of TpnC41C in the IFMs appeared to be reproducibly reduced relative to the accumulation of TpnC4 in control animals. This difference was apparent when compared with a lane in which half as much of the TDT sample was loaded (Figure 3A, lane 9). On the other hand, the accumulation of TpnC41C in a TpnC4 knockdown was not noticeably less than in controls (Figure 3A, lane 4; Eldred et al., 2014). As described in the Discussion, it is possible that these reduced levels of TpnC in the TpnC4 mutants could contribute to the behavioral defects detailed later. Nevertheless, our data overall demonstrate that there is mutual compensation in expression between TpnC41C and TpnC4, although the precise levels of expression in control and compensating animals may differ.
FIGURE 3:

Loss of TpnC4 is not functionally compensated by TpnC41C, despite TpnC41C expression expanding into the IFMs. (A) TpnC isoform composition in TpnC4-knockdown and -knockout mutants. Proteins from whole thoraces (THX) and dissected IFM and TDT muscles were separated by SDS–PAGE, followed by Western blotting and staining for TpnC (top) and α-actinin as a loading control (bottom). Protein samples from w1118 control flies (WT, lanes 1, 2, and 6) were analyzed alongside lysates obtained from either 1151>TpnC4 RNAi flies (C4-KD, lanes 3–5) or TpnC4F2–3 mutant homozygotes (C4-KO, lanes 7–9). Lane 9 has TDT lysate loaded at 50% of the total protein amount used for other lanes. (B) Jump test results. The offspring of the Act79B-Gal4 driver line crossed to the RNAi control line 60100 (VDRC) (79B>WT, dark gray bar) were used as a control to compare to TpnC4 knockdown in jump muscles. Act79B>UAS-TpnC4 RNAi (79B>C4-KD, light gray bar) are the knockdown sample. Heterozygous adults from the cross w1118 x TpnC4F2–3 (w1118xC4-KO, dark green bar) were used to compare with TpnC4F2–3 homozygotes (C4-KO, light green bar). Error bars show SD for average jumping distance in millimeters. Note that the jumping ability is reduced in both knockdown and knockout lines compared with controls, *p < 0.01. (C) Flight test results. The offspring of 1151-Gal4 crossed with the RNAi control line 60100 (1151>WT, dark gray bars) were used as a control for the line with TpnC4 knockdown in both flight and jump muscles, 1151>UAS-TpnC4 RNAi (1151>C4-KD, light gray bar; n = 304 and 174, respectively). Heterozygotes from the cross of w1118 x TpnC4F2–3 (w1118xC4-KO, dark green bar) were used to compare with TpnC4F2–3 homozygotes (C4-KO, light green bar; n = 243 and 89, respectively). Letters on the top show direction of flight: D, downward; H, horizontally; N, no flight at all; U, upward. Note that knockdown and knockout flies of TpnC4 are completely flightless.
We next analyzed the jump and flight capabilities of TpnC4 KD and mutant animals. In this instance, we compared the jumping abilities of control animals, KD animals generated by combining the TDT-specific driver line Act79B-Gal4 with UAS-TpnC4 RNAi, and TpnC4 nulls. We observed a significant reduction in the jumping distance of TpnC4-deficienct animals (Figure 3B). This result was unexpected because we do not detect significant levels of TpnC4 expression in the TDT muscles, and this will be addressed in the Discussion. Of greater importance, when we subjected the TpnC4 mutants to a flight test, we observed that both knockdown and knockout mutants were completely flightless (Figure 3C). These results established that whereas TpnC41C expression could expand into the flight muscles of TpnC4-deficient flies, TpnC41C could not substitute for the lack of TpnC4.
TpnC4 and TpnC41C isoforms are essential for thoracic muscle function
The foregoing experiments showed that two TpnC genes, expressed in large thoracic muscles, can genetically substitute for the expression of each other, and that one isoform, TpnC4, can take over both flight and jump functions. Although TpnC41C does not substitute for TpnC4 in flight muscle function, its expression in TpnC4-deficient flight muscle could be important for supporting muscle structural integrity. Therefore we investigated whether simultaneous removal of TpnC4 and TpnC41C would be detrimental to adult muscle morphology and viability. Again, we used two independent techniques to down-regulate troponin C genes: RNAi knockdown and genetic knockout.
To create a double knockdown, we generated lines containing IFM and TDT-specific drivers (Act88F-Gal4 and Act79B-Gal4, respectively; Bryantsev et al., 2012a), as well as UAS-driven RNAi constructs targeting TpnC4 and TpnC41C transcripts. These lines were crossed to generate Act88F+Act79B>TpnC4 RNAi + TpnC41C RNAi adults.
To generate a double mutant, we initially considered combining the TpnC41C∂#2 and TpnC4F2–3 alleles by recombination. However, because the genes are located within 440 kb of each other in a centromere-proximal part of the second chromosome right arm, we felt it would be more practical to simply generate a TpnC4 mutation on the chromosome that already carries the TpnC41C∂#2 allele. We used the same targeting plasmids as described in Adame et al. (2016) to mutate TpnC4 and obtained two independent mutant lines. Line TpnC∂#25 contained a 2–base pair deletion plus a 9–base pair insertion in exon 3 at the location of the 5′ CRISPR target. The insertion carried a stop codon resulting in translation termination after the sixth amino acid of the polypeptide (Figure 4A). Line TpnC∂#6 contained two single-nucleotide insertions in exon 3, in close proximity to the 5′ CRISPR target. One of the insertions produced a premature stop codon after amino acid 14 of the polypeptide chain (unpublished data). Overall both mutant coding sequences contained in-frame stop codons that were predicted to terminate translation early in the coding sequence of TpnC4. Through these approaches, we were able to assess the effects upon IFM and TDT function of removing all TpnC from those muscles.
