

Abstract
Key points
N‐cadherin formed punctate adherens junctions (AJ) along the borders between vascular smooth muscle cells (VSMCs) in the pressurized rat superior cerebellar artery.
The formation of N‐cadherin AJs in the vessel wall depends on the intraluminal pressure and was responsive to treatment with phenylephrine (PE) (10−5 m) and ACh (10−5 m).
N‐cadherin‐coated beads were able to induce clustering of N‐cadherin‐enhanced green fluorescent protein (EGFP) on the plasma membrane of isolated VSMCs, whereas treatment with PE (10−5 m) or sodium nitroprusside (10−5 m) induced a significant increase or decrease in the N‐cadherin‐EGFP clustering, respectively.
Application of pulling force (∼1 nN) to the N‐cadherin‐coated beads via an atomic force microscope induced a localized mechanical response from the VSMCs that opposed the pulling.
Abstract
N‐cadherin is the major cell–cell adhesion molecule in vascular smooth muscle cells (VSMCs). We tested the hypothesis that N‐cadherin is part of a novel mechanosensory mechanism in VSMCs and plays an active role in both the arteriolar myogenic response and during changes in vascular tone induced by vasomotor agonists. Intact and pressurized rat superior cerebellar arteries were labelled for confocal immunofluorescence imaging. N‐cadherin formed punctate adherens junctions (AJ) along the borders between VSMCs. When the lumen pressure was raised from 50 to 90 mmHg, both the density and the average size of N‐cadherin AJs increased significantly. Similarly, arteriolar constriction with phenylephrine (PE) (10–5 m) induced a significant increase of N‐cadherin AJ density at 50 mmHg, whereas vasodilatation induced by ACh (10–5 m) was accompanied by a significant decrease in density and size of N‐cadherin AJs. An atomic force microscope (AFM) was employed to further examine the mechano‐responsive properties of N‐cadherin adhesion sites in isolated VSMCs. AFM probes with an attached N‐cadherin‐coated microbead (5 μm) induced a progressive clustering of N‐cadherin‐enhanced green fluorescent protein (EGFP) on the VSMC surface. Application of pulling force (∼1 nN) to the N‐cadherin‐coated‐beads with the AFM induced a localized mechanical response from the VSMCs that opposed the pulling. Treatment with PE (10–5 m) or sodium nitroprusside (10–5 m) induced a significant increase or decrease of the N‐cadherin‐EGFP clustering, respectively. These observations provide compelling evidence that N‐cadherin AJs are sensitive to pressure and vasomotor agonists in VSMCs and support a functional role of N‐cadherin AJs in vasomotor regulation.
Keywords: adherens junction, atomic force microscopy, cardiovascular control, cell‐cell adhesion, cerebral circulation, mechanotransduction, myogenic response, vascular tone
Key points
N‐cadherin formed punctate adherens junctions (AJ) along the borders between vascular smooth muscle cells (VSMCs) in the pressurized rat superior cerebellar artery.
The formation of N‐cadherin AJs in the vessel wall depends on the intraluminal pressure and was responsive to treatment with phenylephrine (PE) (10−5 m) and ACh (10−5 m).
N‐cadherin‐coated beads were able to induce clustering of N‐cadherin‐enhanced green fluorescent protein (EGFP) on the plasma membrane of isolated VSMCs, whereas treatment with PE (10−5 m) or sodium nitroprusside (10−5 m) induced a significant increase or decrease in the N‐cadherin‐EGFP clustering, respectively.
Application of pulling force (∼1 nN) to the N‐cadherin‐coated beads via an atomic force microscope induced a localized mechanical response from the VSMCs that opposed the pulling.
Abbreviations
- AFM
atomic force microscope
- AJ
adherens junction
- BSA
bovine serum albumin
- DMEM
Dulbecco's modified Eagle's medium
- ECM
extracellular matrix
- EGFP
enhanced green fluorescent protein
- EMT
epithelial‐mesenchymal transition
- PE
phenylephrine
- PKC
protein kinase C
- SCA
superior cerebellar artery
- SNP
sodium nitroprusside
- VSMC
vascular smooth muscle cell
Introduction
N‐cadherin is a member of the type I classical cadherin subfamily. Proteins in this subfamily are the major cell–cell adhesion molecules that connect adjacent cells via formation of the adherens junction (AJ). N‐cadherins are Ca2+ binding trans‐membrane molecules, and the extracellular domain of N‐cadherins on opposing cells bind with each other via a homotypic, trans‐interaction, leading to the formation of cell–cell adhesions. N‐cadherin‐mediated cell–cell adhesions play an important role in a number of fundamental cellular processes. In neuron development, N‐cadherin complexes are located at puncta adherentia flanking the active zone of synapses, and play an important role in both target recognition and formation of the synapses (Giagtzoglou et al. 2009). In myoblasts, N‐cadherin was shown to be important for muscle cell differentiation and muscle‐specific gene expression (Charrasse et al. 2002; Krauss et al. 2005). In cancer, enhanced N‐cadherin expression and loss of E‐cadherin is a hallmark of the epithelial‐mesenchymal transition (EMT), which promotes cancer cell migration and tumor cell invasion (Mariotti et al. 2007). In vascular smooth muscle cells (VSMCs), there is evidence that N‐cadherin is an important cell–cell adhesion molecule linked to vascular signalling and the control of myogenic vasomotor tone (Jackson et al. 2010), as well as to intimal thickening in atherosclerosis (Jones et al. 2002). Thus, the available data support N‐cadherin as an important adhesion‐based signalling site. We hypothesized that this site is important for the regulation of vascular function.
At cell–cell adhesions, the cytoplasmic tails of type I classical cadherins bind directly with the cytoplasmic proteins β‐catenin and p120‐catenin. Through these molecules, the cadherins form an association with a large number of cytoskeleton proteins, including α‐catenin, cortactin and vinculin (Brigidi & Bamji, 2011; Brasch et al. 2012). These protein interactions physically link cell–cell adhesions sites to intracellular cytoskeletal filaments and signalling pathways, supporting the concept that they transmit mechanical forces and intracellular signals across a cell‐to‐cell network that is linked to the cytoskeleton. Several recent studies have suggested that cadherin adhesion sites play a role mediating mechanotransduction in a variety of cells, including VE‐cadherin in endothelial cells (Conway & Schwartz, 2012), E‐cadherin in epithelial cells (le Duc et al. 2010; Leckband et al. 2011; Tabdili et al. 2012) and N‐cadherin in cardiomyocytes (Matsuda et al. 2005). Based on the available evidence, it is therefore reasonable to speculate that cadherins in VSMCs are mechanically involved in the normal processes that influence vascular tone and hence forces acting at the AJ site. This involvement could take the form of acting both as a mechanosensory element, as well as a mechanoadaptive element related to transmission of force between cells.
N‐cadherin is the major cadherin expressed in VSMCs (George & Beeching, 2006; Resink et al. 2009). However, N‐cadherin has received little attention and relatively few studies have attempted to understand its function and role in VSMC despite its potential for involvement in vascular control and disease. Some controversial data have been reported regarding the role of N‐cadherins in the formation of the neo‐intima in atherosclerosis. For example, Jones et al. (2002) reported that, in rat carotid arteries, the formation of neointima induced by balloon injury was accompanied by a dramatic but transient (weeks 1–3) up‐regulation of N‐cadherin and β‐catenin expression. By contrast, other studies have reported that down‐regulation of N‐cadherin contributed to VSMC proliferation and migration during neo‐intima formation (Uglow et al. 2003; Dwivedi et al. 2008). We have reported previously that the presence of a function blocking N‐cadherin antibody (GC‐4) or N‐cadherin inhibitory peptides (HEV) inhibited the myogenic response of rat skeletal muscle resistance arterioles (Jackson et al. 2010). This suggested that there is a unique mechanosensory role for N‐cadherin. In the present study, we tested the general hypothesis that N‐cadherins are more broadly involved in vasomotor responses. Specifically, we hypothesized that these cell–cell adhesion molecules are sensitive to the contractile state of the vessel wall and that the formation of N‐cadherin AJ is sensitive to changes in intravascular pressure or vasoactive agonists.
