

Abstract
Genetic variation in the platelet endothelial aggregation receptor 1 (PEAR1) gene, most notably rs12041331, is implicated in altered on‐aspirin platelet aggregation and increased cardiovascular event risk. We prospectively tested the effects of aspirin administration at commonly prescribed doses (81, 162, and 324 mg/day) on agonist‐induced platelet aggregation by rs12041331 genotype in 67 healthy individuals. Prior to aspirin administration, rs12041331 minor allele carriers had significantly reduced adenosine diphosphate (ADP)‐induced platelet aggregation compared with noncarriers (P = 0.03) but was not associated with other platelet pathways. In contrast, rs12041331 was significantly associated with on‐aspirin platelet aggregation when collagen and epinephrine were used to stimulate platelet aggregation (P < 0.05 for all associations), but not ADP. The influence of PEAR1 rs12041331 on platelet aggregation is pathway‐specific and is altered by aspirin at therapeutic doses, but not in a dose‐dependent manner. Additional studies are needed to determine the impact of PEAR1 on cardiovascular events in aspirin‐treated patients.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ PEAR1 rs12041331 has been previously implicated in altered baseline and on‐aspirin platelet aggregation. In addition, the impact of genotype on cardiovascular disease risk seems to be aspirin‐dependent.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ We sought to prospectively evaluate the influence of PEAR1 rs12041331 on platelet function in an aspirin dose‐specific and aggregation pathway‐specific manner.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE?
✓ The platelet aggregation pathways impacted by PEAR1 rs12041331 are changed by exposure to aspirin, whereby ADP‐induced platelet aggregation is influenced prior to drug administration, whereas collagen‐induced and epinephrine‐induced platelet aggregation are influenced during aspirin treatment. Furthermore, no genotype‐specific differences in platelet function were observed with increasing doses of aspirin.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ PEAR1 is a critical signaling component of several pathways that regulate platelet aggregation at baseline and in the context of antiplatelet therapy. Better understanding of the mechanisms by which PEAR1 influences platelet function may facilitate novel drug‐target discovery and influence the way patients with coronary artery disease on aspirin are treated.
Dual antiplatelet therapy with aspirin and clopidogrel is the most commonly used regimen for management of patients with acute coronary syndromes undergoing percutaneous coronary intervention. Although generally effective in most patients, significant interindividual variability in dual antiplatelet therapy response has been well documented, leading to suboptimal therapy in some patients, and resulting in an increase in cardiovascular event rates.1, 2, 3, 4 In recent years, substantial effort has been made to identify pharmacogenetic determinants of variable antiplatelet therapy response. Indeed, these investigations have convincingly shown that response to clopidogrel is significantly influenced by genetic variation, most notably CYP2C19*2, which resulted in an update to the clopidogrel label by the US Food and Drug Administration in 2010.3 In contrast, identification of genetic factors that influence aspirin response has remained difficult despite the fact that up to 77% of the variation in aspirin response, as assessed by ex vivo platelet aggregometry, is heritable.5
One of the few recently identified aspirin response candidate genes is platelet endothelial aggregation receptor 1 (PEAR1), which encodes a type 1 membrane protein that has been implicated in megakaryopoiesis, thrombopoiesis, and stabilization of platelet aggregation through enhancement of platelet αIIbβ3 activation.6, 7, 8 To date, several retrospective investigations have reported that genetic variation in PEAR1 significantly influences agonist‐induced platelet aggregation at baseline and after aspirin exposure.4, 9, 10, 11, 12, 13 The most consistently and highly associated of these genetic variants is rs12041331, an intronic variant that is believed to modify PEAR1 expression.4, 9, 10, 11, 13 The effects of aspirin on platelet aggregation according to rs12041331 genotype have not been studied prospectively. In this investigation, we prospectively tested the effects of commonly prescribed aspirin doses (81, 162, and 324 mg/day) on agonist‐induced platelet aggregation according to rs12041331 genotype in 67 healthy individuals using a fixed‐sequence, crossover design. Ex vivo platelet reactivity testing was completed using multiple platelet agonists to gain insights regarding the mechanism(s) by which PEAR1 influences basal and on‐treatment platelet reactivity. Urinary thromboxane B2, a circulating biomarker of platelet function and aspirin responsiveness, was also assessed by PEAR1 genotype in order to complement our platelet aggregation results.
