

Abstract
Chinese liquor is one of the world's oldest distilled alcoholic beverages and an important commercial fermented product in China. The Chinese liquor fermentation process has three stages: making Daqu (the starter), stacking fermentation on the ground, and liquor fermentation in pits. We investigated the bacterial diversity of Maotai and Guotai Daqu and liquor fermentation using high-throughput sequencing of the V4 hypervariable region of the 16S rRNA gene. A total of 70,297 sequences were obtained from the Daqu samples and clustered into 17 phyla. The composition of the bacterial communities in the Daqu from these two soy sauce aroma-style Chinese liquors was the same, although some bacterial species changed in abundance. Between the Daqu and liquor fermentation samples, 12 bacterial phyla increased. The abundance of Lactobacillus and Pseudomonas increased in the liquor fermentation. This study has used high-throughput sequencing to provide new insights into the bacterial composition of the Chinese liquor Daqu and fermentation. Similarities in the distribution of bacteria in the soy sauce aroma-style Chinese liquors Daqu suggest that the abundance of bacteria might be generally concerned to other liquor.
1. Introduction
Fermentation is a well-known ancient technique that uses microorganism to process and preserve food. Chinese liquor is one of the six well-known distillates in the world. It has a long history of production and is produced through unique a fermentation process. It is typically produced from cereals, such as sorghum and rice, via the solid-state fermentation of grain. Chinese liquor has five main styles: strong aroma, light aroma, soy sauce aroma, sweet honey, and miscellaneous. Maotai and Guotai are Chinese liquors famous for their soy sauce aroma. Maotai-flavor liquor is as symbolic a drink in China as whisky is in Scotland and brandy in France [1–3].
Microorganisms usually do the main work of degrading biopolymers, producing alcohol, and forming aromatic compounds. The microbial community of Chinese liquor has been analyzed in previous studies using culture-dependent and culture-independent methods. Culture-dependent studies of the microbial community have used methods such as isolation and enumeration on selective media [4–6]. Culture-independent studies have used methods such as polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) [7–9], amplified fragment length polymorphism [10], and 16S RNA or 26S RNA clone libraries [11].
Chinese liquors are typically produced via solid-state fermentation using a natural fermentation starter termed Daqu. The Daqu starter has long been believed to play a key role in the fermentation of Chinese liquor [12]. Maotai and Guotai liquors share the same unique and complicated spontaneous fermentation process, which includes making Daqu (the starter), stacking fermentation, and liquor fermentation (Figure 1) [13]. In the Daqu-making stage, the maximum temperature of Daqu reaches approximately 65°C, which is typical of high-temperature Daqu. Several studies have shown the diversity of the microbial community in Daqu or fermentation, especially the yeast and fungi [2, 3, 13, 14]. Therefore, it was of interest to analyze the microbial community in Daqu and find the bacteria important to Daqu properties. Little is known regarding the bacterial community composition in Maotai Daqu.
Figure 1.

Flow sheet for making soy sauce aroma-style flavor liquor.
In previous research, culture-independent cloning methods were used for the analysis of microbial communities in Daqu or Chinese liquor fermentation. Here, we applied high-throughput sequencing of the V4 hypervariable region of the 16S rRNA gene to examine in-depth microbial communities from soy sauce aroma-style Chinese liquor to gain insight into the specific fermentative microorganisms. The main objective of this study was to (i) analyze the composition of the microbial communities in the Daqu of soy sauce aroma-style Chinese liquor; (ii) compare them with two different Daqu of soy sauce aroma-style Chinese liquor; and (iii) compare the composition of the microbial communities in the Daqu with those in the liquor fermentation.