Methods
Vessel isolation and myogenic responsiveness and vascular tone
All animal surgery and killing procedures were conducted in accordance with the approved protocols and within the guidelines of the Animal Care and Use Committee of the University of Missouri, and also conformed with NIH guidelines (Guide For The Care And Use Of Laboratory Animals). Sprague–Dawley male rats (weighing 200–250 g; aged 6–8 weeks) were used for all of the experiments. Following anaesthesia (50 mg ml−1, sodium pentobarbital 200 μl 100 g–1, i.p. injection) and death (pneumothorax), the brain was removed and the superior cerebellar artery (SCA) was isolated and cannulated between two micropipettes using a vessel chamber (Living System, Inc., Burlington, VT, USA) containing physiological saline solution (in mm): 145 NaCl, 5.6 KCl, 2 CaCl2, 1 MgSO4, 1.2 NaH2PO4, 3 MOPS, 5 glucose and 1 pyruvic acid. A chamber heater and a pressure control unit (Living System, Inc.) were used to control the vessel temperature (37 °C) and lumen pressure, respectively. Bath solution was changed every 10 min during each experiment. Vessel intraluminal pressure was initially set to 50 mmHg and, after establishing spontaneous myogenic (intrinsic) tone, the pressure was raised stepwise to 70, 90 and 110 mmHg. Each pressure step was maintained for 10 min to allow the vessel achieve stable vascular tone. In experiments designed to test the effect of vasoactive agonists, vessels were allowed to establish stable myogenic tone at pressures of 50 or 90 mmHg. Either phenylephrine (PE) (10 μm) or ACh (10 μm) was then added into the vessel bath to induce vasoconstriction or vasodilatation, respectively. Images of vessel at mid‐diameter were recorded using a video camera, and vessel lumen diameter was measured offline using proprietary software written in MATLAB (MathWorks Inc., Natick, MA, USA).
Cell isolation and cell culture
The isolation of VSMCs has been described previously (Wu et al. 1998). Briefly, SCAs (100–150 μm diameter) were isolated from the brain of Sprague–Dawley rats. Isolated arteries were subjected to a two‐step enzymatic digestion with papain (27 U ml−1, 37 °C, 30 min), followed by collagenase F and collagenase H (37 °C, 3–4 min). The released VSMCs were plated on a glass bottomed culture dish (Willco Wells BV, Amsterdam, The Netherlands) and were cultured in Dulbecco's modified Eagle's medium (DMEM/F‐12, supplemented with 20% fetal bovine serum, 10 mm Hepes, 2 mm L‐glutamine, 1 mm sodium pyruvate, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 0.25 μg ml−1 amphotericin B). Cells were maintained in a humidified incubator (Heraeus Instruments, Inc., Newtown, CT, USA) with 5% CO2 at 37 °C. The identity of the cultured VSMCs was confirmed by positive staining with antibody against smooth muscle α‐actin. Twenty‐four hours prior to an experiment, serum‐free medium was substituted for the culture media. Only primary cultured cells, non‐passaged, were used for the experiments using an atomic force microscope (AFM). Because of the small size of SCAs, only one dish of VSMC cells was isolated for each rat. For the AFM experiments, we studied one cell from each dish and so each cell represents a different rat. All of the cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA).
Instruments
An AFM (Bioscope II System; Bruker Nano, Inc., Santa Barbara CA, USA) was mounted on a Fluoview 1000 confocal microscope (Olympus, Thornwood, NY, USA). AFM data were collected and analysed using a Nanoscope (Bruker Nano, Inc.) and proprietary MATLAB (MathWorks Inc.) software.
Preparation of AFM probes
Glass microbeads (diameter 5 μm) were glued to the AFM probes (Bruker Nano Inc., model: MLCT; spring constant ∼0.01 N m−1) with epoxy resin. Recombinant human N‐cadherin (0.2 mg ml−1; R&D Systems, Minneapolis, MN, USA) or bovine serum albumin (BSA) (0.5 mg ml−1) was incubated with the AFM probes for 10 min at room temperature to functionally coat the AFM probe tip. The probes were then washed with Dulbecco's phosphate‐buffered saline (5×) to remove unbound protein.
AFM force application and force measurement
The application of contact and pulling force, as well as measurement of force with the AFM, was performed as described previously (Sun et al. 2008). Briefly, the AFM was operated in contact mode. After a thermal equilibration period of 30 min, the functionally coated AFM probe was brought into contact with the VSMC surface, and contact with the cell surface was maintained for ∼20 min with a contact force of ∼1 nN. The deflection set point was then manually adjusted to apply a step increase in pulling force (∼1000 pN) to the AFM mounted microbead adhesion site. The AFM height data were recorded continuously and analysed to quantify changes of the vertical position of the microbead. In experiments designed to examine the clustering process of enhanced green fluorescent protein (EGFP)‐conjugated N‐cadherin, the microbead was placed in contact with the VSMC surface for 70 min without application of a pulling force to investigate clustering of N‐cadherin around the microbead adhesion site. To test the effect of PE and sodium nitroprusside (SNP), the microbead was placed in contact with the VSMC surface for 60 min to induce N‐cadherin‐EGFP clustering, and testing reagents were then introduced into the cell bath.
Vessel and cultured VSMC immunofluorescence labelling
For immunolabelling, the SCAs were fixed with 8% paraformaldehyde at steady‐state (2 h) transmural pressures of 50, 70, 90 or 110 mmHg . The vessel was then incubated in glycine buffer (0.1 m glycine in MilliQ water; Millipore, Billerica, MA, USA) for 1 h (2×). This was followed by incubation of the vessel in primary antibody buffer [1% BSA, 0.05% Triton X‐100 in sodium citrate solution buffer (NaCl: 152 mm, sodium citrate 17 mm, pH 7.4) plus primary antibody] with agitation at 37 °C for 48 h. The vessel was then washed 3× with antibody washing buffer (0.05% Triton X‐100 in sodium citrate solution) and incubated in secondary antibody solution overnight (37 °C with agitation). After incubation with the secondary antibody, the vessel was washed 3× (0.05% Triton X‐100 in sodium citrate solution), mounted on a coverslip and imaged using confocal microscopy. Confocal imaging of the vessel was performed using a 60× oil objective with 1.42 NA. Through‐focus images were collected at intervals of 0.3 μm. The co‐localization between N‐cadherin and β‐catenin was quantified using the Pearson coefficient, as calculated using Imaris software (Bitplane AG, Zurich, Switzerland). For immunostaining of cultured cells, VSMCs were fixed by paraformaldehyde (4%) for 20 min at room temperature and labelled with primary antibody (dilution 1:100) in antibody buffer overnight at 4 °C. The cells were then washed five times with antibody washing buffer at room temperature, followed by incubation with Alexa‐488 conjugated secondary antibody (dilution 1:100) for 2 h at room temperature. The cells were again washed five times with antibody washing buffer and then imaged using confocal microscopy.
Transfection of smooth muscle cells
VSMCs were transfected with N‐cadherin‐EGFP using Turbofectin reagent (Origene, Inc., Rockville, MD, USA) in accordance with the manufacturer's instructions. Briefly, 5 μg DNA was mixed with 100 μl of DMEM and 10 μl of Turbofectin. After incubation for 25 min, the mixture was added to the VSMCs. The cells were then returned to the incubator and protein expression was observed after 48 h.