MATERIALS AND METHODS
Subjects
We recruited 67 relatively healthy Amish individuals from Lancaster, Pennsylvania, who had previously participated in the Heredity and Phenotype Intervention (HAPI) Heart study,14 the Pharmacogenomics of Anti‐Platelet Intervention (PAPI) Study,3 and targeted family members by known or predicted PEAR1 rs12041331 genotype between August 2013 and November 2015. Enrolled subjects were rescreened to ensure they met the eligibility criteria, which were identical as those for the original PAPI study.3 The protocol was approved by the University of Maryland, Baltimore Institutional Review Board, and all subjects provided written informed consent.
Study design
The study design used in this investigation is shown in Figure 1. Study procedures were performed at the University of Maryland Amish Research Clinic in Lancaster, PA. All participants safely withdrew from medications, vitamins, and supplements at least 1 week prior to, and for the duration of, the study. Any participant who was taking aspirin withdrew from this mediation at least 14 days prior to protocol initiation.
Figure 1.
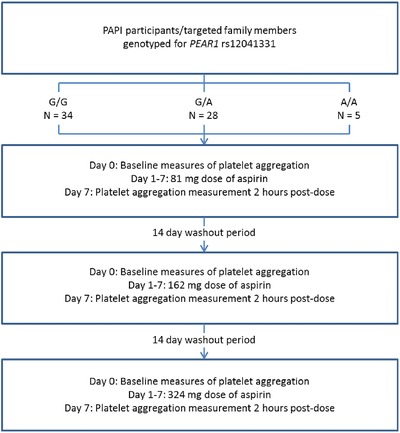
Each study participant underwent 7‐day aspirin interventions at a low (81 mg/day), medium (162 mg/day), and high aspirin dose (324 mg/day), with a minimum 2‐week washout in between each aspirin regimen. Platelet aggregation was measured at day 0 prior to the first aspirin dose and on day 7 following the final aspirin dose of each treatment period. Blood and urine samples were also obtained at day 0 and day 7 of each treatment period. PAPI, Pharmacogenomics of Anti‐Platelet Intervention; PEAR1, platelet endothelial aggregation receptor 1.
During the initial clinic visit, blood samples were obtained from each participant for baseline measures of platelet aggregation and for specimen banking. After samples were obtained, participants received an 81 mg chewable dose of nonenteric‐coated aspirin and instructed to take 81 mg/day for the next 6 days. Subjects returned to the clinic on the 7th day, on‐aspirin measures of platelet aggregation were recorded 2 h after the last 81 mg dose, and samples were banked for future analysis. At this time, participants underwent a washout period of at least 14 days to allow platelet function to return to basal levels, consistent with the average lifespan of platelets (range, 7–10 days).15 After the washout period, these procedures were repeated for a second time with 162 mg/day of aspirin for 7 days. Again, platelet function was assessed and samples were banked pre‐aspirin and post‐aspirin administration. Participants then underwent another 14‐day washout period. These procedures were repeated for a third time with 324 mg/day of aspirin for 7 days, with platelet function testing and specimen banking pre‐aspirin and post‐aspirin administration. Medication adherence was confirmed during the study through use of history logbooks and pill counts. Clinic staff was blinded to PEAR1 genotype throughout the course of the study. Banked samples were stored at –80°C until used.
Genotyping
All subjects were recruited based on known or predicted rs12041331 genotype, which had previously been measured as part of the HAPI Heart or PAPI study for most participants.3, 14 Genotype of each participant was validated by repeat genotyping of rs12041331 using a TaqMan single nucleotide polymorphism genotyping assay (Applied Biosystems, Foster City, CA), according to the manufacturer's instructions. The genotype concordance rate of this polymorphism in a subset of duplicate samples was 100% and the overall call rate was 100%.