2. Materials and Methods
2.1. Samples of Daqu and Fermented Grains
Sampling was performed in two different liquor production factories (Maotai and Guotai) in Guizhou Province, China. Three pits of Daqu samples from Maotai and Guotai were taken in the same years from 2011 to 2013. Samples of fermented grains were obtained from a randomly selected fermentation batch at the same time points (the fourth liquor fermentation) in 2013. The sampling was randomly selected from the mixture of the upper, middle, and bottom stacked layers. All samples were transferred to sterile bags, sealed, and stored at −80°C.
2.2. DNA Extraction and Quantitation
10 g samples were suspended in 50 mL of sterile PBS buffer (0.1 mol/L, pH 7.2–7.4) and vortexed for 15 min at ambient temperature. The suspension was then centrifuged (500 ×g, 4°C) for 5 min, and the pellet was washed three times in PBS buffer. The supernatants were also collected and centrifuged (10,000 ×g, 4°C) for 10 min; the resulting pellets were washed three times in PBS buffer. The resulting pellets were resuspended in PBS buffer and stored at −20°C until DNA extraction. DNA was extracted according to the method of Wang et al. [2, 3].
2.3. MiSeq Sequencing of 16S rRNA Gene Amplicons
The communities of bacteria were analyzed using Illumina MiSeq sequencing of the 16S rRNA gene V4 region amplicons, which can yield accurate taxonomic information and shows few biases for various bacterial taxa [15]. The V4 region of the 16S rRNA gene was amplified with the primer set 515f (5′-GTGCCAGCMGCCGCGGTAA-3′)/806r (5′-GGACTACHVGGGTWTCTAAT-3′), and all PCR amplifications were conducted in triplicate for each sample. This short targeted gene region can provide sufficient resolution for the accurate taxonomic classification of microbial sequences [16]. The initial 10 cycles of PCR amplification were performed. The products were then purified with Agencourt® Ampure® XP (Beckman Coulter, Inc., CA, USA) and used as a template for the second PCR amplification of 20 cycles using the same primer set; however, the reverse primer contained the appropriate adapters and different barcodes to distinguish samples. PCR products were visualized using 1% agarose gels stained with ethidium bromide, and negative controls were always performed to confirm the absence of contamination. True positive amplicons were quantified using a PicoGreen dsDNA Assay kit (Invitrogen, CA, USA), combined equally, and then gel purified. The DNA library was sequenced using the Illumina MiSeq platform according to the manufacturer's instructions [17]. Sequences were analyzed with the QIIME [18] software package and UPARSE pipeline [19]. Quality filtering and processing of MiSeq reads were conducted by QIIME. Default settings for Illumina processing in QIIME were used (r = 3 p = 0.75 total read length; q = 3; n = 0) (p: minimum number of consecutive high-quality base calls to retain read; r: maximum number of consecutive low-quality base calls allowed before truncating a read; n: maximum number of ambiguous (N) characters allowed in a sequence; q: last quality score considered low quality). We ultimately obtained 10,083–17,973 high-quality sequences from the Daqu samples and 12,418–15,302 sequences from the liquor fermentation samples. Then we use UPARSE pipeline to picking operational taxonomic units (OTUs) through making OTU table. Sequences were assigned to OTUs at 97% similarity. We pick representative sequences for each OTU and use the RDP classifier [20] to assign taxonomic data to each representative sequence.
2.4. Statistical Analysis
The datasets generated using 16S rRNA gene sequencing (OTU composition) were further analyzed with the following statistical methods: (i) α-diversity comparison (Chao value and Shannon index) and β-diversity comparison (unweighted UniFrac distances and weighted UniFrac distances); (ii) hierarchical clustering based on the relative abundance of bacteria (the specificity measure (SPM)) and Euclidean distance and complete linkage being used in this clustering analysis; and (iii) significance tests based on unpaired Student's t-tests and Wilcoxon rank-sum test to identify differences between any two compared objects. All statistical analyses described above were performed using the R package vegan/gplot. To quantitatively estimate the relative abundance of a bacterial genus in a sample, the specificity measure (SPM) [21] was introduced as follows and was used in a Heatmap. Each abundance of a bacterial genus was first transformed into vector X:
| (1) |
where n is the number of samples in a profile. At the same time, vector Xi was created to represent the abundance of a bacterial genus in sample i:
| (2) |
The SPM of a bacterial genus in a sample was then determined by calculating the cosine value of intersection angle θ between vectors Xi and X in high-dimension feature space:
| (3) |
where |Xi| and |X| are the length of vectors Xi and X, respectively. The value of SPM ranges from 0 to 1.0. A SPM value close to 1.0 indicates the major contribution of bacterial abundance in a designated sample (e.g., vector Xi) against that in all samples (vector X).