Quantification of protein clusters in the vessel
Protein clusters were quantified using MATLAB. The number of clusters and the average size of clusters were determined by setting an intensity threshold and were normalized to area of the vessel wall (i.e. number of clusters per area of 1000 pixels). An region of interest (40 000–80 000 pixels) was drawn in freehand style in the area of smooth muscle and a histogram of fluorescence intensity was then calculated of all pixels in this region. The peak of the histogram was used to represent the normal fluorescence signal level in the smooth muscle layer and the value was then multiplied by constant factor 3.5 to set the threshold level. The constant factor was determined by testing a series of consecutive numbers (from 2.5 to 4.5) on several vessel image stacks from different pressure conditions; the number 3.5 satisfactorily detected cluster signals from these data sets and thus was selected as the constant factor and applied across all dalta sets. The average size and number of clusters in VSMC were compared statistically. Co‐localization of fluorescence signals for different protein antibodies was analysed with the co‐localization analysis module in Imaris software (Bitplane AG).
Reagents
Recombinant Human N‐cadherin (catalogue number 1388‐NC) was purchased from R&D Systems. N‐cadherin antibody was obtained from Millipore (catalogue number 04‐1126) and β‐catenin antibody was obtained from BD Bioscience (Clontech, Palo Alto, CA, USA) (catalogue number 610153); rabbit control IgG was purchased from Cell Signaling (Beverly, MA, USA) (catalogue number 3900) and mouse control IgG was purchased from BioLegend (San Diego, CA, USA) (catalogue number 401402). Phenylephrine, ACh and sodium nitroprusside were purchased from Sigma‐Aldrich (St Louis, MO, USA). Alexa488‐conjugated goat anti‐rabbit IgG (for N‐cadherin; catalogue number A11008) and Alexa633‐conjugated goat anti‐mouse IgG (for β‐catenin; catalogue number A21052) were purchased from Life Technologies Inc. (Life Technologies, Grand Island, NY, USA).
Statistical analysis
Results were compared using ANOVA. P < 0.05 was considered statistically significant.
Results
N‐cadherin forms AJs in the medial smooth muscle layer of the cannulated rat superior cerebellar artery
To localize N‐cadherin AJs between adjacent VSMCs in the vessel wall, immunofluorescence labelling of N‐cadherin and confocal microscopy were used to visualize the distribution of N‐cadherin in the wall of pressurized and intact rat SCAs. As shown in Fig. 1 A, N‐cadherin was detected as numerous fluorescence punctate sites in the VSMC layer of the intact SCA. For the vessel shown in Fig. 1 A, the vessel was pressurized to 90 mmHg and had developed stable myogenic tone prior to fixation. A large number of the punctate N‐cadherin containing structures were aligned in a pattern analogous to beads‐on‐a‐string along the intercellular boundaries (Fig. 1 A, white arrows). For this experiment, the artery was also co‐labelled for β‐catenin. The distribution of β‐catenin in the VSMC was similar to that observed for N‐cadherin, again with the strongest labelling located along the intercellular borders in the vessel wall (Fig. 1 A). An image overlay showed that a majority of N‐cadherin containing punctate spots localized with areas where β‐catenin fluorescence was observed (Fig. 1 A). The similarities in location of N‐cadherin and β‐catenin in VSMCs are evidence of the presence of mature AJs in the pressurized artery. Some N‐cadherin clusters were not co‐localized with the β‐catenin and were not located at the cell borders. These N‐cadherin clusters presumably represent protein pools that can be directed towards actual AJs. A Pearson co‐localization coefficient of >0.6 was observed between the overlay of N‐cadherin and β‐catenin labelling. Control experiments with rabbit and mouse IgG and secondary antibodies showed no significant labelling or fluorescence when imaged under the same conditions (Fig. 1 B).
Figure 1. N‐cadherin forms AJs between VSMC.
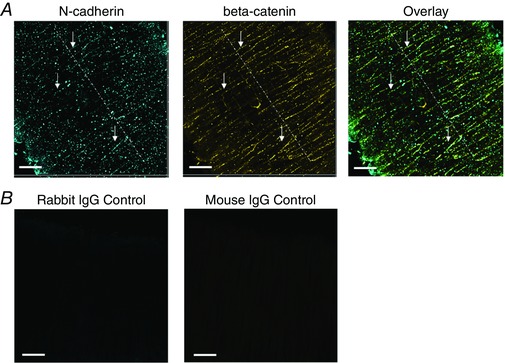
A, N‐cadherin clusters are observed along the VSMC borders between adjacent cells in the vessel wall of SCAs. Many N‐cadherin and β‐catenin clusters appear patterned as a string of beads along cell–cell borders (depicted by white arrows). The SCA was pressurized to 90 mmHg and had myogenic tone. The vessel was fixed under pressure, immunolabelled for N‐cadherin and β‐catenin, and visualized by confocal microscopy. B, vessel labelled with control antibodies. The SCA was pressurized to 90 mmHg and had myogenic tone. The vessel was fixed under pressure, and immunolabelled with rabbit control IgG and mouse control IgG, followed by labelling with the same secondary antibodies. Scale bar = 20 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Pressure induced changes of N‐cadherin labelling
To address the question of whether the N‐cadherin AJ sites are sensitive to changes in intraluminal pressure, SCAs were initially pressurized to 50 mmHg. After the vessel developed stable myogenic tone, the lumen pressure was elevated stepwise (20 mmHg increments) to pressures of 70, 90 and 110 mmHg. As the lumen pressure was increased from 50 to 110 mmHg, the myogenic tone of vessels increased, as indicated by progressive reductions in diameter (Fig. 2 A). N‐cadherin labelling increased dramatically when the lumen pressure increased from 50 to 90 mmHg (Fig. 2 B). For quantitative analysis, the density and average size of the N‐cadherin AJs in the VSMC layer were compared between pressures. Both the density and size of the N‐cadherin AJs significantly increased when the pressure increased from 50 to 90 mmHg (Fig. 2 C and D). As the pressure was increased further to 110 mmHg, the size of the N‐cadherin AJs was maintained at levels similar to 90 mmHg, although a significant drop in the density of N‐cadherin AJs was observed, indicating a biphasic effect of the pressure (Fig. 2 C and D). These data support the hypothesis that N‐cadherin AJs are sensitive to changes in intraluminal pressure in SCA after the development of myogenic tone.
Figure 2. The formation N‐cadherin AJs was sensitive to changes in transmural pressure in SCAs.