Platelet aggregation
Maximal platelet aggregation was assessed by optical aggregometry, as previously described.3 Briefly, platelet‐rich plasma was isolated from blood samples drawn into 3.2% trisodium citrate anticoagulated tubes (Becton‐Dickinson, Franklin Lakes, NJ) and platelet counts were adjusted to 200,000 platelets/μl using platelet‐poor plasma. Platelet function was assessed using a PAP8E Aggregometer (Bio/Data Corporation, Horsham, PA), according to the manufacturer's instructions after stimulation with collagen (2 μg/mL), adenosine diphosphate (ADP; 2 μmol), epinephrine (10 μM), and arachidonic acid (AA; 1.6 mM), and was expressed as the maximal percentage change in light transmittance using platelet‐poor plasma as a referent.
11‐dehydrothromboxane B2 measurement
Measurement of urinary 11‐dehydrothromboxane B2 was performed using a monoclonal 11‐dehydrothromboxane B2 enzyme‐linked immunosorbent assay kit (Cayman Chemical, Ann Arbor, MI), according to the manufacturer's instructions. The 11‐dehydrothromboxane B2 concentration was determined photometrically at 405 nm using a PerkinElmer Victor X3 plate reader (PerkinElmer, Shelton, CT) and expressed as pg/mL. All samples were measured in duplicate and expressed as the mean of both measurements.
Statistical analyses
SAS version 9.2 (SAS Institute, Cary, NC) was used to calculate summary statistics, distributions, and frequencies. Association analyses of PEAR1 rs12041331 were performed using Mixed Model Analysis for Pedigrees (http://edn.som.umaryland.edu/mmap/) under an additive genetic model with adjustments for age, sex, and relatedness among study participants. Mixed Model Analysis for Pedigrees has been extensively used for genomic analyses in the Amish.3, 4, 16, 17, 18, 19, 20 Assuming an effect size similar to what was observed in the PAPI study,4 we estimate 80% power to detect differences in platelet aggregation of at least 6.75%, as calculated by PS Power.
Urinary concentrations of 11‐dehydrothromboxane B2 were standardized to creatinine and a paired t‐test was conducted in R to assess the impact of aspirin (324 mg/day) on thromboxane production. Association analyses between PEAR1 rs12041331 and 11‐dehydrothromboxane B2 levels pre‐aspirin and post‐aspirin administration (324 mg/day) were performed using Mixed Model Analysis for Pedigrees with age and sex as covariates. Differences between baseline platelet aggregation at study initiation and platelet aggregation after 2‐week washout periods were assessed by a repeated‐measures analysis of variance in R. The impact of aspirin dose on platelet aggregation as well as aspirin dose‐by‐genotype interactions were also assessed by repeated‐measures analysis of variance in R. All statistical tests were two‐sided and P < 0.05 was considered statistically significant.
RESULTS
This study used a prospective, genotype‐stratified design in which all subjects were studied under three different aspirin doses (81, 162, and 324 mg/day for 7 days) and platelet aggregation at each dose measured in response to four platelet agonists (ADP, collagen, epinephrine, and AA; Figure 1). Characteristics of the 67 study participants are shown in Table 1. Briefly, these subjects were healthy, middle‐aged (mean age = 50.2 years), and had low prevalence of disease (e.g., obesity [mean body mass index = 28.4], hypertension [6.0%], hypercholesterolemia [26.9%], diabetes [0.0%]) and smoking (10.4%). In total, 34 PEAR1 rs12041331 major allele homozygotes (G/G), 28 heterozygotes (G/A), and 5 minor allele homozygotes (A/A) were recruited. None of the clinical characteristics differed significantly by rs12041331 genotype (Table 1) with the exception of platelet count (P = 0.03), which is consistent with the role of PEAR1 in thrombopoeisis.8
Table 1.