3. Results
3.1. Composition of Bacterial Communities in Different Daqu Determined Using High-Throughput Sequencing
A total of 70,297 high-quality sequences (approximately 260 bp) were obtained from the 6 Daqu samples, with an average of 11,716 sequences per sample. There were no significant differences between the Maotai Daqu high-quality sequences with the Guotai Daqu (t-test, p = 0.530). The OTUs detected in the Guotai Daqu largely overlapped with the Maotai Daqu; the number of OTUs in each of the 6 samples (average 5,418) was similar (t-test, p = 0.691). These OTUs clustered into 17 phyla, 55.5% of which, on average, were classified as Proteobacteria, followed by Firmicutes 39.1% (Figure 2). In terms of relative abundance, Proteobacteria (mainly Gammaproteobacteria) were the most abundant bacterial phylum in the Chinese liquor Daqu (accounting for 37.16–64.07% of the different Daqu samples), and Firmicutes (mainly Bacilli) were also abundant (accounting for 28.69–61.80%; Figure 3). There were no significant differences (t-test, p > 0.05) in the most bacterial relative abundances with two Daqu samples. However, Actinobacteria and Bacteroidetes were significantly different (t-test, p < 0.05). Of all the bacterial families, Bacillaceae, Enterobacteriaceae, and Pseudomonadaceae were the most abundant.
Figure 2.
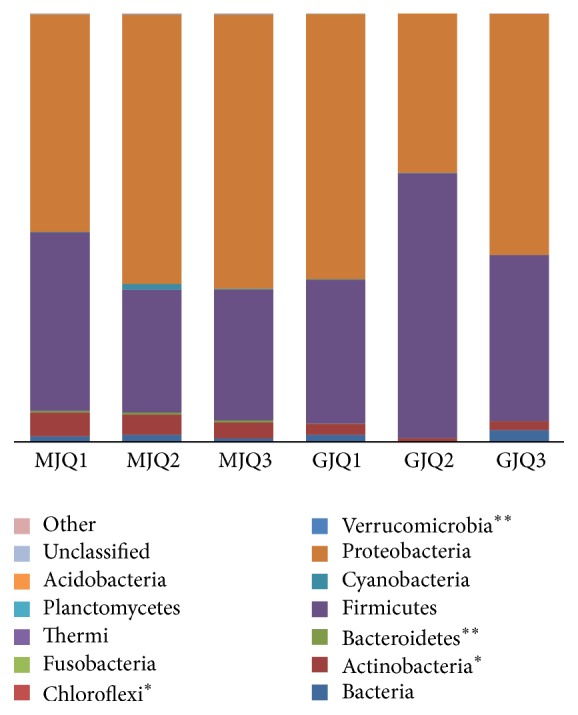
The percentages of OTUs assigned to major bacterial phyla (≥0.01%). Three pits of Maotai-liquor Daqu samples in 2011 to 2013 years are abbreviated as MJQ1, MJQ2, and MJQ3, respectively, and the three Guotai-liquor Daqu samples are GJQ1, GJQ2, and GJQ3 (t-test, ∗p < 0.05, ∗∗p < 0.01). An asterisk indicates a significant difference with major bacterial phyla between the Maotai-liquor Daqu and Guotai-liquor Daqu.