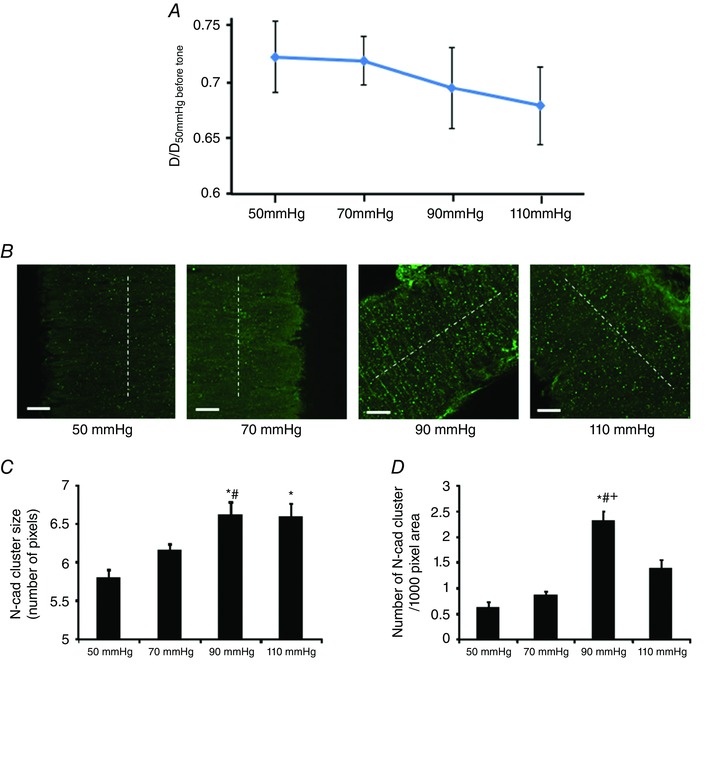
A, SCAs developed myogenic tone before undergoing paraformaldehyde fixation. Vessel diameter was normalized to the initial passive diameter of the vessel at 50 mmHg. Number of vessels: 50 mmHg, n = 5; 70 mmHg, n = 6; 90 mmHg, n = 7; 110 mmHg, n = 7. B, N‐cadherin clusters in the vessel wall. Note the increase of clustering at 90 and 110 mmHg compared to 50 mmHg. White dashed lines indicate the direction of vessel orientation. C and D, both N‐cadherin cluster size and density were significantly changed when the pressure was increased. Pixel size is 0.16 μm 2. Confocal z‐stack images of the vessel wall were collected vy through‐focus imaging, and the image stack was quantitatively analysed to determine the average density and size of N‐cadherin clusters in the VSMC layer. 50 mmHg, n = 5 vessels; 70 mmHg, n = 6 vessels; 90 mmHg, n = 7 vessels; 110 mmHg, n = 7 vessels. One vessel was used for each animal (i.e. the number of vessels n equals the number of animals used for each experiment). * P < 0.05 compared to 50 mmHg; #P < 0.05 compared to 70 mmHg; +P < 0.05 compared to 110 mmHg. Scale bar = 20 μm. Data are presented as the mean ± SE. [Color figure can be viewed at wileyonlinelibrary.com]
Modulation of SCA N‐cadherin AJs by vasoactive agonists
We further determined whether vasoactive agonists regulated N‐cadherin AJs. PE was employed to induce vasoconstriction and ACh was used to induce vasodilatation. The vasoconstriction induced by PE was 10.5 ± 1.1% at 50 mmHg. The vasodilatation induced by ACh was 40.8 ± 4.6% at 50 mmHg and 45.1 ± 4.1% at 90 mmHg. The addition of PE (10−5 m) caused a significant increase in the density and average size of N‐cadherin AJs at 50 mmHg. ACh (10−5 m) caused a significant decrease in the density of the N‐cadherin AJs at both 50 and 90 mmHg, with the average size of N‐cadherin AJs decreasing only at 90 mmHg (Fig. 3 A, C, D and E). These results suggest that the assembly/disassembly of N‐cadherin AJs is sensitive to regulation by vasoactive agonists.
Figure 3. Vasoactive agents modulate the assembly/disassembly of N‐cadherin AJs in the wall of SCAs.
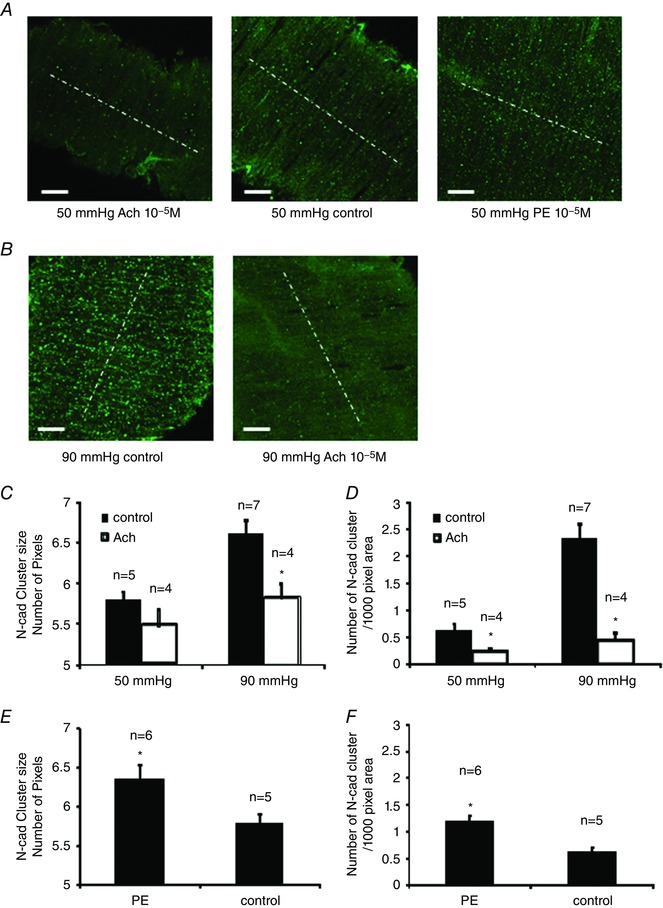
SCAs were pressurized to 50 or 90 mmHg. Control vessels were fixed at 10 min after the vessel pressure was elevated to 90 mmHg or after the vessel tone stabilized at 50 mmHg. For the ACh group, ACh (10−5 m) was added into the bath at 10 min after the vessel tone stabilized at 50 mmHg, or after the vessel pressure was elevated to 90 mmHg. SCAs were fixed within 10 s after the treatment. For PE group, PE (10−5 m) was added into the bath at 10 min after vessel tone stabilized at 50 mmHg. A, N‐cadherin AJs in control vessels and vessels treated with either ACh or PE and vessels were pressurized to 50 mmHg. B, N‐cadherin AJs in control vessels and ACh‐treated vessels pressurized at 90 mmHg. White dashed lines indicate the direction of vessel orientation. C, comparison of N‐cadherin AJ size between control and ACh‐treated groups. D, comparison of N‐cadherin AJ density. E, comparison of N‐cadherin AJ size between control and PE‐treated group. F, comparison of N‐cadherin AJ density between control and PE‐treated group. Pixel size is 0.16 μm 2. One vessel was used for each animal (i.e. the number of vessels n equals the number of animals used for each experiment). * P < 0.05 compared to ACh or PE‐treated groups; n indicates the number of vessels in each group. [Color figure can be viewed at wileyonlinelibrary.com]
N‐cadherin clustering in isolated VSMCs
By combining the AFM with a confocal microscope, we aimed to examine the process of N‐cadherin clustering in isolated VSMCs. We first examined the presence of endogenous N‐cadherin in cultured VSMCs. The VSMCs were isolated from rat SCAs, and the identity of VSMCs was confirmed by successfully immunostaining with an antibody against smooth muscle α‐actin (Fig. 4 B). As shown in Fig. 4 A, the endogenous N‐cadherin formed both linear and punctate AJs in the cultured VSMCs. Fig. 4 C shows that the N‐cadherin‐EGFP expressed in VSMCs was incorporated into the cell adhesion structures, suggesting that the expression of N‐cadherin‐EGFP was functioning in VSMCs. We then used VSMCs that were transfected with an N‐cadherin‐EGFP construct to test the process of N‐cadherin clustering. Using fluorescence microscopy, we first identified a VSMC that expressed N‐cadherin‐EGFP and an N‐cadherin‐coated bead was then brought into contact with the cell surface using an AFM. In these experiments, the contact force (∼1 nN) was applied to maintain the N‐cadherin bead in contact with the cell surface for up to 70 min. During this period, a progressive clustering of N‐cadherin was observed, which reached a plateau after ∼55 min of contact (Fig. 5 A and B). By contrast, application of N‐cadherin beads on control VSMCs induced no clustering of EGFP (Fig. 5 C) and application of BSA‐coated beads also was unable to induce clustering of N‐cadherin‐EGFP in control VSMCs (Fig. 5 D). These results suggest that contact with the N‐cadherin surface was able to induce the N‐cadherin clustering on the cell membrane of VSMCs. Because PE and ACh can regulate the N‐cadherin AJs in isolated SCAs (Fig. 3), we also tested the effect of PE and SNP on the N‐cadherin clustering in isolated VSMCs. In these experiments, an N‐cadherin‐coated bead was first brought into contact with the VSMC surface for 60 min as in control. At t = 60 min, when the N‐cadherin clustering was in a plateau phase, either PE (10−5 m) or SNP (10−5 m) was added to the cell bath, and N‐cadherin clustering was compared before and after treatments. PE significantly increased N‐cadherin clustering (Fig. 5 E and G), whereas SNP significantly decreased N‐cadherin clustering in isolated VSMCs (Fig. 5 F and H). These results are consistent with the observations made in intact SCAs, and further suggest that PE and SNP can regulate N‐cadherin clustering in vascular smooth muscle.