Characteristics of study participants
| PEAR1 rs12041331 genotype | |||||
|---|---|---|---|---|---|
| Trait, units | Total | G/G | G/A | A/A | P valuea |
| Number | 67 | 34 | 28 | 5 | |
| Age ± SD, year | 50.2 ± 1.2 | 49.7 ± 1.7 | 51.6 ± 1.9 | 45.4 ± 4.6 | 0.86 |
| Sex (n = Female) (%) | 28 (42) | 15 (44) | 10 (36) | 3 (60) | 0.96 |
| BMI ± SD (kg/m2) | 28.4 ± 0.6 | 28.0 ± 0.9 | 29.1 ± 0.9 | 27.4 ± 2.2 | 0.41 |
| Hypertensionb (%) | 4 (6) | 2 (6) | 2 (7) | 0 (0) | 0.83 |
| Hypercholesterolemiac (%) | 18 (27) | 9 (26) | 9 (32) | 0 (0) | 0.6 |
| Self‐reported diabetes (%) | 0 | 0 | 0 | 0 | N/A |
| Current smoker (%) | 7 (10) | 3 (9) | 4 (14) | 0 (0) | 0.96 |
| Hematocrit ± SD (%) | 40.3 ± 0.4 | 40.3 ± 0.5 | 40.3 ± 0.6 | 39.9 ± 1.0 | 0.85 |
| White blood count ± SD (n × 1,000/μl) | 5.8 ± 0.2 | 5.5 ± 0.2 | 6.1 ± 0.3 | 5.8 ± 0.6 | 0.12 |
| Platelet count ± SD (n × 100,000/μl) | 223.1 ± 5.8 | 206.7 ± 7.3 | 243.8 ± 8.8 | 219.2 ± 20.8 | 0.03 |
A/A, minor allele homozygotes; BMI, body mass index; G/A, heterozygotes; G/G, major allele homozygotes; N/A, not applicable; PEAR1, platelet endothelial aggregation receptor 1.
a P values represent statistical difference of each trait by PEAR1 rs12041331 genotype. bDefined as systolic blood pressure greater than 140 mm Hg or diastolic blood pressure greater than 90 mm Hg or taking prescription medication for previously diagnosed hypertension. cDefined as low‐density lipoprotein cholesterol >160 mg/dL or taking prescription medication for previously diagnosed hypercholesterolemia.
To confirm that basal platelet function was restored after each aspirin dose intervention, we compared platelet aggregation measures obtained during the initial visit (prior to any aspirin administration) to measures obtained after the first (∼2 weeks after the 81 mg dosing period) and second (∼2 weeks after the 162 mg dosing period) washout periods. Indeed, no difference in agonist‐induced platelet aggregation was observed between these visits (Supplementary Table S1). Similarly, we assessed the impact of aspirin dose on agonist‐induced platelet aggregation. As expected, near‐complete inhibition of AA‐induced platelet aggregation was achieved at 81 mg/day for 7 days with only a modest change at the higher aspirin doses (i.e., at 162 or 324 mg/day) for the same length of time (P = 0.03; Supplementary Table S2). In addition, the aspirin dose did not significantly influence mean ADP‐induced, collagen‐induced, and epinephrine‐induced platelet aggregation values (P = 0.57, 0.56, and 0.07, respectively).
Prior to aspirin administration, we tested whether PEAR1 rs12041331 was associated with maximal percentage of platelet aggregation after stimulation with ADP, collagen, epinephrine, and AA. Given that urinary levels of 11‐dehydrothromboxane B2 were also measured before and after administration of 324 mg of aspirin (see below), baseline measures of platelet aggregation recorded prior to administration of 324 mg of aspirin are reported in Figure 2 and genotype‐stratified analysis of all baseline data are shown in Supplementary Figure S1. We observed that subjects who carried the minor allele of rs12041331 had significantly reduced ADP‐stimulated platelet aggregation compared with noncarriers (mean maximal platelet aggregation = 20.2, 37.0, and 41.8 for rs12041331 genotypes A/A, G/A, and G/G, respectively; P = 0.03). In contrast, we observed no evidence of association between rs12041331 and collagen, as well as AA‐induced platelet aggregation (P = 0.20 and 0.21, respectively). Although a reduction in epinephrine‐induced platelet aggregation was observed in participants who were homozygous for the rs12041331 minor allele (maximal platelet aggregation = 33.6, 64.0, and 60.8 for PEAR1 rs12041331 genotypes A/A, G/A, and G/G, respectively), this comparison did not reach statistical significance (P = 0.14).
