

Abstract
A hallmark of cystic fibrosis (CF) lung disease is neutrophilic airway inflammation. Elevated neutrophil counts have been associated with decreased forced expiratory volume in 1 second and poor clinical measures in patients with CF. Interleukin 8 (IL‐8), epithelial neutrophil activating protein 78 (ENA‐78), tumor necrosis factor alpha (TNF‐α), granulocyte macrophage colony‐stimulating factor (GM‐CSF), and granulocyte colony‐stimulating factor (G‐CSF) contribute to neutrophil activation and disease pathogenesis in the airways of patients with CF. Drugs that modify the production of these chemokines in the airways could potentially benefit CF patients. Thus, we determined the effects of fenofibrate on their production in cell populations obtained from the airways. Human small airway epithelial cells and CF bronchial epithelial cells were treated with IL‐1β to induce inflammation. We cotreated the cells with fenofibrate at concentrations ranging from 10 to 50 μM to determine if this drug could attenuate the inflammation. IL‐8, ENA‐78, TNF‐α, GM‐CSF, and G‐CSF production were measured from the cell culture supernates by ELISA. ANOVA statistical testing was conducted using SPSS 17.0. IL‐1β increased the production of each of the chemokines by several fold. Fenofibrate reduced IL‐1β induced production of each of these neutrophilic chemokines at the concentrations used. IL‐1β increases the production of neutrophilic chemokines in airway epithelial cells. Cotreatment with fenofibrate blunts these processes. Fenofibrate should be explored as a therapeutic option to modulate the abundant neutrophilic inflammation observed in CF.
Keywords: cystic fibrosis, fenofibrate, neutrophils, chemokines, drug repurposing
Introduction
A hallmark of cystic fibrosis (CF) lung disease is neutrophilic airway inflammation.1, 2 Neutrophils are recruited and activated in response to inflammatory chemokine production. Neutrophils subsequently increase the activation of transcription factors that produce inflammatory chemokines. Thus begins a vicious cycle of inflammation and airway destruction.3, 4, 5, 6 Inflammatory chemokine production is also stimulated by pulmonary microbial infections which occur in the vast majority of patients with CF. In normal healthy small airways, this response is beneficial, but in the airways of patients with CF neutrophilic inflammation exacerbates disease progression with hyperresponsive neutrophils contributing to airway remodeling.7, 8, 9 Specifically, our group recently reported that an elevated neutrophil to lymphocyte ratio (NLR) in the first 3 months of life was associated with a decreased forced expiratory volume in 1 second at age 12 in patients with CF.10 Furthermore, the NLR was elevated, as expected, with acute respiratory exacerbations/hospitalizations.10 Our data supporting the role of the neutrophil in the long term pathogenesis of CF‐induced bronchiectasis has been suggested by multiple groups previously.1, 3, 9, 11, 12, 13, 14 However, reliable therapeutic approaches to attenuate neutrophilic inflammation in the CF airway remain elusive.
There are several inflammatory chemokines known to stimulate chemotaxis of neutrophils. Included are interleukin 8 (IL‐8), epithelial neutrophil activating protein 78 (ENA‐78), granulocyte macrophage colony‐stimulating factor (GM‐CSF), granulocyte colony‐stimulating factor (G‐CSF), and tumor necrosis factor alpha (TNF‐α). While IL‐8 and ENA‐78 are relatively specific to neutrophilic inflammation, GM‐CSF and G‐CSF are markers of both neutrophilic and general inflammation and TNF‐α is a driver of broad inflammatory responses. Increased ENA‐78 production is seen during severe chronic obstructive pulmonary disease (COPD) exacerbations, mild asthma exacerbations, severe asthma disease, pleural effusions, and in CF airway remodeling.15, 16, 17 Furthermore, IL‐8 and TNF‐α were shown to be increased in the sputum of CF patients as compared to asthma and COPD patients and healthy controls.14, 18, 19 Moreover, GM‐CSF and G‐CSF have been negatively correlated with lung function in CF patients.20, 21, 22 Drugs that could modulate the production of these chemokines have the potential to influence the pathogenesis of CF associated respiratory pathologies and improve prognosis.
Fenofibrate is a ligand for peroxisome proliferator‐activated receptor α (PPAR‐α), a nuclear receptor transcription factor. Binding of this ligand to its receptor causes upregulation of lipoprotein lipase and apolipoprotein A‐I and A‐II. Both apolipoprotein A‐I and A‐II are involved with reverse cholesterol transport. Binding of fenofibrate to PPAR‐α also results in down‐regulation of apolipoprotein C‐III. The overall effect is a reduction in triglycerides and a modest increase in high density lipoprotein. Several nonlipid related effects of the fibrates have surfaced, suggesting multiple mechanisms of action. Our laboratory and others have shown that fibrates exhibit anti‐inflammatory properties in endothelial cells, monocytes, macrophages, T lymphocytes, vascular smooth muscle cells, and adipocytes.23, 24, 25, 26, 27, 28, 29, 30 However, there is a paucity of data regarding the effect of fibrates on the production of neutrophilic chemokines in the airways. Therefore, we investigated the effects of fenofibrate on IL‐8, ENA‐78, GM‐CSF, G‐CSF, and TNF‐α production in human small airway epithelial cells (SAEC) and bronchial epithelial cells obtained from patients with CF.