Figure 4. Both endogenous N‐cadherin and N‐cadherin‐EGFP were recruited to AJs in cultured VSMC.
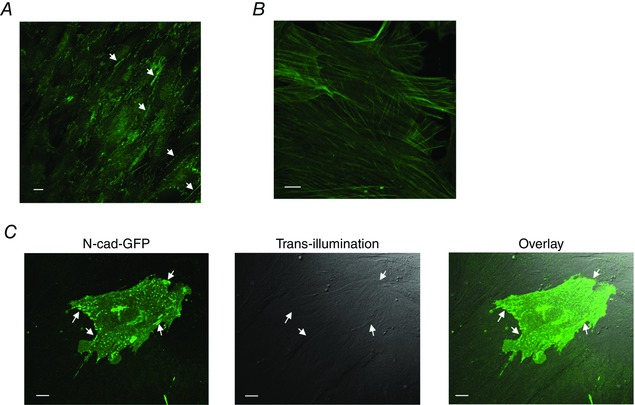
A, immunofluorescence labelling of cell endogenous N‐cadherin in primary cultured VSMCs isolated from rat SCAs. Through‐focus stack images were collected using confocal fluorescence microscopy, and the stacked images were summed together to show the presence of N‐cadherin adherens junctions (white arrows). B, immunofluorescence labelling of smooth muscle α‐actin in primary cultured VSMCs. C, VSMCs were transfected with N‐cadherin‐EGFP. N‐cadherin‐EGFP was incorporated into adhesion structures between overlapping VSMCs. Left: a single cerebral VSMC in confluent culture exhibiting transfection with N‐cadherin‐EGFP was imaged using confocal fluorescence microscopy. White arrows point to the clusters of N‐cadherin‐EGFP. Middle: trans‐illumination image of the same area showing the transfected VSMC and neighbouring VSMCs that are not expressing N‐cadherin‐GFP. The neighbouring cells were overlapping and in contact with the cell expressing N‐cadherin‐EGFP. Right: overlay of the fluorescence image and trans‐illumination image. White arrows indicate the adhesion structures that incorporated N‐cadherin‐EGFP. Scale bar = 10 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 5. Formation and vasoactive agonist induced modulation of N‐cadherin‐EGFP clustering on the surface of VSMC.
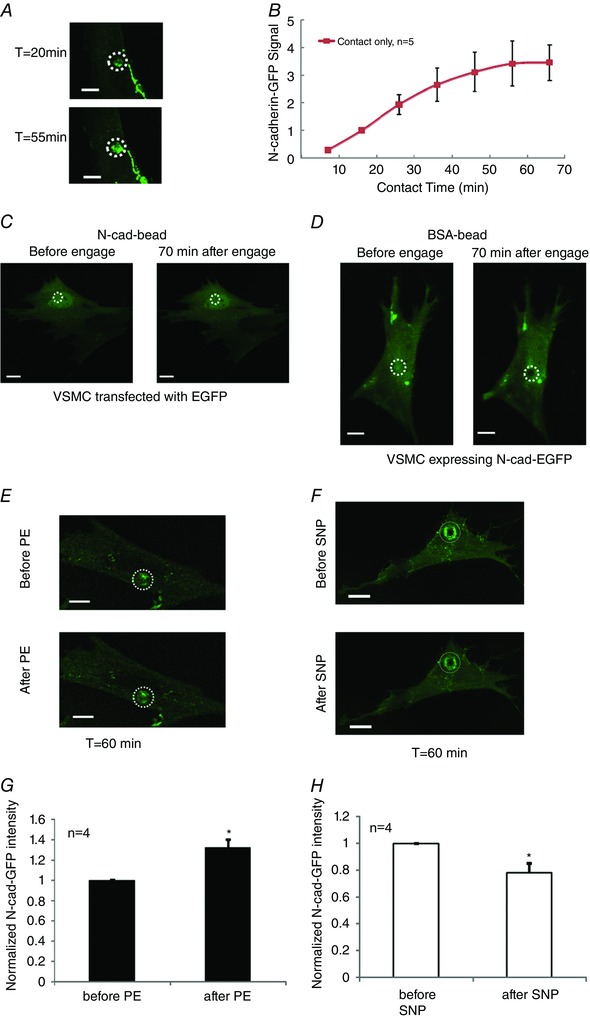
A, N‐cadherin clustering to an N‐cadherin‐coated bead on the cell membrane of VSMCs. T: time after the N‐cadherin‐coated bead was applied to the cell. Isolated VSMCs were transfected to express N‐cadherin‐EGFP, and the EGFP signal was recorded using confocal microscopy. Confocal z‐stack images were collected at the indicated time point. The z‐axis stack image was summed together to show the total N‐cadherin clustering at the bead adhesion site. A white circle depicts the bead location. B, quantitation of N‐cadherin clustering over time. Fluorescence signal was normalized by the signal at t = 16 min. C and D, negative controls of N‐cadherin clustering on VSMC cell membrane. C, an N‐cadherin‐coated bead induced no clustering of EGFP on the VSMC surface. Isolated VSMCs were transfected to express enhanced GFP (EGFP). An N‐cadherin‐coated bead was applied to the surface of VSMCs using an AFM, and the experiment was performed as described above. The experiment was repeated with four cells, similar results were observed. D, BSA‐coated beads induced no meaningful N‐cadherin clustering on the VSMC surface. Isolated VSMCs were transfected to express N‐cadherin‐EGFP, a BSA‐coated bead was applied to the surface of VSMC, and the experiment was performed as described above. The experiment was repeated with three cells, and similar results were observed. E and F, vasoactive agents modulate the assembly/disassembly of N‐cadherin AJs in VSMCs isolated from SCA. Clustering of N‐cadherin‐EGFP was induced by engaging the N‐cadherin‐coated bead to the cell surface for 60 min. Vasoactive agents were then added into the cell bath. The EGFP signal was recorded using confocal microscopy before and after treatment with vasoactive agents. E, N‐cadherin clustering before and after PE (10−5 m) was applied to the bath. F, N‐cadherin clustering before and after SNP (10−5 m) was applied to the bath. The z‐stack images was summed together to show the total N‐cadherin clustering at the bead adhesion site. G, quantitative comparison of N‐cadherin‐EGFP fluorescence signal before and after PE treatment, fluorescence signal was normalized by the signal recorded before treatment. H, quantitative comparison of N‐cadherin‐GFP fluorescence signal before and after SNP treatment. One cell was used for each animal (i.e. the number of cells n equals the number of animals used for each experiment). A white circle depicts the bead locations. * P < 0.05 compared to control (or before treatment). Scale bar = 10 mm. [Color figure can be viewed at wileyonlinelibrary.com]
Force applied through N‐cadherin adhesions induced a VSMC contractile response
To further characterize the mechanosensory properties of N‐cadherin AJs, an AFM was used to apply mechanical force at sites of N‐cadherin adhesion on the surface of an N‐cadherin‐EGFP expressing VSMCs and to measure the cell mechano‐response to the applied force. Using the AFM, a N‐cadherin‐coated microbead was brought into contact with the surface of a single isolated VSMC (Fig. 6 A). When applying a microbead to the VSMC surface with the AFM, a contact force (∼1 nN) was applied to keep the microbead in contact with the cell. After 20 min of contact, the AFM probe was raised in the z‐axis, thus effectively applying a pulling force on the N‐cadherin‐mediated adhesion structure (Fig. 6 B). In response to the upward pull, the VSMC responded by applying an opposing resistive force to the microbead (Fig. 6 C). As the resistance force from the VSMC increased progressively over a 5 min observation period, the microbead was pulled downward towards the cell (Fig. 6 C). In control experiments using BSA‐coated beads, the same pulling force applied by the AFM lifted the beads away from cell surface (i.e. no resistance force was observed). These results are consistent with N‐cadherin adhesion being linked with the intracellular contractile filaments and the transmission of the mechanical force across the cell membrane to trigger a cellular response opposing the pulling force.