Figure 3.

Abundance of the dominant taxonomy (relative abundance of phylum > 0.1% and of family > 1.0%) determined by 16S rRNA gene sequencing. Numbers in cells are percentages of relative abundance, highest abundances are red, middle values are in white, and lowest values are in blue. An asterisk indicates a significant different between the Guotai-liquor Daqu and the Maotai-liquor Daqu. (t-test, ∗p < 0.05;∗∗p < 0.01).
The Chao value and Shannon index, which reflect the α-diversity of bacterial communities, showed no significant differences between the Guotai Daqu and the Maotai Daqu (t-test, p = 0.140 and p = 0.117, and Wilcoxon rank-sum test, p = 0.210 and p = 0.213, resp.). The unweighted UniFrac distances and weighted UniFrac distances which reflect the β-diversity of bacterial communities showed no significant differences between the Guotai Daqu and the Maotai Daqu (Wilcoxon rank-sum test, p = 0.128 and p = 0.462) (Table 1).
Table 1.
α-Diversity and β-diversity of bacterial communities. (a) The Chao value and Shannon index; (b) the unweighted UniFrac distances and weighted UniFrac distances. The unweighted UniFrac distances are indicated by ∗∗, and weighted UniFrac distances are indicated by ∗.
(a).
| a-diversity | MJQ1 | MJQ2 | MJQ3 | GJQ1 | GJQ2 | GJQ3 | p(t−) | p(w−) |
|---|---|---|---|---|---|---|---|---|
| Chao value | 3655.8 | 3872.88 | 3427.75 | 3038.48 | 3067.18 | 3608.63 | 0.140 | 0.210 |
| Shannon index | 6.782 | 6.876 | 6.863 | 6.199 | 5.944 | 6.818 | 0.117 | 0.213 |
(b).
| MJQ1 | MJQ2 | MJQ3 | GJQ1 | GJQ2 | GJQ3 | |
|---|---|---|---|---|---|---|
| MJQ1 | 0 | 0.119∗ | 0.187∗ | 0.200∗ | 0.190∗ | 0.183∗ |
| MJQ2 | 0.315∗∗ | 0 | 0.201∗ | 0.177∗ | 0.178∗ | 0.189∗ |
| MJQ3 | 0.397∗∗ | 0.418∗∗ | 0 | 0.171∗ | 0.141∗ | 0.181∗ |
| GJQ1 | 0.441∗∗ | 0.434∗∗ | 0.404∗∗ | 0 | 0.081∗ | 0.172∗ |
| GJQ2 | 0.457∗∗ | 0.465∗∗ | 0.411∗∗ | 0.302∗∗ | 0 | 0.171∗ |
| GJQ3 | 0.465∗∗ | 0.469∗∗ | 0.396∗∗ | 0.327∗∗ | 0.308∗∗ | 0 |
However, the 14 genera whose abundances were greater than 0.01% of the total bacteria in the Guotai and the Maotai were significantly different (t-test, p < 0.05; Figure 4). In terms of relative abundance, 13 bacterial genera were significantly higher in the Maotai Daqu, and only Lactobacillus was significantly higher in the Guotai Daqu. Although there were no significant differences in the number of detected bacterial taxa, the samples clustered into two groups based on the relative abundances of the major genera, indicating clear differences between Maotai Daqu and Guotai Daqu (Figure 4). Including Thermoactinomyces, Saccharopolyspora, Acinetobacter, and Pseudomonas, 17 bacterial genera in the Maotai Daqu were higher, and 6 genera bacterial (from Corynebacterium to Sebaldella) in the Guotai Daqu were higher.
Figure 4.

Heatmap showing the differences among the six investigated communities based on the abundance of the domain genura. (relative abundance > 0.01%) An asterisk indicates a significant different between Goutai and Maotai Daqu. (t-test, ∗p < 0.05, ∗∗p < 0.01).