Here we show that fenofibrate significantly reduces the production of the aforementioned chemokines in airway epithelial cells from healthy donors and those with CF.
Methods
We cultured SAEC and CF human bronchial epithelial cells (DHBEC‐CF) from Lonza Inc., (Walkersville, MD, USA). The healthy donor SAEC are from a distal region of the lung where the bronchioles measure 1 mm in diameter. The CF bronchial epithelial cells are from epithelia just above the bifurcation in the lungs. These two cells types were chosen because they were the most similar tissues available for purchase with one being from a donor without lung disease and one being from a donor with CF. In brief, SAECs at passage three were seeded at 25,000 cells/cm2 and cultured to 80% confluence in growth media (SAGM, Lonza Inc., Walkersville, MD, USA) at 37°C and 5% CO2. DHBEC‐CF, also on third passage, were seeded at 35,000 cells/cm2 and cultured to the same confluency under the same conditions in their respective growth media. We assessed cell viability with trypan blue staining of cells across fenofibrate concentrations from 1 to 200 μM. Concentrations of fenofibrate that reduced cell viability to less than 80% of control were eliminated from subsequent studies. Fenofibrate was dissolved in dimethyl sulfoxide (Sigma‐Aldrich, St. Louis, MO, USA). The DMSO concentration was always less than or equal to 0.1% during cell treatment.
We tested the concentration dependent effects of fenofibrate on IL‐1β stimulated production of ENA‐78, IL‐8, GM‐CSF, G‐CSF, and TNF‐α proteins. IL‐1β was utilized to stimulate the epithelial cells because ENA‐78, IL‐8, GM‐CSF, G‐CSF and TNF‐α are highly inducible by this cytokine,27, 31, 32 and IL‐1β is elevated in the sputum of patients with CF.12 We also examined the effects of fenofibrate on the expression of several subunits of the nuclear factor of light chain enhancer of activated B‐cells (NFκB). NFκB is a transcription factor involved in rapid signal transduction for a host of events including inflammation, immunity, growth, and apoptosis. Treatment groups included control (DMSO), IL‐1β 2 ng/mL, fenofibrate 10–50 μM, and fenofibrate 10–50 μM plus IL‐1β. IL‐1β was added 2 hours after fenofibrate. Treatment durations were 24 hours. Fenofibrate and IL‐1β were acquired from Sigma‐Aldrich. Each experiment was performed in triplicate.
We also tested the effects of fenofibrate on the gene expression of ENA‐78, IL‐8, and RELA. Our experimental groups included unstimulated control (DMSO), IL‐1β 2 ng/mL, fenofibrate 10 μM, and fenofibrate plus IL‐1β. We selected the lowest fenofibrate concentration associated with significant findings in the protein studies to serve as the treatment concentration for gene expression studies.
ENA‐78, IL‐8, GM‐CSF, G‐CSF, and TNF‐α protein concentrations were measured in duplicate by ELISA (R+D Systems, Minneapolis, MN, USA) and normalized to total protein content as previously described.27, 33 The standard curves for each cytokine were generated using Gen 5 Microplate Data Collection and Analysis Software (Biotek, Winooski, VT, USA) by plotting the corrected absorbance (± standard deviation) of the supplied standard against their known concentrations.
RNA was isolated using RNeasy Mini Kits (Qiagen Inc., Valencia, CA, USA). Complementary DNA conversion was performed by high capacity cDNA reverse transcription using 500 ng of total RNA (7500 Fast Real‐Time PCR Applied Biosystems, Foster City, CA, USA). RNA and cDNA quality and quantity were assessed using nanodrop (ThermoFisher Scientific, Waltham, MA, USA). Real‐time quantitative PCR was performed using primer and probe sets for ENA‐78 (CXCL5), IL‐8, NFκB1, NFκB2, RELA, and RELB genes as well as the reference gene, 18s (Taqman Gene Expression Assays, Applied Biosystems).
ENA‐78, IL‐8, TNF‐α, GM‐CSF, G‐CSF, and TNF‐α protein concentrations are reported as mean ± standard error of the mean. Proteins were compared by one‐way ANOVA with Bonferroni's correction for multiple comparisons. Relative gene expression was reported using the 2‐ΔΔCt method and significance (p < 0.05) was tested using the Student's t‐test. Statistical analyses were performed using SPSS version 17.0 (SPSS Inc., Chicago, USA).