Figure 6. N‐cadherin mediated VSMC mechano‐response to pulling force.
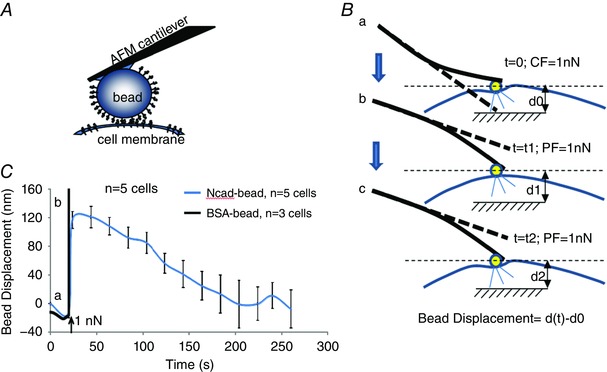
A, schematic of the AFM experiment on isolated VSMCs. An N‐cadherin‐coated bead was mounted on the AFM cantilever and was applied to cell surface. B, schematic of the AFM pulling experiment. An AFM cantilever is a soft spring and was applied to maintain a force sufficient to maintain contact (CF) and keep the N‐cadherin‐coated bead on the VSMC surface; when the cantilever was lifted vertically in the z‐axis, a constant pulling force (PF) was applied to the cell at the bead contact site (a to b). The cell responded and the bead was pulled downward or displaced by the cell (b to c). C, bead displacement measurements indicated that VSMC resisted the pulling force applied to the N‐cadherin‐bead by locally generating force to oppose the upward z‐axis displacement. By contrast, force applied to the BSA‐coated bead directly lifted the bead away from cell surface. Bead displacement was calculated as shown in (B). One cell was used for each animal (i.e. the number of cells n equals the number of animals used for each experiment). [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
The molecular mechanisms underlying how VSMC‐extracellular matrix (ECM) adhesion sites and cell–cell adhesions are involved in regulation of vascular tone and intrinsic control related to the myogenic response of small arteries remains incompletely understood. Despite this, evidence of their importance continues to emerge (Resink et al. 2009; Jackson et al. 2010; Mui et al. 2015). The ability of resistance vessels to constrict and dilate is fundamental to the regulation of blood pressure and organ/tissue blood flow (i.e. blood flow autoregulation) (Harper & Bohlen, 1984; Jackson & Duling, 1989; Iliescu et al. 2008). In this regard, further characterization is required regarding the function and behaviour of N‐cadherin and AJs in VSMCs of small arteries and their potential importance as sensory and signal transducing sites for vascular mechanotransduction and the regulation of vasomotor function. In the present study, we have confirmed the presence of N‐cadherin clusters at the border between VSMCs in rat SCAs. N‐cadherin also co‐localized with β‐catenin, suggesting that these punctate structures are mature AJs between adjacent VSMCs. The novel findings of the present study are that N‐cadherin AJs were responsive to changes of intraluminal pressure and vasoactive agonists. This was confirmed by changes in both the size and number of the N‐cadherin AJs as the intraluminal pressure was varied or by exposure to vasoactive agonists (PE and ACh). We further confirmed our observations in the intact vessel at the level of the single VSMC. An AFM was used to bring an N‐cadherin‐coated bead into contact with the VSMC membrane and the formation of N‐cadherin clusters under the bead was observed. In agreement with the observation from intact SCAs, PE also increased the N‐cadherin clustering in isolated VSMCs, whereas SNP decreased the N‐cadherin clustering. Furthermore, when a pulling force was applied to the N‐cadherin adhesion site via the AFM, we observed a local mechanical contraction of the VSMC to oppose the pull. These observations support the hypothesis that N‐cadherin AJs play an important role in the force transmission within the vessel wall.
For a blood vessel to constrict or dilate, VSMCs are required to co‐ordinate their individual contractions or relaxations with each other, forming a tissue‐scale mechanical syncytium. We propose that this co‐ordination depends on attachment sites both between cells and through the ECM. Earlier electron microscopy studies described dense bodies/plaques along the VSMC borders (Bond & Somlyo, 1982; Sleek & Duling, 1986), which are probably important sites for this co‐ordination during changes in vascular tone. One of the well‐studied molecular structures that has been implicated in this role is the integrin‐mediated focal adhesion; although studied less, cadherin‐mediated AJs have also been hypothesized as important sites of mechanotransduction and force transmission (le Duc et al. 2010; Yonemura et al. 2010; Yonemura, 2011) and thus may play a similar role (Fig. 7). Studies of integrin‐mediated focal adhesions in the vessel wall and in isolated VSMCs support the hypothesis that integrin‐mediated VSMC‐ECM adhesions play an important role in the regulation of the vessel myogenic tone (Mogford et al. 1997; Davis et al. 2001; Martinez‐Lemus et al. 2005; Sun et al. 2008). There is more recent evidence that VSMC cell–cell adhesions mediated by N‐cadherin also serve an important role in the development of myogenic tone and the myogenic response (Jackson et al. 2010). In a previous study, we demonstrated that application of function‐blocking antibody or peptide mimicking N‐cadherin binding motif inhibited the myogenic response of rat cremaster arteriole (Jackson et al. 2010). In the present study, we provide further evidence indicating that, in intact arterioles and single isolated VSMCs, there are changes in N‐cadherin‐mediated adhesions associated with alterations in vascular tone induced by myogenic means or by vasoactive agonists. We also found that the cadherin attachment site was sensitive to pulling with the AFM and that this resulted in localized contractile events (Fig. 6). Localized responses to pulling such as these have been reported previously by our laboratory when a pulling force is applied to integrin‐fibronectin focal adhesions (Sun et al. 2008; Sun et al. 2012). Our collective observations in the present study suggest that N‐cadherin AJs are sensitive to mechanical forces. Importantly, similar to the integrin‐mediated focal adhesions, they mediate VSMC mechanotransduction in response to changes in intravascular pressure and, furthermore, can adapt to stimulation by vasoactive agonists. Based on the available evidence, we propose that both integrin focal adhesions and N‐cadherin AJs are important sites in VSMCs for mechanotransduction and force transmission during changes in vascular tone (Fig. 7).
Figure 7. Schematic model illustrates formation of N‐cadherin AJs between VSMCs in the vessel wall of SCAs.