3.2. Composition of Bacterial Communities in Liquor Fermentation Determined Using High-Throughput Sequencing
A total of 27,720 high-quality sequences were obtained from the 2 liquor fermentation samples, corresponding to 10,200 OTUs. These OTUs clustered into 27 phyla, the most abundant of which was Proteobacteria (67.04% and 70.76%), followed by Firmicutes (20.74% and 24.11%, Figure 5). The OTUs clustered into 17 phyla in the Daqu sample and then in the liquor fermentation sample plus an additional 10 phyla. Twelve bacterial phyla were more abundant in the liquor fermentation process, including Acidobacteria, Bacteroidetes, Chlorobi, Chloroflexi, Proteobacteria, and Planctomycetes. However, Actinobacteria and Firmicutes were less abundant in the liquor fermentation process relative to the Daqu samples.
Figure 5.
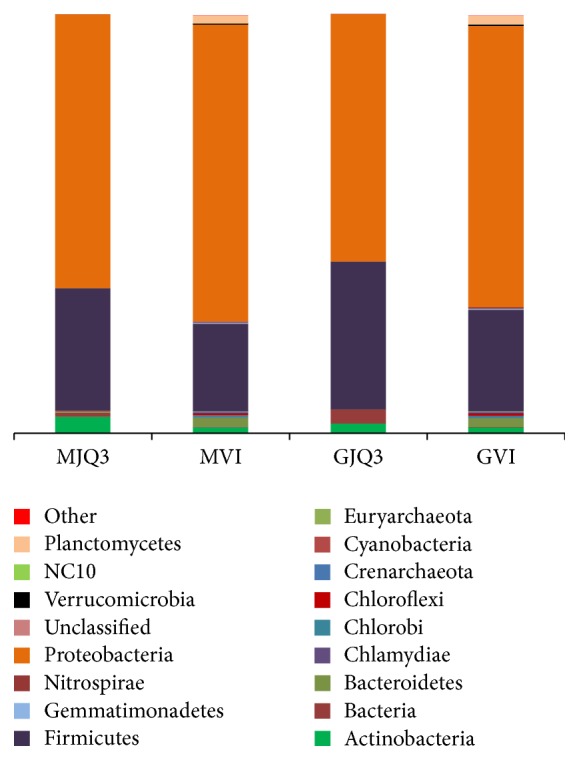
The percentages of OTUs assigned to major bacterial phyla (≥0.01%). The Maotai-liquor fermentation sample are abbreviated as MVI, and the Guotai-liquor fermentation as GVI. The Daqu sample in 2013 are abbreviated as MJQ3 and GJQ3.
These OTUs clustered into 314 genera, and the genera whose abundances were greater than 0.01% of the total bacteria were different between the Daqu and liquor fermentation samples (Figure 6). Lactobacillus abundance increased from an average of 1.35% to 19.78% in the liquor fermentation and Pseudomonas from 5.07% to 33.52%. In the liquor fermentation process, 18 bacterial genera increased, including Pseudomonas, Lactobacillus, Agrobacterium, Rhodoplanes, Ochrobactrum, and Nitrospira, and 5 bacterial genera decreased, Lactococcus, Enterococcus, Saccharopolyspora, Bacillus, and Pediococcus.
Figure 6.

Heatmap showing the difference among the liquor fermentation based on the abundance of the domain genera. (relative abundance > 0.01%).
4. Discussion
Prior studies on the Chinese liquor fermentation process have focused on a limited number of isolated samples or microbial diversity examined using 16S rRNA gene library analysis and PCR-DGGE. Previously, the higher bacterial diversity as measured by the Shannon index (H′ = 1.19) was found in a high-temperature Daqu (9-H-S-W) [11]. In the current study, the average Shannon index of the Daqu was H' = 6.58. Using high-throughput sequencing of the V4 hypervariable region of the 16S rRNA gene to examine microbial communities in-depth, the abundance of the bacterial community in Daqu was higher than previously reported [2, 3, 6, 9]. Some of the bacterial genera observed in our study were not previously reported.