Results
Fenofibrate effects on neutrophilic chemokine production
Compared to controls IL‐1β increased the production of ENA‐78 in SAECs by over 15‐fold (1,044 pg/mg vs. 15,373 pg/mg), TNF‐α by over fourfold (219 pg/mg vs. 951 pg/mg), IL‐8 by over 30‐fold (8,444 pg/mg vs. 259,854 pg/mg) GM‐CSF by over sixfold (9,438 pg/mg vs. 57,960 pg/mg), and G‐CSF by over 13‐fold (3,551 pg/mg vs. 49,102 pg/mg; p < 0.000001 for all). Attenuation of this IL‐1β‐induced production of ENA‐78, TNF‐α, IL‐8, GM‐CSF, and G‐CSF was measured across a range of fenofibrate concentrations (10, 25, and 50 μM). Fenofibrate significantly reduced IL‐1β‐induced production of ENA‐78, GM‐CSF, and G‐CSF at concentrations 10–50 μM (Figure 1). Fenofibrate also attenuated the IL‐1β stimulated production of IL‐8 and TNF‐α (Figures 2 and 3, respectively).
Figure 1.
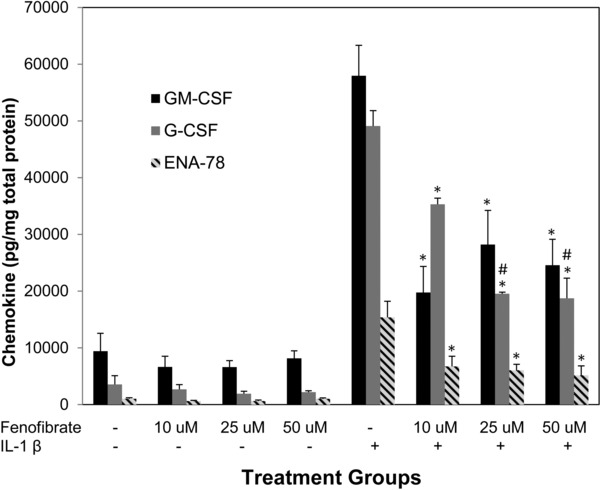
Reduction of IL‐1β induced chemokine production in SAECs with fenofibrate cotreatment. Protein production of several neutrophilic chemokines is induced by IL‐1β at a concentration of 2 ng/mL and attenuated by fenofibrate across varying concentrations. *indicates p‐value < 0.01 versus IL‐1β. #indicates p‐value < 0.001 versus fenofibrate 10 μM + IL1β.
Figure 2.
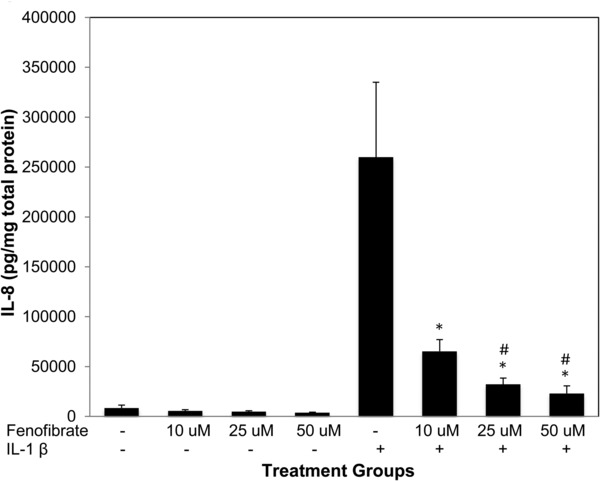
Fenofibrate reduces IL‐1β induced IL‐8 production in SAECs. IL‐8 production is induced by IL‐1β (2 ng/mL) compared to negative controls. Fenofibrate is able to decrease the production of IL‐8 in a dose‐dependent fashion. *indicates p‐value < 0.01 versus IL‐1β. #indicates p‐value < 0.001 versus fenofibrate 10 μM + IL1β.
Figure 3.
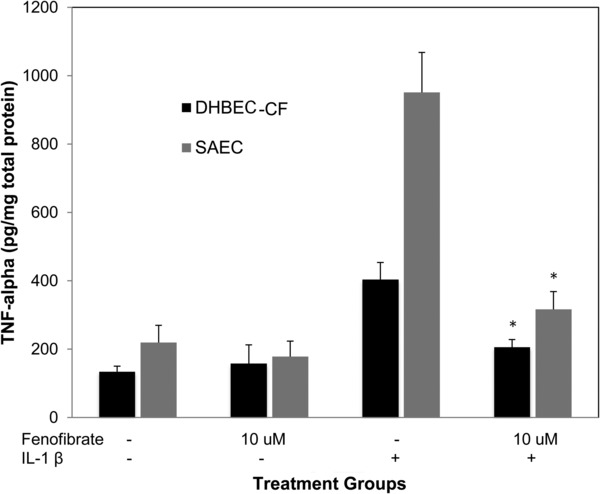
Fenofibrate reduces IL‐1β induced TNF‐α production in SAECs and DHBEC‐CF. TNF‐α production in DHBEC‐CF and SAECs is induced by IL‐1β 2 ng/mL and attenuated by fenofibrate 10 μM. *indicates p‐value < 0.0001 versus IL‐1β.