FIGURE 4:

TpnC41C and TpnC4 double mutants cannot jump and are flightless. (A) Design and analysis of a TpnC4 mutation that was created in the TpnC41C∂#2 background. Top, GBrowse image from flybase.org showing the TpnC4 locus and its annotated transcripts. Bottom, positions of CRISPR targets (X-marks) and primers used for analysis of mutations (arrows), showing the sequence analysis of TpnC4 in WT and TpnC4∂#25 that was generated in the TpnC41C∂#2 genetic background. This new TpnC4 allele has a 9–base pair insertion (red) that includes an in-frame stop codon. The allele also has a 2–base pair deletion. Underlined sequence in WT corresponds to the site targeted by CRISPR/Cas9 mutagenesis. (B) TpnC isoform composition in TpnC double-knockdown and double-knockout mutants. Proteins from whole thoraces (THX) and dissected IFM and TDT muscles were separated by SDS–PAGE, followed by Western blotting and immunostaining with TpnC antibody (top) and α-actinin antibody as a loading control (bottom). Protein samples from w1118 control flies (lines 1, 2, and 6) were analyzed alongside lysates obtained from females carrying Act79B-Gal4 and Act88F-Gal4 drivers together with UAS-TpnC41C RNAi and UAS-TpnC4 RNAi (C4/C41-KD, lines 3–5) or alongside sample from TpnC41C∂#2 TpnC4∂#25 double-mutant homozygotes (C4/C41-KO, line 7). Vertical gray lines between lanes 3 and 4 and lanes 6 and 7 indicate that the samples were analyzed on the same blot, but intervening lanes were removed. (C) Jump test results. The TpnC4/TpnC41C double-knockdown line (C4/C41-KD) and the TpnC41C∂#2 TpnC4∂#25 double-knockout line (C4/C41-KO) showed no jumping ability compared with w1118 controls (WT, dark grey bar). Error bars show SD for average jumping distance in millimeters. (D) Flight test results. Heterozygous females carrying both Act79B-Gal4 and Act88F-Gal4 drivers (88F/79B, dark gray bars) were used as a control for the line with double TpnC4/TpnC41C knockdown (88F/79B>C4/C41-KD, light gray bars; n = 58 and 34, respectively); w1118 flies (dark green bars) were used as a control for the TpnC41C∂#2 TpnC4∂#25 homozygotes (C4/C41-KO, light green bars; n = 79 and 29, respectively); letters on the top show direction of flight: D, downward; H, horizontally; N, no flight at all; U, upward. Note that the knockdown and knockout mutants were unable to jump or fly.
For the double-knockdown approach, analysis of protein lysates from IFM and TDT muscles showed complete elimination of TpnC4 and a marked decrease of TpnC41C (Figure 4B, lanes 4 and 5). Although these double-knockdown flies survived until adulthood, they were flightless and unable to jump (Figure 4, C and D).
For the double-mutant approach, we observed that homozygotes of the two independent TpnC41C TpnC4 double-knockout lines were able to reach late pupal stages, but only a few adults could successfully eclose. The surviving adult flies showed very low mobility, could not jump, and were flightless (Figure 4, C and D). These results highlight the biological importance of the TpnC isoform switching observed because when only one isoform was disabled, the animals showed strong viability and jumping ability. Thus the TpnC genes complemented each other for viability and jumping ability, but there was no complementation for flight muscle function.
Myofibril structure of TpnC double mutants
It was reported that reduced expression of TpnC4 in flight muscle resulted in abnormalities in sarcomere structure and loss of Z- and M-line integrity (Eldred et al., 2014). This phenotype represented the effects of switching TpnC isoforms between the IFM and TDT. In contrast, depletion of TpnC41C did not show any changes in the organization of the fibrillar muscle (Eldred et al., 2014; Singh et al., 2014). To identify the effects of a complete lack of adult TpnCs in the IFM and TDT, we analyzed myofibril structure in each muscle fiber type in double TpnC4/TpnC41C knockout mutants. To specifically visualize the formation of the Z-line and M-line of the sarcomere, we stained muscle cryosections of pharate adults with antibodies recognizing the muscle structural proteins Sallimus (Sls; also known as kettin) to label the Z-line and Obscurin to label the M-line. The overall organization of myofibrils was evaluated by staining for F-actin using a fluorescent conjugate of phalloidin.
In wild-type IFMs, the myofibrils were cylindrical, and bright phalloidin staining represented the Z-lines, which accumulate Sls protein (Figure 5, A–A″). In the double-mutant animals, the overall sarcomere architecture was retained but the Z-lines were frequently indistinct, and the myofibrils did not retain a consistent diameter along their length (Figure 5, B–B″). In addition, there was background Sls staining in the mutant, suggesting that some of the Sls might have been relocalized from myofibrils to the sarcoplasm. Similarly, in the TDTs, wild-type fibers show striking organization into parallel arrays of myofibrils, with Z-lines of adjacent myofibrils kept in close register (Figure 5, C–C″). In the double-mutant animals, however, there was significant disorganization of the TDT myofibrils, with reduced and patchy Z-line-specific signal, plus diffuse Sls accumulation (Figure 5, D–D″).
