
Fig. 1.
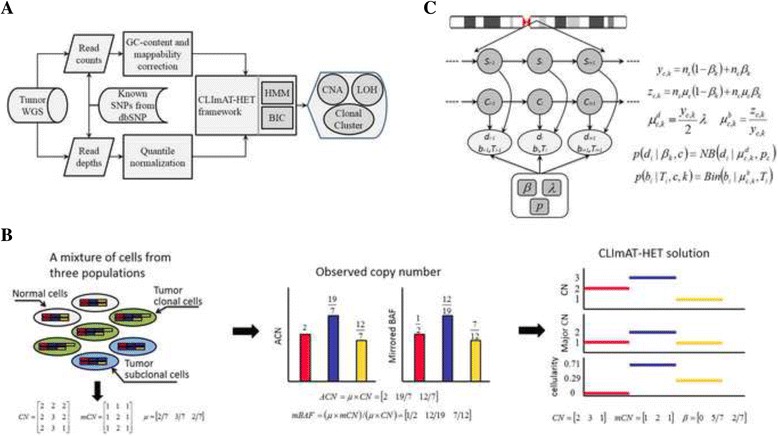
Overview of the CLImAT-HET statistical framework. a CLImAT-HET analysis workflow. 1) Read counts and read depths of known SNP positions are extracted from whole-genome sequencing data of tumor sample; 2) read counts signals are preprocessed to correct GC-content and mappability bias, quantile normalization of read depths is performed to eliminate allelic bias; 3) read counts and read depth signals are jointly analyzed using an integrated hidden Markov model, and the model complexity is iteratively evaluated for different number of clonal clusters using Bayesian information criterion; 4) and finally the clonal/subclonal CNA and LOH segments as well as the cellularity of each clonal cluster are inferred. b Representation of the intra-tumor heterogeneity and CLImAT-HET solution. The observed copy number signals are generated from three types of cell populations: normal cells, tumor cells with an amplification, and tumor cells with the amplification event and an additional deletion event. CLImAT-HET infers the total and major copy number as well as the corresponding cellularity of each event. c The factorial hidden Markov model adopted in CLImAT-HET. The hidden Markov model has two underlying Markov chains with one chain depicting aberration events and another delineating corresponding clonal clusters