
Fig. 5.
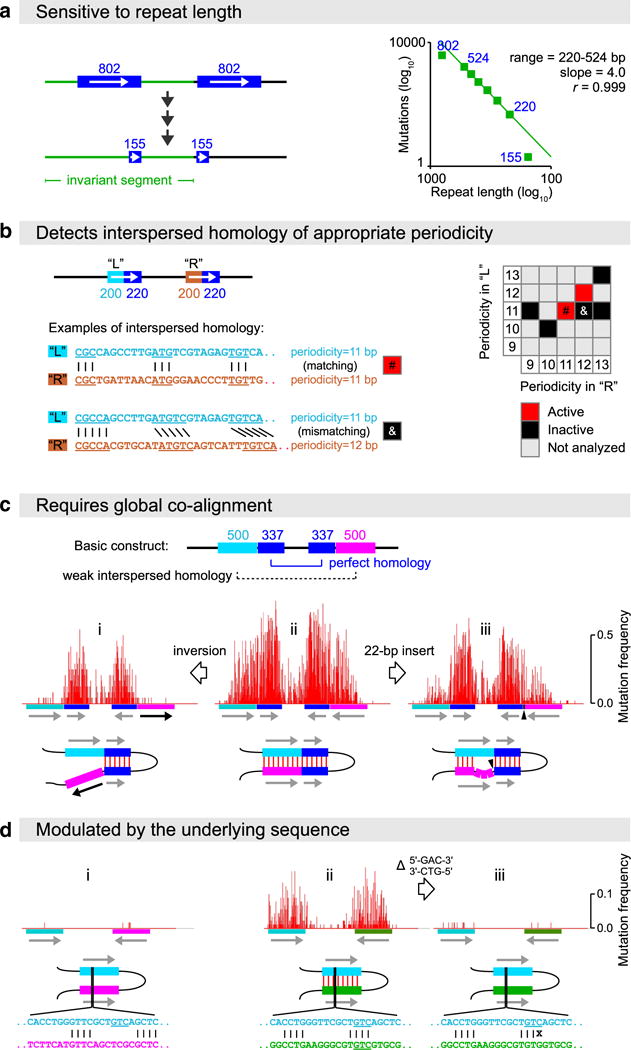
Elucidating the homology requirements for RIP. a RIP mutation of closely positioned direct repeats of graded lengths (802, 524, 460, 400, 337, 279, 220, and 155 base-pairs) as reported by Gladyshev and Kleckner (2014). C-to-T and G-to-A mutations are counted together in the longest continuous region shared by all the repeat constructs (the invariant segment, shown in green). b Short interspersed islands of homology can be detected by RIP only when arrayed with an appropriate matching periodicity along the participating DNA segments (Gladyshev and Kleckner 2014). c Efficient recognition of homology for RIP requires the global co-alignment of participating DNA segments (Gladyshev and Kleckner 2014). The basic construct (ii) includes a 337-bp segment of perfect homology (dark blue) and an adjoining 500-bp segment of weak interspersed homology, in which 4-bp homologous units are arrayed with a matching periodicity of 11 base-pairs (light blue/magenta). Disrupting their global alignment significantly attenuates RIP. d When homologous interactions are weakened, the underlying DNA sequence plays a prominent role. i a 500-bp segment of interspersed homology, same as in (c), does not trigger much RIP by itself. ii another instance of the same homology pattern (featuring homologous units of 4 base-pairs arrayed with a 11-bp periodicity over the same total length of 500 base-pairs) triggers a nearly 100-fold stronger RIP response. This effect is attributed to the inclusion of 5′-GAC-3′/5′-GTC-3′ triplets (underlined) into the homologous units in (ii) but not (i) or (iii) (Gladyshev and Kleckner 2016)