FIGURE 5:

TpnC is required for myofibril assembly. Confocal images of cryosectioned IFM and TDT muscles from WT and TpnC41C∂#2 TpnC4∂#25 double-knockout mutants stained for F-actin, Sallimus (Sls), and Obscurin. (A-A″, B–B″, C–C″, D–D″) Staining with phalloidin (green) and anti-Sls antibody (magenta). (A-A″, B–B″) IFM. (C–C″, D–D″) TDT myofibrils. (E–E″, F–F″, G–G″, H–H″) Immunostaining with phalloidin (green) and anti-Obscurin antibody (magenta). (E–E″, F–F″) IFM. (G–G″, H–H″) Images of TDT. Scale bar, 5 μm. All images represent longitudinal sections of the muscles. Note that myofibril organization is disrupted in the mutants compared with controls.
The M-line in wild-type IFMs corresponds to the absence of strong phalloidin staining and was visualized as a distinct signal using anti-Obscurin immunostaining (Figure 5, E–E″). In the double TpnC mutant homozygotes, the M-line staining in the IFMs was still detected but was somewhat disorganized compared with the result in control animals (Figure 5, F–F″). Similarly in the TDTs, anti-Obscurin immunostaining labeled the organized register of M-lines in control animals (Figure 5, G–G″), and whereas mutant animals showed evidence of M-line assembly, there was still some disorganization to the muscles (Figure 5, H–H″). These findings indicate that the presence of TpnC in flight and jump muscles is required for normal myofibril assembly.
The TpnC isoform switching is regulated at the transcriptional level
Since our cloned TpnC4-lacZ and TpnC41C-lacZ reporters contained the necessary regulatory sequences to direct correct fiber-specific expression in nonmutant animals (Figure 1), we asked whether the isoform switching occurred at the transcriptional level and whether it was controlled via the regulatory sequences that we had identified. We achieved this by combining the TpnC-lacZ reporter for one gene with a knockdown for the other gene and vice versa (described in more detail in Materials and Methods). Upon down-regulation of TpnC41C, the expression of the TpnC4-lacZ reporter was activated in the TDTs, where normally this reporter was not expressed (Figure 6, A and B). This result indicated that the accumulation of TpnC4 in the TDTs of TpnC41C KD animals resulted from a change in the pattern of transcription for TpnC4. Similarly, when TpnC4 was down-regulated in the IFMs, expression of the TpnC41C-lacZ reporter was detected in these muscles, although potentially at reduced levels compared with that in the jump muscles (Figure 6, C and D). Collectively these results indicated that the enhancers of both troponin genes contain regulatory sequences required for their expression in both IFMs and TDTs. However, normal expression of one TpnC isoform apparently inhibits transcription of the paralogous gene.
FIGURE 6:
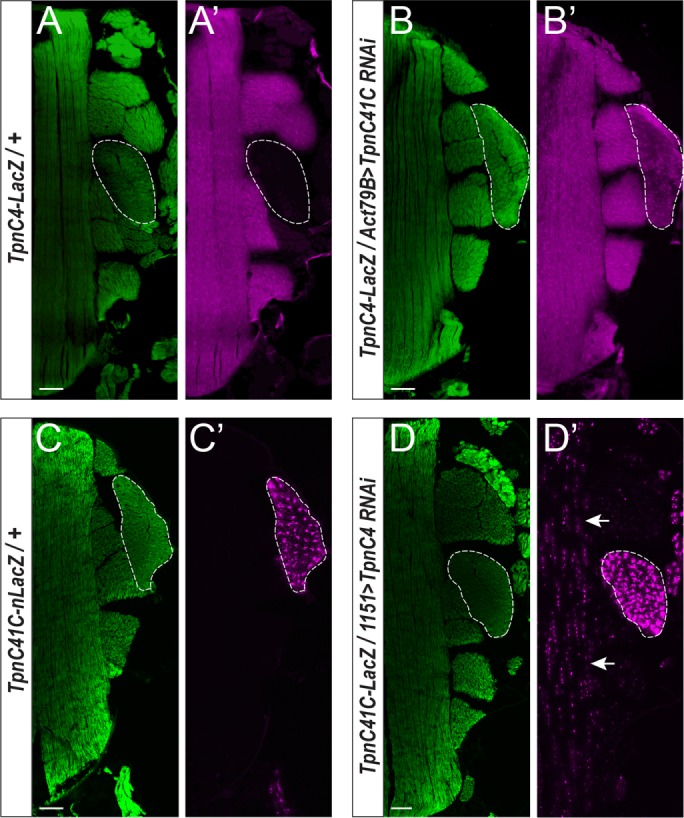
The TpnC isoform switch is regulated at the transcriptional level. (A–D, A′–D′) Frozen sections of control and experimental flies were stained with phalloidin (green) and with antibody to β-Gal (magenta) and imaged using confocal microscopy. (A, A′) Control line carrying a genomic insertion of TpnC4-lacZ shows reporter expression in the IFMs and not the TDT (TDT is outlined with a dashed line). (B, B′) TpnC41C-knockdown flies carrying the TpnC4-lacZ reporter show lacZ expression expanding into the TDT. (C, C′) Control line carrying genomic insertion of TpnC41C-nLacZ shows reporter expression in the TDT. (D, D′) TpnC4-knockdown flies carrying the TpnC41C-nlacZ reporter show lacZ expression expanding into the IFM (arrows). Note that in C′ and D′, the β-Gal reporter carries a nuclear localization signal and accumulates in muscle nuclei, creating a punctate pattern. Scale bars, 50 µm.
DISCUSSION
We showed that there is partial functional redundancy between TpnC isoforms expressed in the adult Drosophila thorax. When either TpnC4 or TpnC41C level was reduced, flies were viable, and expression of the complementary gene expanded to compensate. At the functional level, whereas the flight muscle isoform, TpnC4, could rescue jump muscle function in TpnC41C mutants, the reverse was not the case. These studies are important for understanding how genetic redundancy might occur and they also define the degree to which these different TpnC isoforms are interchangeable.