In DHBEC‐CF, compared to controls, IL‐1β increased the production of ENA‐78 by nearly twofold (1,153 pg/mg vs. 2,300 pg/mg), TNF‐α by over threefold (134 pg/mg vs. 404 pg/mg) GM‐CSF by 3.5‐fold (1,927 pg/mg vs. 6,774 pg/mg), and G‐CSF by approximately eightfold (1,655 pg/mg vs. 12,842 pg/mg; p < 0.002 for all). Because the DHBEC‐CF were available in smaller quantities than the SAECs we only performed IL‐1β cotreatments with the minimum effective concentration of fenofibrate (10 μM). Fenofibrate significantly reduced IL‐1β‐induced production of TNF‐α, GM‐CSF, G‐CSF, and ENA‐78 at the low concentration of 10 μM (Figures 3, 4, 5).
Figure 4.
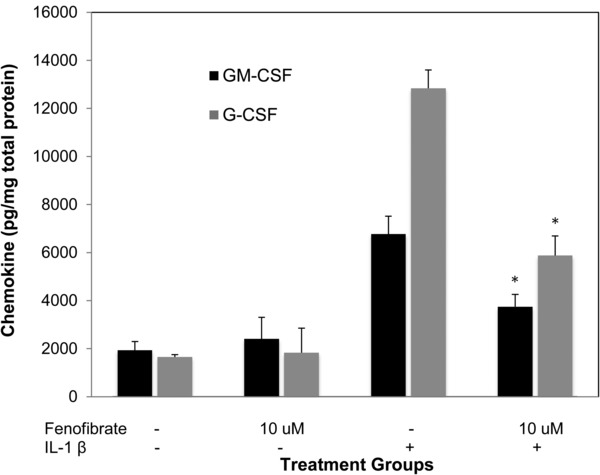
Fenofibrate reduces IL‐1β induced GM‐CSF and G‐CSF production in DHBEC‐CF. GM‐CSF and G‐CSF production in DHBEC‐CF is induced by IL‐1β 2 ng/mL and attenuated by fenofibrate 10 μM. *indicates p‐value < 0.0001 versus IL‐1β.
Figure 5.
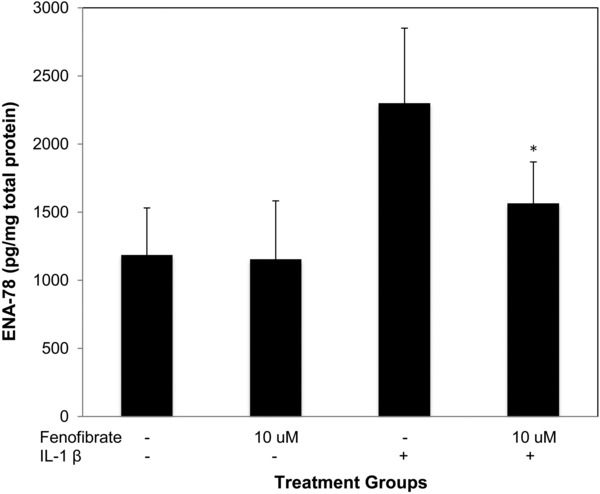
Fenofibrate reduces IL‐1β induced ENA‐78 production in DHBEC‐CF. ENA‐78 production in DHBEC‐CF is induced by IL‐1β 2 ng/mL and attenuated by fenofibrate 10 μM. * indicates p‐value < 0.02 versus IL‐1β.
Gene expression data
The effect of fenofibrate on the gene expression of ENA‐78 (CXCL5) and IL‐8 is shown in Figure 6. IL‐1β induces the expression of both of these genes while fenofibrate alone downregulates them both. Fenofibrate attenuates the effects of IL‐1β on the gene expression for ENA‐78 and IL‐8. Finally, IL‐1β induces RELA gene expression (Figure 7). Treatment of SAECs with IL‐1β 2 ng/mL increases RELA expression by twofold. Fenofibrate cotreatment attenuates the IL‐1β induced increase in RELA expression. The mRNA expression of the other NFκB subunits, NFκB1 NFκB2 and RELB were not significantly modulated by treatment with IL‐1β and fenofibrate (data shown in Figures S1 and S2).
Figure 6.