Knowledge of the microbiota of Chinese liquor fermentation is still far from complete, especially the microbiota of Daqu. Therefore, this study was initiated to understand the composition of the microbial community in two representative soy sauce aroma-style Chinese liquors. In the soy sauce aroma-style Chinese liquor, Daqu is made from ground wheat and is produced in Guizhou Province in China, applying high-temperature fermentation conditions for Daqu production. It is expected that the relative abundance of several of the microorganisms identified in the Daqu correlate with different Chinese liquors. Several studies indicated that the bacterial community in Daqu is affected by certain factors, including raw materials, environmental conditions (e.g., soil and air), moisture content, oxygen condition and “mother Daqu” (Daqu that was produced 1 year ago). Maotai-flavor and Guotai-flavor liquor have the same unique, complicated spontaneous fermentation process and the same environmental conditions. Their Daqu may thus have the same microbial communities. The Chao value and Shannon index, which reflect the α-diversity of bacterial communities, of the Daqu samples were not significantly different between the Guotai Daqu and the Maotai Daqu. Furthermore, the same bacterial species were present in the Daqu of both liquors. However, the abundance of 13 bacterial genera in the Maotai Daqu was significantly higher than in the Guotai Daqu: Thermoactinomyces, Erwinia, Saccharopolyspora, Acinetobacter, Planifilun, Brachybacterium, Acetobacter, Akkermansia, Saccharomonospora, Sphingobacterium, Desemzia, Amycolatopsis, and Halomonas. Only the abundance of Lactobacillus was significantly higher in the Guotai Daqu than in the Maotai Daqu (Figure 4). Based on our analysis of different Daqu samples from soy sauce aroma-style Chinese liquor, we concluded that the composition of the bacterial communities in the different Chinese liquor were the same. Nevertheless, some bacterial species have significantly different abundances between the different Daqu samples. Some types of Daqu contained highly similar bacteria species, whereas some bacterial species abundances were significantly different. The relative abundance of bacteria may be important for the Daqu.
Several studies mentioned the importance of lactic acid bacteria (LAB) during the production of Daqu, including Enterococcus, Lactobacillus, Leuconostoc, Pediococcus, Streptococcus, and Weissella. In this study, various genera of lactic acid bacteria (LAB) were identified in Maotai and Guotai Daqu. In the current study, LAB were only found at high abundance during the beginning of the Fenjiu-Daqu production process [22], and LAB except Weissella were found in low abundance [11]. We observed a high abundance of Weissella and Pediococcus, and the abundance of LAB except Lactobacillus was not significantly different. Some species of Lactobacillus inhibit the growth of Bacillus [23]. However, in this study, there were no significant differences in Bacillus between the two Daqu samples. Bacillus is a well-known producer of proteases and amylases [24] and produces more than 70 metabolites, most of which are flavor compounds and flavor precursors (Yan et al. 2013). The production of these molecules is important for the aroma of fermented products. This explained that the abundance of Bacillus in Maotai and Guotai Daqu was not significantly different, although Lactobacillus was. In the liquor fermentation process, the abundance of LAB excluding Lactobacillus decreased, and Lactobacillus abundance increased from the average 1.35% to 19.78%. With the dramatic increase of Lactobacillus, the abundance of Bacillus decreased. At the initial stage of stacking fermentation, Lactobacillus quickly propagates and becomes the main bacteria [2, 3].
In this study, the abundance of the phyla Actinobacteria, Bacteria, and Firmicutes decreased between both types of Daqu and the fermentation processes, while Proteobacteria and others increased. The genus Lactobacillus of the family Lactobacillaceae and the Pseudomonas of the family Pseudomonadaceae increased. When the fermentation began, several chemicals were enriched, making the habitat suitable for some bacteria, such as Lactobacillus and Pseudomonas. During the Fen liquor fermentation process, the bacterial community diversity decreases, and only the family Lactobacillaceae increases [25]. The family Lactobacillaceae is a contributor to the fermentation reaction.