Functional compensation between TpnC isoforms
Both our work and that of Eldred et al. (2014) demonstrate that, when TpnC41C expands its expression into the flight muscles of TpnC4-deficient flies, the flies are flightless. The inability of TpnC41C to rescue loss of TpnC4 flight ability might have a number of reasons. In one scenario, there might be corresponding changes in the levels or isoforms of other myofibrillar proteins that are not consistent with normal flight muscle function. Although we cannot test this possibility exhaustively, we analyzed by Western blotting Troponin T (TpnT) accumulation in control and mutant flight muscles and did not observe a change in TpnT isoform abundance (unpublished data). Note that flight and jump muscles accumulate different isoforms of TpnT that differ in apparent relative mass (Benoist et al., 1998; Domingo et al., 1998).
In a second scenario, it could be argued that even though TpnC41C accumulates in the TpnC4-knockout flight muscles, there are insufficient levels of accumulation to rescue muscle function. Some support for this possibility comes from the observation that TpnC41C levels in the IFMs of TpnC4 knockouts appeared reduced. On the other hand, TpnC41C levels in the IFMs of RNAi-induced TpnC4 knockdowns did not appear different from controls (Figure 3A, lane 4), and the same can be observed in the results of Eldred et al. (2014).
A third possibility, which we feel to be most likely, is that functional differences in the TpnC proteins affect their ability to rescue muscle function. Herranz et al. (2005) used a bioinformatic approach to predict functional differences between TpnC4 and TpnC41C isoforms and suggested that they would differ in their Ca2+-binding ability. Moreover, Eldred et al. (2014) detected significant differences in isometric tension and power production in the flight muscles expressing TpnC41C that would seem unlikely to result simply from a reduction in TpnC levels. Accordingly, we propose that whereas TpnC4 can substitute for loss of TpnC41C in the TDTs, the reciprocal is not true.
It is also interesting to note that knockdown and knockout TpnC4 mutants jumped less well than wild type, even though TpnC4 was not detected in the TDTs. One explanation for this result is that there are low levels of TpnC4 in the jump muscles that are required for maximal muscle function, although these levels must be very low since we do not detect TpnC4 in the jump muscles of control animals on Western blots. Alternatively, the differences in jumping ability might simply reflect differences in genetic background because different control lines can have significantly different jumping abilities (see, e.g., Cripps et al., 1994).
Requirement of TpnC for myofibril assembly
IFM myofibrillar defects were previously observed in animals that had undergone knockdown of TpnC4 (Eldred et al., 2014; Singh et al., 2014). Since knockdown or knockout of TpnC4 results in up-regulation of TpnC41C expression in the IFMs, the observed defects might arise through one of two mechanisms: either TpnC41C lacks some molecular function that is required for normal IFM myofibril structure, or TpnC41C has an antimorphic effect upon flight muscle structure. Moreover, it was not clear from the previous studies whether muscle structure was evaluated in animals that had already used the muscles (i.e., after emerging from the pupal case). The latter condition might be important because mechanical strain can provide an additional test on myofiber integrity (Firdaus et al., 2015). Thus the contribution of TpnC to myofibril assembly, rather than myofibril maintenance, remained unclear.
Here we demonstrate that myofibrils from double-null pharate adults showed severe defects in muscle structure and function. Are these muscles completely devoid of TpnC protein, which, if true, would indicate a requirement for TpnC in myofibril assembly? This question is germane because expression of TpnC4 and TpnC41C compensates for the absence of each other in the flight and jump muscles. Therefore it might be that in their absence, other TpnC proteins are produced in the double-null IFMs and TDTs, although no other TpnC species were observed in our Western blots. Qiu et al. (2003) demonstrate that the MAC352 antibody that we use cross-reacts with TpnC4 and TpnC41C (as we also show here), as well as with a third TpnC, named TpnC47D. In addition, we observed that MAC352 stains preparations of Drosophila embryos (R.M.C., unpublished observation), in which the predominant isoform is TpnC73F (Herranz et al., 2004). Cross-reactivity with the remaining TpnC isoform, TpnC25D, has not been tested. Thus, if another TpnC gene were expressed in the double-null muscles, because our Western blots were negative for TpnC protein, the only possibility is that TpnC25D is expressed there.
Given that MAC352 also cross-reacts with a number of Lethocerus TpnC species (Qiu et al., 2003), the most reasonable conclusion is that MAC352 cross-reacts with all Drosophila TpnCs. Therefore the IFM and TDT of the double-null mutants that we describe here are devoid of TpnC, making this study one of the first to evaluate the requirement of TpnC to muscle assembly. Our study complements an earlier finding that point mutations in the Caenorhabditis elegans TpnC gene pat-10 cause defects in muscle organization (Terami et al., 1999).
We also note with interest that whereas the TpnC double-knockout animals are largely flightless (70–80% in the N category upon flight testing; Figure 4D), the TpnC4-knockout animals are completely flightless (>95% in the N category; Figure 3C). This indicates that TpnC41C-expressing flight muscles are even less functional than double-null flight muscles, also supporting the idea that TpnC41C might be having an antimorphic effect in the flight muscles. Whether this antimorphic effect is on myofibril assembly or function (or both) remains to be determined.