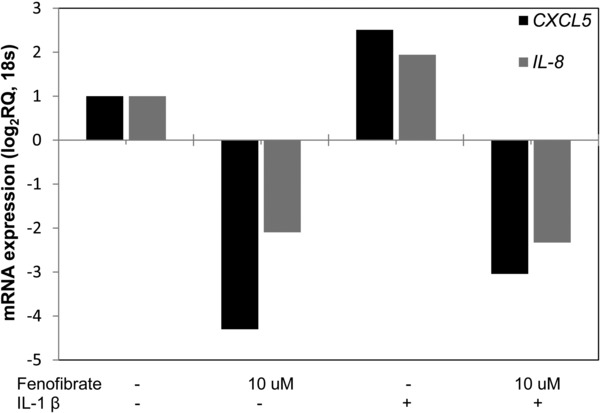
Fenofibrate reduces expression of ENA‐78 (CXCL5) and IL‐8 in SAECs. Treatment of SAECs with IL‐1β 2 ng/mL increases CXCL5 and IL‐8 gene expression. Fenofibrate reduces expression in unstimulated cells and blocks the induction of these genes upon addition of IL‐1β.
Figure 7.
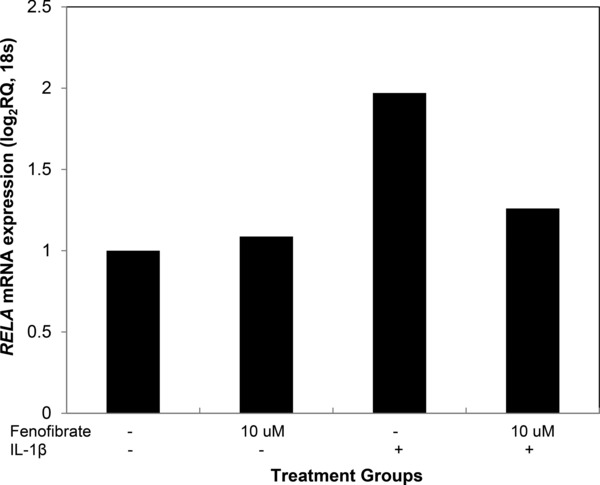
Fenofibrate reduces the stimulated expression of RELA in SAECs. Treatment of SAECs with IL‐1β 2ng/mL increases RELA mRNA expression by twofold. Cotreatment with Fenofibrate attenuates the induction of RELA mRNA expression.
Discussion
In this study, we show that fenofibrate blunts the production of clinically relevant neutrophilic chemokines in airway epithelial cells relevant to the pathogenesis of CF associated lung pathologies. Neutrophilic inflammation is generally accepted as a distinguishing factor of CF associated lung pathologies. This has been shown to exacerbate disease progression and may serve as druggable target for new therapeutic strategies. Evidence of fenofibrate's anti‐inflammatory effects continues to emerge. However, because it is primarily used in adults with mixed dyslipidemia, these anti‐inflammatory effects have been largely characterized in patients with cardiovascular diseases and cardiovascular cell models. The anti‐inflammatory effects of fibrates may be useful in the treatment of respiratory diseases like CF. However, the anti‐inflammatory effects of fibrates in respiratory epithelium had not been previously explored. In this study, we used normal SAEC and bronchial epithelial cells from CF patients to characterize fenofibrate's neutrophilic anti‐inflammatory effects in the respiratory epithelium.
In this study, IL‐1β significantly increased the production of neutrophilic chemokines ENA‐78, IL‐8, GM‐CSF, and G‐CSF in both airway cell model systems. IL‐1β is a proinflammatory cytokine critical for the inflammatory response against infection and allergens. IL‐1β was utilized because it is elevated in the sputum of patients with CF and it induces ENA‐78, IL‐8, GM‐CSF, and G‐CSF. Fenofibrate blunted the IL‐1β stimulated ENA‐78, IL‐8, GM‐CSF, and G‐CSF production in both the normal cell model, and more importantly, the CF cell model. Fenofibrate also reduced the IL‐1β stimulated TNF‐α production in both normal and CF airway epithelial cells. This demonstrates a broader anti‐inflammatory effect.
To gain additional insight into the mechanism of the observed anti‐inflammatory effects, we examined fenofibrate's effect on the NFκB family of transcription factors; NFκB1 (p105, p50), NFκB2 (p100, p52), RELA (p65), and RELB. RELA is the p65 subunit of NFκB and has been shown to regulate the gene expression of ENA‐78, IL‐8, and TNF‐α.31, 34 We observed that RELA was inducible by IL‐1β in our experiments and this effect was attenuated by cotreatment with fenofibrate. None of the other subunits were significantly induced by IL‐1β or modulated by fenofibrate. Therefore, our data suggests that this may explain the anti‐inflammatory effects that were observed in our experiments. This observation is supported by previous reports where PPAR‐α agonists were shown to decrease inflammatory signaling through the attenuation of NFκB RELA (p65) transcriptional activity in vivo.29
These results are in keeping with asthma model studies investigating chemokine production in mouse bronchoalveolar lavage fluid after allergen‐induced airway inflammation. These studies showed increases in neutrophilic and general inflammatory chemokines upon stimulation with lipopolysaccharide and ovalbumin challenge to the airways as well as suppression of the inflammatory response by fenofibrate.35, 36, 37 Our study illustrates that fenofibrate robustly attenuates the production of neutrophilic chemokines in addition to a general anti‐inflammatory effect in small airway epithelium and in bronchial epithelial cells obtained from CF patients. This effect was apparent even at the lowest treatment concentration of fenofibrate (10 μM). This concentration correlates with the lower end of clinically observed fenofibrate plasma concentrations (approximately 8–30 μM). Thus, this data suggests that fenofibrate may be associated with beneficial anti‐inflammatory effects in patients that have airway inflammation. Inflammation in small airways appears early in the pathogenesis of CF lung pathologies.2, 4 In CF pathogenesis, inflammation is driven by neutrophils attracted to the tissues by chemokines in the bronchial epithelium. These aberrant inflammatory processes have been associated with a reduction of PPARα mRNA in the lymphocytes of CF patients and with increased lung fibrosis in animal models. PPARα knockout mice developed significantly worse levels of fibrosis and inflammatory markers.38, 39 Our data suggests that fenofibrate could stimulate PPARα expression and attenuate the neutrophilic inflammation that contributes to fibrosis in CF.