Figure 2.
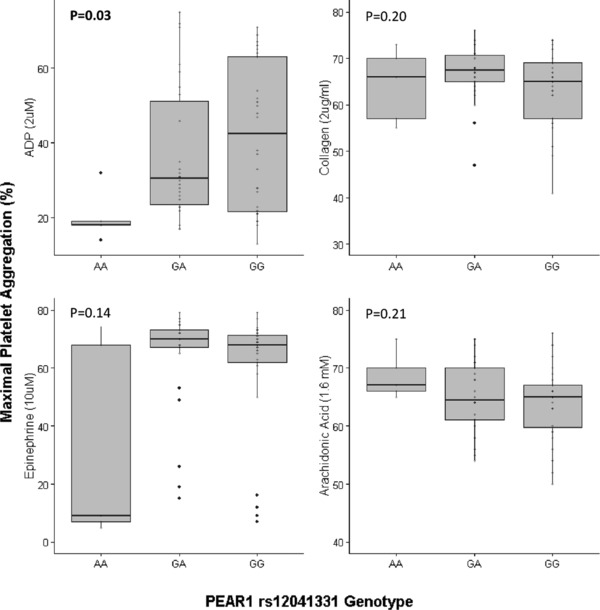
Baseline agonist‐induced ex vivo platelet aggregation measurements were obtained in platelet‐rich plasma prior to administration of the first aspirin dose. Platelet aggregation is expressed as the percentage of maximal aggregation using platelet‐poor plasma as a referent. Adenosine diphosphate (ADP; 2 μM), collagen (2 μg/mL), epinephrine (10 μM), and arachidonic acid (AA; 1.6 mM) were used to stimulate platelet aggregation. The horizontal line within each box indicates the median; the top and bottom borders of each box indicate the interquartile range (IQR). The whiskers extending from each box indicate plus/minus 1.5 IQRs, and the points beyond the whiskers indicate outliers beyond 1.5 IQRs.
Interestingly, we observed different patterns of association between rs12041331 and agonist‐induced platelet aggregation after aspirin treatment (Figure 3). Unlike pre‐aspirin measurements, we observed no evidence of association between rs12041331 and ADP‐stimulated platelet aggregation after administration of 81, 162, and 324 mg of aspirin for 7 days (P = 0.21, 0.28, and 0.95, respectively). In contrast, we observed strong evidence of association between this polymorphism and collagen‐stimulated platelet aggregation regardless of aspirin dose (P = 0.03, 7.0 × 10−3, and 0.01 for aspirin doses of 81, 162, and 324 mg, respectively). Furthermore, subjects who carried the rs12041331 minor allele had significantly reduced on‐aspirin epinephrine‐induced platelet aggregation compared with noncarriers (P = 0.04, 0.03, and 0.04 for aspirin doses of 81, 162, and 324 mg, respectively). Similar to pre‐aspirin measurements, AA‐induced platelet aggregation was not influenced by rs12041331 regardless of aspirin dose (P = 0.86, 0.66, and 0.68 for aspirin doses of 81, 162, and 324 mg, respectively).
Figure 3.