Mechanisms by which duplicate genes diverge in their expression
One common feature of subfunctionalization is the divergence in expression patterns for duplicate genes. As TpnC41C and TpnC4 are the most recent TpnC gene duplicates in the Drosophila lineage (Herranz et al., 2005), it seems likely that a primordial gene, before duplication, was expressed in both IFM and TDT muscles or their equivalents. After duplication, each gene acquired different patterns of expression and different functions, such that they are now not functionally identical. Bioinformatic studies by Herranz et al. (2005) determined that the Apis mellifera genome contains only a single TpnC gene, which is classified as most similar to Drosophila TpnC41C and TpnC4. It would be interesting to determine whether this gene is expressed in both flight and jump muscles of the honeybee, and, if so, how the sequence of the encoded protein contributes to the unique properties of the different muscles.
Our data demonstrate that a likely mechanism for divergence in expression is through the repression of gene expression in the tissue in which the gene is not normally expressed. There are two lines of evidence for this. First, we show that each gene is capable of expanding its expression into the noncognate muscle in the absence of expression of the other gene. This result indicates that there are still transcriptional mechanisms that can promote broader expression of each gene; however, these mechanisms are suppressed under normal conditions.
Second, we recently characterized the enhancer that controls TDT-specific expression of TpnC41C. In that study, we identified a repressor element that, when mutated, resulted in an expansion of TpnC41C-lacZ expression from the TDT into the flight muscles (Chechenova et al., 2015). The identity of the factor that mediates the repression has not been identified. It is tempting to speculate that transcription factors that promote flight muscle fate, such as Spalt-major, Extradenticle, and Homothorax (Schönbauer et al., 2011; Bryantsev et al., 2012b), might do so at least in part by suppressing jump muscle–specific genes. However, consensus binding sites for those factors are not found in the short repressor element that we defined (Chechenova et al., 2015).
More broadly, a working model for how the divergence in expression of TpnC genes has been achieved, at least for TpnC41C, could be through the acquisition of this repressor element. Our model also predicts that another repressor element must be present in the TpnC4 enhancer that we identify in this study, although, given that the 1.3-kb regulatory region for TpnC4 that we identified has not been further delimited, we do not have insight into the factors that might regulate the expression of this gene.
How is loss of expression of one TpnC gene detected in the cell and transduced into an alteration in gene regulation? The mechanism of this phenomenon, termed paralogous compensation, is yet to be determined and is a relatively understudied process (reviewed in Diss et al., 2014). Defining how this process works at the mechanistic level for TpnC genes studied here might provide significant insight into gene regulatory mechanisms and the processes that account for divergence in expression of duplicated genes. Moreover, investigating this process could generate broader insight into how muscle protein homeostasis is achieved.
MATERIALS AND METHODS
Drosophila methods and fly stocks
All fly stocks and crosses were cultured at 25°C, and crosses with Gal4 driver lines were carried out at 29°C. Targeted gene expression was accomplished using the UAS/Gal4 system (Brand and Perrimon, 1993). Tissue-specific Gal4 driver lines used were Act79B-Gal4 for expression in jump muscles (Bryantsev et al., 2012a), 1151-Gal4 for adult myoblasts (Roy and Vijayraghavan, 1997), and Act88F-Gal4 for flight muscle–specific expression (Bryantsev et al., 2012a). In experiments with down-regulation of single TpnC41C or TpnC4 genes, UAS-RNAi lines 27053 (Bloomington Drosophila Stock Center [BDSC]) and 102031 (Vienna Drosophila RNAi Center [VDRC]) were used, respectively. Line 36303 (BDSC), which carries an attP landing site but no inserted UAS-RNAi element, was used in crosses with Act79B-Gal4 or 1151-Gal4 driver lines to produce control offspring for UAS-TpnC41C RNAi in the jump and flight tests. Line 60100 (VDRC), which similarly carries only attP landing sites, was crossed with Act79B-Gal4 or 1151-Gal4 driver lines, and the progeny was used as control for UAS-TpnC41C RNAi in the jump and flight tests. The laboratory strain w1118 was used as control for all protein samples. In crossing schemes, balancer lines used were w; CyO/In(2LR) Gla, Gla Bc (line 5439, BDSC), w; CyO/Bl; TM2/TM6 (3704, BDSC), and Mef244–5/CyO, Cy Kr>GFP (Lovato et al., 2009).
The stable lines Act79B>UAS-TpnC4 RNAi, 1151>UAS-TpnC4 RNAi, and Act88F>UAS-TpnC41C RNAi were created by crossing the Gal4 and UAS-RNAi lines listed here with balancer-containing lines listed earlier. In experiments with simultaneous down-regulation of TpnC41C and TpnC4, we analyzed female progeny from Act88F>UAS-TpnC41C RNAi crossed with Act79B>UAS-TpnC4 RNAi. As a control for these flies in flight testing, we used female progeny of the cross between Act88F-Gal4 and Act79B-Gal4. The TpnC4F2–3-knockout line was described in Adame et al. (2016). The line CAS-0002, expressing the Cas9 nuclease used for genome editing (Kondo and Ueda, 2013), was obtained from the National Institute of Genetics, Japan.
Drosophila transgenic lines expressing cytoplasmic lacZ or nuclear nlacZ controlled by TpnC41C promoter regions were created earlier (Chechenova et al., 2015). Transgenic lines expressing cytoplasmic lacZ under control of TpnC4 regulatory sequences were created by P-element–mediated transformation (Rubin and Spradling, 1982) of w1118 with the TpnC4(5+6)-CHAB construct (described later). In experiments to assess TpnC4-lacZ expression in TpnC41C KDs, we first crossed female Act79B-Gal4 with male TpnC4-lacZ. The resulting male offspring were crossed with UAS-TpnC41C RNAi adults to obtain TpnC4-lacZ/Act79B>TpnC41C RNAi females, which were used in the experiment. The male progeny of the latter cross were used as controls (TpnC4-lacZ/+). In experiments to assess TpnC41C-lacZ expression in TpnC4 KDs, we crossed lines TpnC41C-lacZ and 1151>UAS-TpnC4 RNAi adults, and female progeny were used in the experiment (TpnC41C-lacZ/1151>TpnC4 RNAi). Male offspring of this cross were used as controls (TpnC41C-lacZ/+).