Different microbial communities in Daqu facilitate the selection of starters for creation of unique flavors. Further research is required to gain deeper insight into the microbial communities of the different types of Daqu and the function of unknown microorganisms in these communities. This work may increase liquor producers' understanding of the bacterial community in Daqu, and the relative abundance of bacteria in Daqu may be another important topic for liquor production.
Acknowledgments
This work was supported by the Social Development Project of Guizhou province (sz20113068, sy20123035), the International Cooperation Project of Guizhou province (G20137020), Major Projects of Guizhou Education Department (2012005), Program for Changjiang Scholars and Innovative Research Team in University of China (IRT1227), and Science and Technology Platform Construction Project of Guizhou Province (Z20114005).
Competing Interests
The authors declare that they have no competing interests.
Authors' Contributions
Jing Tang and Xiaoxin Tang contributed equally to this work.
References
- 1.Fan W., Qian M. C. Characterization of aroma compounds of Chinese “Wuliangye” and “Jiannanchun” liquors by aroma extract dilution analysis. Journal of Agricultural and Food Chemistry. 2006;54(7):2695–2704. doi: 10.1021/jf052635t. [DOI] [PubMed] [Google Scholar]
- 2.Wang C.-L., Shi D.-J., Gong G.-L. Microorganisms in Daqu: a starter culture of Chinese Maotai-flavor liquor. World Journal of Microbiology and Biotechnology. 2008;24(10):2183–2190. doi: 10.1007/s11274-008-9728-0. [DOI] [Google Scholar]
- 3.Wang H.-Y., Zhang X.-J., Zhao L.-P., Xu Y. Analysis and comparison of the bacterial community in fermented grains during the fermentation for two different styles of Chinese liquor. Journal of Industrial Microbiology and Biotechnology. 2008;35(6):603–609. doi: 10.1007/s10295-008-0323-z. [DOI] [PubMed] [Google Scholar]
- 4.Li C., Mu L., Wang J. Y., Lei Z. H., Chen J. Y., Han B. Z. Physiochemical and microbiological analysis of Fen-type Daqu. China Brew. 2009;1:140–142. [Google Scholar]
- 5.Zheng X.-W., Yan Z., Nout M. J. R., et al. Microbiota dynamics related to environmental conditions during the fermentative production of Fen-Daqu, a Chinese industrial fermentation starter. International Journal of Food Microbiology. 2014;182-183:57–62. doi: 10.1016/j.ijfoodmicro.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Zheng X.-W., Yan Z., Han B.-Z., et al. Complex microbiota of a Chinese ‘Fen’ liquor fermentation starter (Fen-Daqu), revealed by culture-dependent and culture-independent methods. Food Microbiology. 2012;31(2):293–300. doi: 10.1016/j.fm.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Wang H.-Y., Gao Y.-B., Fan Q.-W., Xu Y. Characterization and comparison of microbial community of different typical Chinese liquor Daqus by PCR-DGGE. Letters in Applied Microbiology. 2011;53(2):134–140. doi: 10.1111/j.1472-765X.2011.03076.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L., Wu C., Ding X., Zheng J., Zhou R. Characterisation of microbial communities in Chinese liquor fermentation starters Daqu using nested PCR-DGGE. World Journal of Microbiology and Biotechnology. 2014;30(12):3055–3063. doi: 10.1007/s11274-014-1732-y. [DOI] [PubMed] [Google Scholar]
- 9.Zheng Y., Zheng X.-W., Han B.-Z., Han J.-S., Nout M. J. R., Chen J.-Y. Monitoring the ecology of Bacillus during Daqu incubation, a fermentation starter, using culture-dependent and culture-independent methods. Journal of Microbiology and Biotechnology. 2013;23(5):614–622. doi: 10.4014/jmb.1211.11065. [DOI] [PubMed] [Google Scholar]