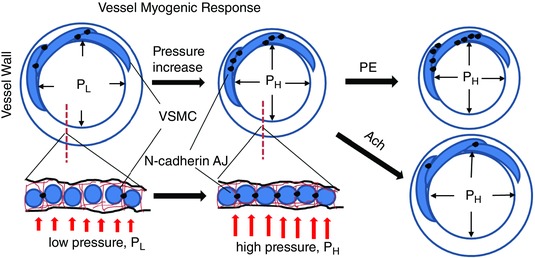
Upper: cross‐sectional view of vessel wall. Lower: transection view of the vessel wall. Increase of transmural pressure induces the SCA myogenic response and vasoconstriction. This process leads to enhanced cell–cell contacts, resulting in increase of the cell–cell adhesions and mechanical coupling. PE enhanced the formation of N‐cadherin AJs between VSMCs, whereas ACh induced the disassembly of AJs. PL, low pressure; PH, high pressure. [Color figure can be viewed at wileyonlinelibrary.com]
An important observation in the present study is that the N‐cadherin clustering is dynamic. The cadherin clustering involves both binding between the EC1 domains of opposing cadherins (i.e. trans‐interaction) and the interactions among EC1 and EC2 domains of parallel cadherin molecules (i.e. cis‐interaction). The trans‐interaction bridges cadherins on the opposing cells membrane (forming cell–cell adhesion), whereas the cis‐interaction brings parallel cadherins into the structure (strengthening the cell–cell adhesion) (Brasch et al. 2012; Sivasankar, 2013). In the present study, N‐cadherin clustering occurs upon contact of a N‐cadherin‐coated microbead in an isolated VSMC cell (Fig. 5), suggesting that simple physical contact and association between adjacent VSMCs may be sufficient to trigger formation of N‐cadherin AJs. Additionally, these clusters in the intact vessel were responsive to changes of intravascular pressure or the presence of vasoactive agonists (Fig. 7). This was indicated by changes in the number and size of the cadherin clusters. The underlying mechanisms behind this clustering and the associated dynamics are not yet clear and will require further study. From our observations, it can be initiated by simple contact, which presumably brings adjacent N‐cadherin molecules into sufficiently close proximity to initiate binding. Whether the mechanism for these dynamic changes is the result of an alteration in intracellular signalling at the intracellular site of the AJ and/or whether it is secondary to the increase/decrease of cell–cell contacts associated with the change of vessel tone remains to be clarified. Of relevance to the latter perspective, a recent study showed that endothelial cells were flattened by an increase in intravascular pressure and it was argued that the flattening/thinning of the endothelial cell leads to suppression of calcium diffusion near the inositol trisphosphate‐receptor microdomain in the endothelial layer (Wilson et al. 2015). It will be interesting to examine whether the increased intravascular pressure can also induce shape changes in VSMCs within the vascular wall such that there is increased contact with neighbouring cells. This hypothetically could initiate a contact induced increase in N‐cadherin cluster formation. Similarly, in the PE/ACh induced change of vasomotor tone, the associated geometric change in the shape of VSMC (e.g. cell shortening, fattening or stretching) may also contribute to a change of cell–cell contact area that increases or decreases N‐cadherin clusters.
Of possible relevance, evidence from studies of E‐ and N‐cadherin suggests that clusters form through processes involving both membrane diffusional movements and the cytosolic delivery of E‐cadherin vesicles to the cell–cell adhesion sites. The important roles of kinesin and microtubule in the delivery of E‐ and N‐cadherin at cell–cell contacts have been suggested in several studies (Mary et al. 2002; Stehbens et al. 2006). N‐cadherin has been shown to form a complex with p120‐ and β‐catenin before being delivered to cell surface (Wahl et al. 2003). Furthermore, Chen et al. (2003) showed that p120 catenin moved along the microtubule through binding with kinesin. Collectively, this evidence suggests that p120‐catenin and kinesin play a key role in mediating the trafficking of N‐cadherin to cell surface via microtubule.
In the present study, in addition to pressure‐induced changes, we also investigated whether the formation of N‐cadherin AJs could be modulated in situations where the contractile tone of the vessel was altered by vasoactive agonists. We observed that the vasodilatation induced with ACh was accompanied by disassembly of N‐cadherin AJs in the vessel wall (Fig. 3), whereas vasoconstriction with PE was accompanied by increase of N‐cadherin AJs in the vessel wall. The observation that PE‐induced increases and SNP‐induced decreases of N‐cadherin clustering in isolated VSMCs could be used to argue for the existence of an ‘inside‐out’ signalling mechanism, although a mechanically dominated mechanism cannot be ruled out, as discussed above. In this respect, Wang et al. (2015) showed that the recruitment of β‐catenin to N‐cadherin AJs is enhanced by ACh stimulation of human airway smooth muscle cells, providing additional evidence of inside‐out signalling modulating the function of N‐cadherin AJs in airway smooth muscle. Interestingly, in their study, the recruitment of β‐catenin was dependent on actin polymerization but not on myosin II activity or microtubule polymerization. It was also shown that modulation of protein kinase C (PKC) activity in the VSMC can lead to changes of N‐cadherin clustering. In both epithelial cells (Shafer et al. 1999) and keratinocytes (Lewis et al. 1994), PKC activity was shown to be required for E‐cadherin to form AJs, whereas inhibition of PKC induced the disassembly of E‐cadherin AJs. PE is known to activate PKC through activation of Gq‐protein, whereas NO/SNP activates guanylyl cyclase and decreases intracellular calcium levels, thus down‐regulating PKC. (Wier & Morgan, 2003) Thus, it is possible that PE‐ and SNP‐induced changes in N‐cadherin clustering involve PKC activity in VSMC. In summary, the modulation of AJs by vasoactive agonists is of importance because it may provide novel mechanistic information relevant to normal regulation of vascular tone and changes in vascular tone and structure in disease.
In conclusion, for the first time, we have demonstrated that N‐cadherin AJs dynamically align along the VSMC cell borders in response to intraluminal pressure and the modulation of vascular tone by vasodilatating and vasoconstricting agonists. These data extend our previous largely pharmacological observations and provide more mechanistic support for the hypothesis that N‐cadherin is part of a novel and dynamic mechanosensory mechanism in VSMCs that plays an active role in the arteriolar myogenic response, as well as during changes in vascular tone.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
ZS and GAM were responsible for the conception of the study. ZS, ML, MAH and GAM were responsible for the design of the study. ZS, ML and ZL were responsible for data acquisition. ZS, ML, ZL, MAH and GAM were responsible for data analysis. ZS, MAH and GAM were responsible for data interpretation. ZS, MAH and GAM were responsible for drafting and revising the manuscript. All authors have approved the final version of the manuscript submitted for publication. All authors agree to being accountable for all aspects of the work. All authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This work was supported by the National Institutes of Health (1P01HL095486 to GAM).
Contributor Information
Zhe Sun, Email: sunzh@missouri.edu.