The data that we present herein are exciting and provide preliminary support for additional studies using fenofibrate to favorably modulate inflammatory processes in the airway. However, our study is not without limitations. Specifically, studies should be replicated using an air–liquid interface culture protocol similar to that described both by Krunkosky and Chen in both normal and CF bronchial epithelium.5, 40 This protocol is designed to more efficiently reflect physiological conditions for airway epithelium. Furthermore, we lack clinical data in CF patients to support the experimental data that we presented. However, we believe that our approach of establishing a mechanism of proposed function in cell models is the safest way of identifying drugs that can be repurposed for vulnerable CF patients. Because neutrophilia contributes to CF lung pathologies early in life we needed to perform these preliminary studies in vitro before proposing fenofibrate clinical studies in pediatric CF patients.
Conclusion
In summary, fenofibrate blunts IL‐1β stimulated neutrophilic inflammation in both SAEC from donors without lung diseases and from bronchial epithelial cells from CF patients. Furthermore, our data demonstrated that fenofibrate's anti‐inflammatory effects were robust across the range of clinically observed fenofibrate concentrations suggesting potential low‐dose options in pediatric CF patients. Fenofibrate should be explored as a therapeutic option to modulate neutrophilic inflammation in CF.
Acknowledgments/Funding
This project was supported by UAMS College of Pharmacy Start‐Up Funds (ETP), UAMS College of Pharmacy Voldeng Fellowships (AJS, RAF), and National Institutes of Health Awards: UL1TR000039 & KL2TR000063 (ETP).
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1. NFκB1 and NFκB2 are not induced by IL‐1β in SAECs. Treatment of SAECs with IL‐1 2 ng/mL alone or together with fenofibrate 10 μM does not induce the transcription factors NFκB1 and NFκB2.
Figure S2. RELB is not induced by IL‐1β in SAECs. Treatment of SAECs with IL‐1 2 ng/mL alone or together with fenofibrate 10 μM does not induce the RELB.
References
- 1. Abraham E. Neutrophils and acute lung injury. Crit Care Med. Apr 2003; 31(4 Suppl): S195–199. [DOI] [PubMed] [Google Scholar]
- 2. Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. Jan 22 2015; 372(4): 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Basran A, Jabeen M, Bingle L, Stokes CA, Dockrell DH, Whyte MK, Walmsley SR, Higgins KR, Vogel SN, Wilson HL, et al. Roles of neutrophils in the regulation of the extent of human inflammation through delivery of IL‐1 and clearance of chemokines. J Leukoc Biol. Jan 2013; 93(1): 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chmiel JF, Berger M, Konstan MW. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol. Aug 2002; 23(1): 5–27. [DOI] [PubMed] [Google Scholar]
- 5. Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, Adler KB. Effects of TNF‐alpha on expression of ICAM5‐1 in human airway epithelial cells in vitro. Signaling pathways controlling surface and gene expression. Am J Respir Cell Mol Biol. Jun 2000; 22(6): 685–692. [DOI] [PubMed] [Google Scholar]
- 6. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, Cosmi L, Lunardi C, Annunziato F, Romagnani S, et al. Evidence for a cross‐talk between human neutrophils and Th17 cells. Blood. Jan 14 2010; 115(2): 335–343. [DOI] [PubMed] [Google Scholar]
- 7. McKeon DJ, Cadwallader KA, Idris S, Cowburn AS, Pasteur MC, Barker H, Haworth CS, Bilton D, Chilvers ER, Condliffe AM. Cystic fibrosis neutrophils have normal intrinsic reactive oxygen species generation. Eur Respir J. Jun 2010; 35(6): 1264–1272. [DOI] [PubMed] [Google Scholar]
- 8. Pohl K, Hayes E, Keenan J, Pohl K, Hayes E, Keenan J, Henry M, Meleady P, Molloy K, Jundi B, et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood. Aug 14 2014; 124(7): 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, Murray CP, Stick SM; AREST CF Investigators . Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. May 23 2013; 368(21): 1963–1970. [DOI] [PubMed] [Google Scholar]