In the flight and jump tests, 1- to 2-d–old males and females were used. For fly lines carrying two Gal4 drivers (Act88F-Gal4 and Act 79B-Gal4), only female progeny were tested. We analyzed 30–300 animals per experimental set in flight tests and 20–50 animals in jump tests. Flight testing was performed according to Drummond et al. (1991). The jump test was performed as described in Zumstein et al. (2004) with some modifications. Briefly, flies were anesthetized with CO2, and their wings were clipped off; flies were then returned to vials and left at room temperature for at least 20 h to recover. Jump tests were performed on a piece of white paper; flies were stimulated to jump by touch with a paintbrush from the rear, and the points of takeoff and landing were marked with a pencil. The distances of three successful jumps were measured and recorded for each fly, and the average distances and SDs calculated from at least 20 male and female flies per line are plotted on the graphs. Statistical analyses were performed using two-tailed Student's t tests.
CRISPR/Cas9 genome editing
To generate a line with a deletion of the TpnC41C gene, we created two plasmids expressing different single guide RNAs (sgRNAs) by cloning annealed oligonucleotides into the BbsI site of pBFv-U6.2 (Kondo and Ueda 2013). Plasmid pBFv-U6.2-TC41(-652) contained the annealed oligonucleotides 5′-CTTCGCTATTCAGCTAGTGAGAGC-3′ and 5′-AAACGCTCTCACTAGCTGAATAGC-3′ to target a site in the 5′ upstream region of TpnC41C. Plasmid pBFv-U6.2-TC41(+69) contained the annealed oligonucleotides 5′-CTTCGTATTTTCGACTGCTTAGAT-3′ and 5′-AAACATCTAAGCAGTCGAAAATAC-3′ to target a site in the 3′UTR of TpnC41C. These two plasmids were mixed in a 1:1 ratio and injected into embryos of CAS-0002 line following the protocols of Rubin and Spradling (1982). To purify incorporated mutations, we performed consecutive crosses of the progeny with balancer lines. Candidate lines containing homozygous TpnC41C deletions were analyzed by Western blotting and probed for TpnC protein. Among 14 lines tested, only the line TpnC41C∂#2 showed loss of the corresponding protein. Genomic DNA from this line was amplified using the primers 5′-GTGCGTATCAGCCTATTG-3′ and 5′-GTCTCCTCTAGCTCTTGG-3′. Sequencing of the obtained PCR product confirmed deletion of the corresponding genomic region.
To generate a line with double TpnC4/TpnC41 deletion, we first created a TpnC41C∂#2 line that also carried a third chromosome expressing Cas9 under control of vasa regulatory sequences from BDSC line 51324. Embryos from this line were injected with two pBFv-U6.2–based plasmids expressing sgRNA for deletion of the TpnC4 third exon (described in Adame et al., 2016). Injected progeny were analyzed by consecutive crossing with balancer lines and assessed for deletion of TpnC4 using Western blotting. Of all lines tested, we selected two (TpnC4∂#6 and TpnC4∂#25) that were defective in both flight and jumping ability. Sequencing of the affected genomic regions revealed that in both lines we obtained frameshift mutations resulting in premature termination of TpnC4 protein synthesis (Figure 4).
DNA methods
Standard protocols for PCR amplification and either conventional cloning or Gateway recombination were used for plasmid construction. Drosophila genomic DNA was isolated according to Huang et al. (2009) with minor modifications. The TpnC4-lacZ reporter construct was created as follows: a 1.3-kb genomic region containing ∼200 base pairs of the most immediate upstream region of the TpnC4 gene and the entire first intron up to the first coding exon was amplified using the primers C4(5 + 6)-F 5′GGGGACAAGTTTGTACAAAAAAGCAGGCTACAAACAATGAGATCACACA and C4(5 + 6)-R 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTAAGGTAATATCTTCTTCACTG. The underlined sequences within each primer indicate attB sequences compatible with Gateway integration technology (Hartley et al., 2000). The amplified fragment was recombined into pDONR-CHAB, which is a modified pCHAB vector (Thummel and Pirrotta, 1992) that had its multiple cloning site replaced with the Gateway recombination cassette, flanked with attP1 and attP2 sequences, which were subcloned from pDONR221 (Invitrogen). For recombination, we used BP Cloning Mix II (Invitrogen) and followed the manufacturer's protocol. The resulting construct, pC4(5+6)-CHAB, was used for producing transgenic flies. Plasmid pBFv-U6.2, used for genome editing, was obtained from the National Institute of Genetics, Japan.
Protein methods and Western blotting
Dissected IFM and TDT muscles were homogenized in Laemmli SDS sample buffer and heated at 100°C for 5 min, and lysates were centrifuged at 14,000 × g for 2 min. Protein concentration in clarified extracts was measured using the RC DC Protein Assay Kit (Bio-Rad). Protein extracts were loaded on 4–20% gradient TGX gels (Bio-Rad) in a volume corresponding to 9–10 µg of total protein per well. Separated proteins were transferred to nitrocellulose membrane and probed with MAC352 antibody to TpnC (concentration 1:1,000) or MAC276 antibody (concentration 1:100,000) to α-actinin (Abcam). An anti-rat horseradish peroxidase–immunoglobulin G conjugate (Invitrogen) was used as the secondary antibody (concentration 1:1,000), and chemiluminescence signal was detected using the Amersham Biosciences ECL Western blot analysis system (GE Healthcare).