Gerald A. Meininger, Email: meiningerg@missouri.edu
References
- Bond M & Somlyo AV (1982). Dense bodies and actin polarity in vertebrate smooth muscle. J Cell Biol 95, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch J, Harrison OJ, Honig B & Shapiro L (2012). Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol 22, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigidi GS & Bamji SX (2011). Cadherin‐catenin adhesion complexes at the synapse. Curr Opin Neurobiol 21, 208–214. [DOI] [PubMed] [Google Scholar]
- Charrasse S, Meriane M, Comunale F, Blangy A & Gauthier‐Rouviere C (2002). N‐cadherin‐dependent cell–cell contact regulates Rho GTPases and beta‐catenin localization in mouse C2C12 myoblasts. J Cell Biol 158, 953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Kojima S, Borisy GG & Green KJ (2003). p120 catenin associates with kinesin and facilitates the transport of cadherin‐catenin complexes to intercellular junctions. J Cell Biol 163, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway D & Schwartz MA (2012). Lessons from the endothelial junctional mechanosensory complex. F1000 Biol Rep 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Davis GE, Hill MA & Meininger GA (2001). Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol 280, H1427–H1433. [DOI] [PubMed] [Google Scholar]
- Dwivedi A, Sala‐Newby GB & George SJ (2008). Regulation of cell‐matrix contacts and beta‐catenin signalling in VSMC by integrin‐linked kinase: implications for intimal thickening. Basic Res Cardiol 103, 244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SJ & Beeching CA (2006). Cadherin: catenin complex: a novel regulator of vascular smooth muscle cell behaviour. Atherosclerosis 188, 1–11. [DOI] [PubMed] [Google Scholar]
- Giagtzoglou N, Ly CV & Bellen HJ (2009). Cell adhesion, the backbone of the synapse: ‘vertebrate’ and ‘invertebrate’ perspectives. Cold Spring Harb Perspect Biol 1, a003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SL & Bohlen HG (1984). Microvascular adaptation in the cerebral cortex of adult spontaneously hypertensive rats. Hypertension 6, 408–419. [DOI] [PubMed] [Google Scholar]
- Iliescu R, Cazan R, McLemore GR, Jr. , Venegas‐Pont M & Ryan MJ (2008). Renal blood flow and dynamic autoregulation in conscious mice. Am J Physiol Renal Physiol 295, F734–F740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson PA & Duling BR (1989). Myogenic response and wall mechanics of arterioles. Am J Physiol 257, H1147–H1155. [DOI] [PubMed] [Google Scholar]
- Jackson TY, Sun Z, Martinez‐Lemus LA, Hill MA & Meininger GA (2010). N‐cadherin and integrin blockade inhibit arteriolar myogenic reactivity but not pressure‐induced increases in intracellular Ca. Front Physiol 1, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Sabatini PJ, Lee FS, Bendeck MP & Langille BL (2002). N‐cadherin upregulation and function in response of smooth muscle cells to arterial injury. Arterioscler Thromb Vasc Biol 22, 1972–1977. [DOI] [PubMed] [Google Scholar]
- Krauss RS, Cole F, Gaio U, Takaesu G, Zhang W & Kang JS (2005). Close encounters: regulation of vertebrate skeletal myogenesis by cell–cell contact. J Cell Sci 118, 2355–2362. [DOI] [PubMed] [Google Scholar]
- le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D & de Rooij J (2010). Vinculin potentiates E‐cadherin mechanosensing and is recruited to actin‐anchored sites within adherens junctions in a myosin II‐dependent manner. J Cell Biol 189, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckband DE, le Duc Q, Wang N & de Rooij J (2011). Mechanotransduction at cadherin‐mediated adhesions. Curr Opin Cell Biol 23, 523–530. [DOI] [PubMed] [Google Scholar]
- Lewis JE, Jensen PJ, Johnson KR & Wheelock MJ (1994). E‐cadherin mediates adherens junction organization through protein kinase C. J Cell Sci 107, 3615–3621. [DOI] [PubMed] [Google Scholar]
- Mariotti A, Perotti A, Sessa C & Ruegg C (2007). N‐cadherin as a therapeutic target in cancer. Expert Opin Investig Drugs 16, 451–465. [DOI] [PubMed] [Google Scholar]
- Martinez‐Lemus LA, Crow T, Davis MJ & Meininger GA (2005). alphavbeta3‐ and alpha5beta1‐integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am J Physiol Heart Circ Physiol 289, H322–H329. [DOI] [PubMed] [Google Scholar]
- Mary S, Charrasse S, Meriane M, Comunale F, Travo P, Blangy A & Gauthier‐Rouviere C (2002). Biogenesis of N‐cadherin‐dependent cell–cell contacts in living fibroblasts is a microtubule‐dependent kinesin‐driven mechanism. Mol Biol Cell 13, 285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Takahashi K, Nariai T, Ito T, Takatani T, Fujio Y & Azuma J (2005). N‐cadherin‐mediated cell adhesion determines the plasticity for cell alignment in response to mechanical stretch in cultured cardiomyocytes. Biochem Biophys Res Commun 326, 228–232. [DOI] [PubMed] [Google Scholar]
- Mogford JE, Davis GE & Meininger GA (1997). RGDN peptide interaction with endothelial alpha5beta1 integrin causes sustained endothelin‐dependent vasoconstriction of rat skeletal muscle arterioles. J Clin Invest 100, 1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui KL, Bae YH, Gao L, Liu SL, Xu T, Radice GL, Chen CS & Assoian RK (2015). N‐cadherin induction by ECM stiffness and FAK overrides the spreading requirement for proliferation of vascular smooth muscle cells. Cell Rep 10, 1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resink TJ, Philippova M, Joshi MB, Kyriakakis E & Erne P (2009). Cadherins and cardiovascular disease. Swiss Med Wkly 139, 122–134. [DOI] [PubMed] [Google Scholar]
- Shafer SH, Puhl HL, Phelps SH & Williams CL (1999). Activation of transfected M1 or M3 muscarinic acetylcholine receptors induces cell–cell adhesion of Chinese hamster ovary cells expressing endogenous cadherins. Exp Cell Res 248, 148–159. [DOI] [PubMed] [Google Scholar]
- Sivasankar S. (2013). Tuning the kinetics of cadherin adhesion. J Invest Dermatol 133, 2318–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleek GE & Duling BR (1986). Coordination of mural elements and myofilaments during arteriolar constriction. Circ Res 59, 620–627. [DOI] [PubMed] [Google Scholar]
- Stehbens SJ, Paterson AD, Crampton MS, Shewan AM, Ferguson C, Akhmanova A, Parton RG & Yap AS (2006). Dynamic microtubules regulate the local concentration of E‐cadherin at cell–cell contacts. J Cell Sci 119, 1801–1811. [DOI] [PubMed] [Google Scholar]
- Sun Z, Li Z & Meininger GA (2012). Mechanotransduction through fibronectin‐integrin focal adhesion in microvascular smooth muscle cells: is calcium essential? Am J Physiol Heart Circ Physiol 302, H1965–H1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Martinez‐Lemus LA, Hill MA & Meininger GA (2008). Extracellular matrix‐specific focal adhesions in vascular smooth muscle produce mechanically active adhesion sites. Am J Physiol Cell Physiol 295, C268–C278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabdili H, Langer M, Shi Q, Poh YC, Wang N & Leckband D (2012). Cadherin‐dependent mechanotransduction depends on ligand identity but not affinity. J Cell Sci 125, 4362–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uglow EB, Slater S, Sala‐Newby GB, Aguilera‐Garcia CM, Angelini GD, Newby AC & George SJ (2003). Dismantling of cadherin‐mediated cell–cell contacts modulates smooth muscle cell proliferation. Circ Res 92, 1314–1321. [DOI] [PubMed] [Google Scholar]
- Wahl JK, 3rd , Kim YJ, Cullen JM, Johnson KR & Wheelock MJ. (2003). N‐cadherin‐catenin complexes form prior to cleavage of the proregion and transport to the plasma membrane. J Biol Chem 278, 17269–17276. [DOI] [PubMed] [Google Scholar]
- Wang T, Wang R, Cleary RA, Gannon OJ & Tang DD (2015). Recruitment of beta‐catenin to N‐cadherin is necessary for smooth muscle contraction. J Biol Chem 290, 8913–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier WG & Morgan KG (2003). Alpha1‐adrenergic signalling mechanisms in contraction of resistance arteries. Rev Physiol Biochem Pharmacol 150, 91–139. [DOI] [PubMed] [Google Scholar]
- Wilson C, Saunter CD, Girkin JM & McCarron JG (2015). Pressure‐dependent regulation of Ca2+ signalling in the vascular endothelium. J Physiol 593, 5231–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA & Davis MJ (1998). Modulation of calcium current in arteriolar smooth muscle by alphav beta3 and alpha5 beta1 integrin ligands. J Cell Biol 143, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S (2011). Cadherin‐actin interactions at adherens junctions. Curr Opin Cell Biol 23, 515–522. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Wada Y, Watanabe T, Nagafuchi A & Shibata M (2010). alpha‐Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol 12, 533–542. [DOI] [PubMed] [Google Scholar]