- 10. O'Brien CE, Price ET. The blood neutrophil to lymphocyte ratio correlates with clinical status in children with cystic fibrosis: a retrospective study. PloS One. 2013; 8(10): e77420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Byrnes CA, Vidmar S, Cheney JL, Carlin JB, Armstrong DS, Cooper PJ, Grimwood K, Moodie M, Robertson CF, Rosenfeld M, et al. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax. Jul 2013; 68(7): 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eickmeier O, Huebner M, Herrmann E, Zissler U, Rosewich M, Baer PC, Buhl R, Schmitt‐Grohé S, Zielen S, Schubert R. Sputum biomarker profiles in cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) and association between pulmonary function. Cytokine. May 2010; 50(2): 152–157. [DOI] [PubMed] [Google Scholar]
- 13. Waters V, Stanojevic S, Atenafu EG, Waters V, Stanojevic S, Atenafu EG, Lu A, Yau Y, Tullis E, Ratjen F. Effect of pulmonary exacerbations on long‐term lung function decline in cystic fibrosis. Eur Respir J. Jul 2012; 40(1): 61–66. [DOI] [PubMed] [Google Scholar]
- 14. Xiao W, Hsu YP, Ishizaka A, Kirikae T, Moss RB. Sputum cathelicidin, urokinase plasminogen activation system components, and cytokines discriminate cystic fibrosis, COPD, and asthma inflammation. Chest. Oct 2005; 128(4): 2316–2326. [DOI] [PubMed] [Google Scholar]
- 15. Liu GN, Shi HZ, Xie ZH, Shen HH, Huang HQ, Deng JM, Liang QL, Wu YB. Epithelial neutrophil‐activating peptide‐78 recruits neutrophils into pleural effusion. Eur Respir J. Jul 2009; 34(1): 184–190. [DOI] [PubMed] [Google Scholar]
- 16. Qiu Y, Zhu J, Bandi V, Guntupalli KK, Jeffery PK. Bronchial mucosal inflammation and upregulation of CXC chemoattractants and receptors in severe exacerbations of asthma. Thorax. Jun 2007; 62(6): 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tasaka S, Mizoguchi K, Funatsu Y, Namkoong H, Yamasawa W, Ishii M, Hasegawa N, Betsuyaku T. Cytokine profile of bronchoalveolar lavage fluid in patients with combined pulmonary fibrosis and emphysema. Respirology. Jul 2012; 17(5): 814–820. [DOI] [PubMed] [Google Scholar]
- 18. Greally P, Hussein MJ, Cook AJ, Sampson AP, Piper PJ, Price JF. Sputum tumour necrosis factor‐alpha and leukotriene concentrations in cystic fibrosis. Arch Dis Child. Mar 1993; 68(3): 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koehler DR, Downey GP, Sweezey NB, Tanswell AK, Hu J. Lung inflammation as a therapeutic target in cystic fibrosis. Am J Respir Cell Mol Biol. Oct 2004; 31(4): 377–381. [DOI] [PubMed] [Google Scholar]
- 20. Jensen PO, Moser C, Kharazmi A, Presler T, Koch C, Hoiby N. Increased serum concentration of G‐CSF in cystic fibrosis patients with chronic Pseudomonas aeruginosa pneumonia. J Cyst Fibros. Aug 2006; 5(3): 145–151. [DOI] [PubMed] [Google Scholar]
- 21. Liou TG, Adler FR, Keogh RH, Li Y, Jensen JL, Walsh W, Packer K, Clark T, Carveth H, Chen J, et al. Sputum biomarkers and the prediction of clinical outcomes in patients with cystic fibrosis. PloS One. 2012; 7(8): e42748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moser C, Jensen PO, Pressler T, Moser C, Jensen PØ, Pressler T, Frederiksen B, Lanng S, Kharazmi A, Koch C, Høiby N. Serum concentrations of GM‐CSF and G‐CSF correlate with the Th1/Th2 cytokine response in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. APMIS. Jun 2005; 113(6): 400–409. [DOI] [PubMed] [Google Scholar]
- 23. Belfort R, Berria R, Cornell J, Cusi K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J Clin Endocrinol Metab. Feb 2010; 95(2): 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krysiak R, Okopien B. The effect of fenofibrate on lymphocyte cytokine release in patients with impaired fasting glucose and impaired glucose tolerance: a preliminary report. Atherosclerosis. Nov 2010; 213(1): 325–328. [DOI] [PubMed] [Google Scholar]
- 25. Krysiak R, Stachura‐Kulach A, Okopien B. Metabolic and monocyte‐suppressing actions of fenofibrate in patients with mixed dyslipidemia and early glucose metabolism disturbances. Pharmacol Rep. Jan‐Feb 2010; 62(1): 120–130. [DOI] [PubMed] [Google Scholar]