Cryosectioning, immunofluorescence, and histochemistry
Cryosectioning, histochemistry, and immunostaining of frozen sections were performed as described in Morriss et al. (2012). Rabbit anti-Obscurin antibody (used at a concentration of 1:200) was generously provided by Belinda Bullard (University of York, York, United Kingdom). Mouse anti–β-Gal antibody, used at a concentration of 1:1,000, was obtained from Promega. MAC155 antibody to Sallimus (kettin), used at a concentration of 1:400, was obtained from Abcam. Alexa Fluor–conjugated phalloidin and secondary antibodies were obtained from Molecular Probes and used at dilution 1:500 and 1:2000, respectively. Confocal images were produced using a Zeiss LSM780 confocal microscope.
Acknowledgments
This work was supported by National Institutes of Health/National Institute of General Medical Sciences Grant GM061738 to R.M.C. S.M. was supported by National Institutes of Health/National Institute of General Medical Sciences Grant T34 GM008751 and a Stephanie Ruby Undergraduate Fellowship. A.L.B. acknowledges support from internal grants provided by the College of Science and Mathematics and the Office of Vice-President for Research at Kennesaw State University. We also acknowledge technical support from the Molecular Biology Facility and the Cell Biology Facility at the Department of Biology, University of New Mexico, which is supported by National Institutes of Health Grant P20 GM103452 from the Institute Development Award Program of the National Institute of General Medical Sciences.
Abbreviations used:
- BDSC
Bloomington Drosophila Stock Center
- βGal
β-galactosidase
- CRISPR
clustered regularly interspaced short palindromic repeats
- IFM
indirect flight muscle
- KD
knockdown
- KO
knockout
- MHC
myosin heavy-chain
- RNAi
RNA interference
- shRNA
short hairpin RNA
- TDT
tergal depressor of the trochanter muscle
- TpnC
Troponin C
- TpnT
Troponin T
- UAS
upstream activating sequence
- UTR
untranslated region
- VDRC
Vienna Drosophila RNAi Center
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0498) on January 11, 2017.
REFERENCES
- Adame V, Chapapas H, Cisneros M, Deaton C, Deichmann S, Gadek C, Lovato TL, Chechenova MB, Cripps RM. An undergraduate laboratory class using CRISPR/Cas9 technology to mutate Drosophila genes. Biochem Mol Biol Educ. 2016;3:263–275. doi: 10.1002/bmb.20950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DL, Leinwand LA. Postnatal myosin heavy chain isoform expression in normal mice and mice null for IIb or IId myosin heavy chains. Dev Biol. 2001;229:383–395. doi: 10.1006/dbio.2000.9974. [DOI] [PubMed] [Google Scholar]
- Benoist P, Mas JA, Marco R, Cervera M. Differential muscle-type expression of the Drosophila troponin T gene. A 3-base pair microexon is involved in visceral and adult hypodermic muscle specification. J Biol Chem. 1998;273:7538–7546. doi: 10.1074/jbc.273.13.7538. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Bryantsev AL, Baker PW, Lovato TL, Jaramillo MS, Cripps RM. Differential requirements for Myocyte enhancer factor-2 during adult myogenesis in Drosophila. Dev Biol. 2012a;361:191–207. doi: 10.1016/j.ydbio.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryantsev AL, Duong S, Brunetti TM, Chechenova MB, Lovato TL, Nelson C, Shaw E, Uhl JD, Gebelein B, Cripps RM. Extradenticle and Homothorax control adult muscle fiber identity in Drosophila. Dev Cell. 2012b;23:664–673. doi: 10.1016/j.devcel.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chechenova MB, Maes S, Cripps RM. Expression of the Troponin C at 41C gene in adult Drosophila tubular muscles depends upon both positive and negative regulatory inputs. PLoS One. 2015;10:e0144615. doi: 10.1371/journal.pone.0144615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 2008;9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- Cripps RM, Becker KD, Mardahl M, Kronert WA, Hodges D, Bernstein SI. Transformation of Drosophila melanogaster with the wild-type myosin heavy-chain gene: rescue of mutant phenotypes and analysis of defects caused by overexpression. J Cell Biol. 1994;126:689–699. doi: 10.1083/jcb.126.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diss G, Ascencio D, DeLuna A, Landry CR. Molecular mechanisms of paralogous compensation and the robustness of cellular networks. J Exp Zool. 2014;322B:488–499. doi: 10.1002/jez.b.22555. [DOI] [PubMed] [Google Scholar]
- Domingo A, Gonzalez-Jurado J, Maroto M, Diaz C, Vinos J, Carrasco C, Cervera M, Marco R. Troponin-T is a calcium-binding protein in insect muscle: in vivo phosphorylation, muscle-specific isoforms and developmental profile in Drosophila melanogaster. J Muscle Res Cell Motil. 1998;19:393–403. doi: 10.1023/a:1005349704790. [DOI] [PubMed] [Google Scholar]
- Drummond DR, Hennessey ES, Sparrow JC. Characterisation of missense mutations in the Act88F gene of Drosophila melanogaster. Mol Gen Genet. 1991;226:70–80. doi: 10.1007/BF00273589. [DOI] [PubMed] [Google Scholar]