- 10.Yan L. C., Zhang S. C., Ma X. W., Tang E. J., Huang Z. X., Chen Y. Q. Phylogenetic analysis of 16S rDNA sequence and PCR: RFLP of Bacillus from Fumao-flavor Daqu. Biotechnology. 2012;22:54–58. [Google Scholar]
- 11.Zheng X.-W., Yan Z., Nout M. J. R., et al. Characterization of the microbial community in different types of Daqusamples as revealed by 16S rRNA and 26S rRNA gene clone libraries. World journal of microbiology & biotechnology. 2015;31(1):199–208. doi: 10.1007/s11274-014-1776-z. [DOI] [PubMed] [Google Scholar]
- 12.Zhang W. X., Qiao Z. W., Xiang W. L., et al. Micro-ecological research progress of the cellar of Chinese flavour liquor. Liquor Making. 2004;31:31–35. [Google Scholar]
- 13.Wu Q., Chen L., Xu Y. Yeast community associated with the solid state fermentation of traditional Chinese Maotai-flavor liquor. International Journal of Food Microbiology. 2013;166(2):323–330. doi: 10.1016/j.ijfoodmicro.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Wu Q., Chen B., Xu Y. Regulating yeast flavor metabolism by controlling saccharification reaction rate in simultaneous saccharification and fermentation of chinese maotai-flavor liquor. International Journal of Food Microbiology. 2015;200:39–46. doi: 10.1016/j.ijfoodmicro.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Bates S. T., Berg-Lyons D., Caporaso J. G., Walters W. A., Knight R., Fierer N. Examining the global distribution of dominant archaeal populations in soil. The ISME Journal. 2011;5(5):908–917. doi: 10.1038/ismej.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z., Lozupone C., Hamady M., Bushman F. D., Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Research. 2007;35(18, article e120) doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Q., Bi Y., Deng Y., et al. Impacts of the Three Gorges Dam on microbial structure and potential function. Scientific Reports. 2015;5, article 8605 doi: 10.1038/srep08605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso J. G., Kuczynski J., Stombaugh J., et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods. 2013;10(10):996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 20.Caporaso J. G., Lauber C. L., Walters W. A., et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America. 2011;108, S1:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan J.-B., Hu S.-C., Wang H., Zou Q., Ji Z.-L. PaGeFinder: quantitative identification of spatiotemporal pattern genes. Bioinformatics. 2012;28(11):1544–1545. doi: 10.1093/bioinformatics/bts169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei Z. Preliminary analysis of the change of microbes in the fermentation process of Fenjiu Daqu. Liquor Making Science Technology. 2011;6:65–68. [Google Scholar]
- 23.Katina K., Sauri M., Alakomi H.-L., Mattila-Sandholm T. Potential of lactic acid bacteria to inhibit rope spoilage in wheat sourdough bread. LWT—Food Science and Technology. 2002;35(1):38–45. doi: 10.1006/fstl.2001.0808. [DOI] [Google Scholar]
- 24.Karataş H., Uyar F., Tolan V., Baysal Z. Optimization and enhanced production of α-amylase and protease by a newly isolated Bacillus licheniformis ZB-05 under solid-state fermentation. Annals of Microbiology. 2013;63(1):45–52. doi: 10.1007/s13213-012-0443-6. [DOI] [Google Scholar]
- 25.Li X.-R., Ma E.-B., Yan L.-Z., et al. Bacterial and fungal diversity in the traditional Chinese liquor fermentation process. International Journal of Food Microbiology. 2011;146(1):31–37. doi: 10.1016/j.ijfoodmicro.2011.01.030. [DOI] [PubMed] [Google Scholar]