- 26. Okopien B, Krysiak R, Herman ZS. Effects of short‐term fenofibrate treatment on circulating markers of inflammation and hemostasis in patients with impaired glucose tolerance. J Clin Endocrinol Metab. May 2006; 91(5): 1770–1778. [DOI] [PubMed] [Google Scholar]
- 27. Price ET, Welder GJ, Zineh I. Modulatory effect of fenofibrate on endothelial production of neutrophil chemokines IL‐8 and ENA‐78. Cardiovasc Drugs Ther. Apr 2012; 26(2): 95–99. [DOI] [PubMed] [Google Scholar]
- 28. Ryoo S, Won M, Kim DU, Kim L, Han G, Park SK, Mukaida N, Maeng P, Yoo HS, Hoe KL. PPARalpha activation abolishes LDL‐stimulated IL‐8 production via AP‐1 deactivation in human aortic smooth muscle cells. Biochem Biophys Res Commun. May 28 2004; 318(2): 329–334. [DOI] [PubMed] [Google Scholar]
- 29. Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, et al. Activation of human aortic smooth‐muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. Jun 25 1998; 393(6687): 790–793. [DOI] [PubMed] [Google Scholar]
- 30. Ye P, Li JJ, Su G, Zhang C. Effects of fenofibrate on inflammatory cytokines and blood pressure in patients with hypertriglyceridemia. Clin Chim Acta. Jun 2005; 356(1‐2): 229–232. [DOI] [PubMed] [Google Scholar]
- 31. Chang MS, McNinch J, Basu R, Simonet S. Cloning and characterization of the human neutrophil‐activating peptide (ENA‐78) gene. J Biol Chem. Oct 14 1994; 269(41): 25277–25282. [PubMed] [Google Scholar]
- 32. Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, Akira S. Transcription factors NF‐IL6 and NF‐kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci USA. Nov 1 1993; 90(21): 10193–10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zineh I, Luo X, Welder GJ, Debella AE, Wessel TR, Arant CB, Schofield RS, Chegini N. Modulatory effects of atorvastatin on endothelial cell‐derived chemokines, cytokines, and angiogenic factors. Pharmacotherapy. Mar 2006; 26(3): 333–340. [DOI] [PubMed] [Google Scholar]
- 34. Roebuck KA, Carpenter LR, Lakshminarayanan V, Page SM, Moy JN, Thomas LL. Stimulus‐specific regulation of chemokine expression involves differential activation of the redox‐responsive transcription factors AP‐1 and NF‐kappaB. J Leukoc Biol. Mar 1999; 65(3): 291–298. [DOI] [PubMed] [Google Scholar]
- 35. Delayre‐Orthez C, Becker J, Auwerx J, Frossard N, Pons F. Suppression of allergen‐induced airway inflammation and immune response by the peroxisome proliferator‐activated receptor‐alpha agonist fenofibrate. Eur J Pharmacol. Feb 26 2008; 581(1‐2): 177–184. [DOI] [PubMed] [Google Scholar]
- 36. Delayre‐Orthez C, Becker J, Guenon I, Lagente V, Auwerx J, Frossard N, Pons F. PPARalpha downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res. 2005; 6: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trifilieff A, Bench A, Hanley M, Bayley D, Campbell E, Whittaker P. PPAR‐alpha and ‐gamma but not ‐delta agonists inhibit airway inflammation in a murine model of asthma: in vitro evidence for an NF‐kappaB‐independent effect. Br J Pharmacol. May 2003; 139(1): 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cuzzocrea S, Mazzon E, Di Paola R, Peli A, Bonato A, Britti D, Genovese T, Muià C, Crisafulli C, Caputi AP. The role of the peroxisome proliferator‐activated receptor‐alpha (PPAR‐alpha) in the regulation of acute inflammation. J Leukoc Biol. May 2006; 79(5): 999–1010. [DOI] [PubMed] [Google Scholar]
- 39. Reynders V, Loitsch S, Steinhauer C, Wagner T, Steinhilber D, Bargon J. Peroxisome proliferator‐activated receptor alpha (PPAR alpha) down‐regulation in cystic fibrosis lymphocytes. Respir Res. 2006; 7: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen Y, Nickola TJ, DiFronzo NL, Colberg‐Poley AM, Rose MC. Dexamethasone‐mediated repression of MUC5AC gene expression in human lung epithelial cells. Am J Respir Cell Mol Biol. Mar 2006; 34(3): 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1. NFκB1 and NFκB2 are not induced by IL‐1β in SAECs. Treatment of SAECs with IL‐1 2 ng/mL alone or together with fenofibrate 10 μM does not induce the transcription factors NFκB1 and NFκB2.
Figure S2. RELB is not induced by IL‐1β in SAECs. Treatment of SAECs with IL‐1 2 ng/mL alone or together with fenofibrate 10 μM does not induce the RELB.