- Eldred CC, Katzemich A, Patel M, Bullard B, Swank DM. The roles of troponin C isoforms in the mechanical function of Drosophila indirect flight muscle. J Muscle Res Cell Motil. 2014;35:211–223. doi: 10.1007/s10974-014-9387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott CJH, Brunger HL, Stark M, Sparrow JC. Direct measurement of the performance of the Drosophila jump muscle in whole flies. Fly. 2007;1:68–74. doi: 10.4161/fly.3979. [DOI] [PubMed] [Google Scholar]
- Firdaus H, Mohan J, Naz S, Arathi P, Ramesh SR, Nongthomba U. A cis-regulatory mutation in Troponin-I of Drosophila reveals the importance of proper stoichiometry of structural proteins during muscle assembly. Genetics. 2015;200:149–165. doi: 10.1534/genetics.115.175604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyrberg EA, Fyrberg CC, Biggs JR, Saville D, Beall CJ, Ketchum A. Functional nonequivalence of Drosophila actin isoforms. Biochem Genet. 1998;36:271–287. doi: 10.1023/a:1018785127079. [DOI] [PubMed] [Google Scholar]
- Hartley JL, Temple GF, Brasch MA. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz R, Diaz-Castillo C, Nguyen TP, Lovato TL, Cripps RM, Marco R. Characterization of the whole Troponin C gene repertoire in Drosophila melanogaster. Gene Exp Patterns. 2004;4:183–190. doi: 10.1016/j.modgep.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Herranz R, Mateos J, Marco R. Diversification and independent evolution of troponin C genes in insects. J Mol Evol. 2005;60:31–44. doi: 10.1007/s00239-004-0031-x. [DOI] [PubMed] [Google Scholar]
- Huang AM, Rehm EJ, Rubin GM. Quick preparation of genomic DNA from Drosophila. Cold Spring Harb Protoc. 2009 doi: 10.1101/pdb.prot5198. 2009, pdb.prot5198. [DOI] [PubMed] [Google Scholar]
- Katju V. In with the old, in with the new: the promiscuity of the duplication process engenders diverse pathways for novel gene creation. Int J Evol Biol. 2012 doi: 10.1155/2012/341932. 2012, 341932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Ueda R. Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics. 2013;195:715–721. doi: 10.1534/genetics.113.156737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber RL. Skeletal Muscle Structure, Function, and Plasticity. Baltimore, MD: Lippincott Williams & Wilkins; 2010. [Google Scholar]
- Lovato TL, Adams MM, Baker PW, Cripps RM. A molecular mechanism of temperature sensitivity for mutations affecting the Drosophila muscle regulator Myocyte enhancer factor 2. Genetics. 2009;183:107–117. doi: 10.1534/genetics.109.105056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss GR, Bryantsev AL, Chechenova M, LaBeau EM, Lovato TL, Ryan KM, Cripps RM. Analysis of skeletal muscle development in Drosophila. Methods Mol Biol. 2012;798:127–152. doi: 10.1007/978-1-61779-343-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu F, Lakey A, Agianian B, Hutchings A, Butcher GW, Labeit S, Leonard K, Bullard B. Troponin C in different insect muscle types: identification of an isoform in Lethocerus, Drosophila and Anopheles that is specific to asynchronous flight muscle in the adult insect. Biochem J. 2003;371:811–821. doi: 10.1042/BJ20021814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, VijayRaghavan K. Homeotic genes and the regulation of myoblast migration, fusion, and fibre-specific gene expression during adult myogenesis in Drosophila. Development. 1997;124:3333–3341. doi: 10.1242/dev.124.17.3333. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Sartorius CA, Lu BD, Acakpo-Satchivi L, Jacobsen RP, Byrnes WC, Leinwand LA. Myosin heavy chains IIA and IId are functionally distinct in the mouse. J Cell Biol. 1998;141:943–953. doi: 10.1083/jcb.141.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönbauer C, Distler J, Jährling N, Radolf M, Dodt HU, Frasch M, Schnorrer F. Spalt mediates an evolutionarily conserved switch to fibrillar muscle fate in insects. Nature. 2011;479:406–409. doi: 10.1038/nature10559. [DOI] [PubMed] [Google Scholar]
- Singh SH, Kumar P, Ramachandra NB, Nongthomba U. Roles of the troponin isoforms during insect flight muscle development in Drosophila. J Genet. 2014;93:379–388. doi: 10.1007/s12041-014-0386-8. [DOI] [PubMed] [Google Scholar]
- Terami H, Williams BD, Kitamura S, Sakube Y, Matsumoto S, Doi S, Obinata T, Kagawa H. Genomic organization, expression and analysis of the Troponin C gene pat-10 of Caenorhabditis elegans. J Cell Biol. 1999;146:193–202. doi: 10.1083/jcb.146.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel CS, Pirrotta V. New pCaSpeR P element vectors. Drosoph Inf Serv. 1992;71:150. [Google Scholar]
- Weiss A, Leinwand LA. The mammalian myosin heavy chain gene family. Annu Rev Cell Dev Biol. 1996;12:417–439. doi: 10.1146/annurev.cellbio.12.1.417. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dean AM, Brunet F, Long M. Evolving protein functional diversity in new genes of Drosophila. Proc Natl Acad Sci USA. 2004;101:16246–16250. doi: 10.1073/pnas.0407066101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumstein N, Forman O, Nongthomba U, Sparrow JC, Elliott CJH. Distance and force production during jumping in wild-type and mutant Drosophila melanogaster. J Exp Biol. 2004;207:3515–3522. doi: 10.1242/jeb.01181. [DOI] [PubMed] [Google Scholar]