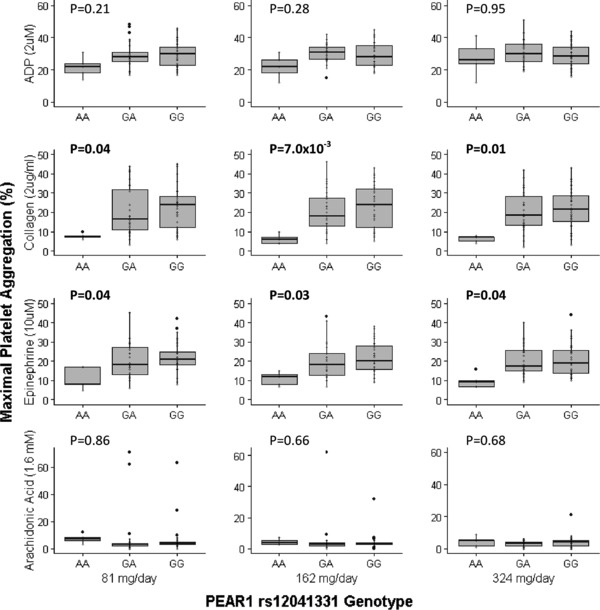
On‐aspirin agonist‐induced ex vivo platelet aggregation measurements were obtained in platelet‐rich plasma on day 7 of each treatment period 2 h following administration of the final aspirin dose. Aspirin doses of 81 mg, 162 mg, and 324 mg were administered for 7 days. Platelet aggregation is expressed as the percentage of maximal aggregation using platelet‐poor plasma as a referent. Adenosine diphosphate (ADP; 2 μM), collagen (2 μg/mL), epinephrine (10 μM), and arachidonic acid (AA; 1.6 mM) were used to stimulate platelet aggregation. The horizontal line within each box indicates the median; the top and bottom borders of each box indicate the interquartile range (IQR). The whiskers extending from each box indicate plus/minus 1.5 IQRs and the points beyond the whiskers indicate outliers beyond 1.5 IQRs.
We also performed interaction analyses to evaluate the influence of increasing doses of aspirin on agonist‐induced platelet aggregation in the context of PEAR1 rs12041331 genotype. Regardless of the agonist used, we observed no interaction between aspirin dose and rs12041331 genotype (P = 0.89, 0.15, 0.77, and 0.55 when ADP, collagen, epinephrine, and AA were used as an agonist, respectively), indicating an aspirin dose above 81 mg had no major impact on degree of reduction in agonist‐induced platelet aggregation by PEAR1 rs12041331 genotype (Supplementary Table S3).
Finally, we assessed urinary 11‐dehydrothromboxane B2 levels pre‐aspirin and post‐aspirin administration in the total population as well as by rs12041331 genotype (Table 2). As expected, levels of 11‐dehydrothromboxane B2 were significantly reduced after aspirin exposure (P = 2.2 × 10−16), with near‐complete inhibition of 11‐dehydrothromboxane B2 achieved after aspirin exposure for 7 days. The 11‐dehydrothromboxane B2 levels were not significantly different by rs12041331 genotype before (P = 0.09) or after aspirin exposure (P = 0.16).
Table 2.
Urinary 11‐dehyrothromboxane B2 levels by PEAR1 rs12041331 genotype
| G/G homozygotes | G/A heterozygotes | A/A homozygotes | P value | |
|---|---|---|---|---|
| Pre‐aspirin administration | 2463 ± 261 | 2154 ± 217 | 1748 ± 432 | 0.16 |
| Post‐aspirin administrationa | 474 ± 59 | 361 ± 33 | 365 ± 92 | 0.09 |
PEAR1, platelet endothelial aggregation receptor 1.
Urinary 11‐dehydrothromboxane B2 levels ± SDs are expressed as pg/mg creatinine.
Measurements were recorded before and after administration of 324 mg/day aspirin for 7 days.
DISCUSSION
Although aspirin is the most frequently used antiplatelet medication globally, there is a high degree of interindividual variation in patient response to this agent when considering cyclooxygenase (COX)‐independent pathways of platelet function and occurrence of major adverse cardiovascular events. This is reflected in the lack of consensus regarding the appropriate dose of aspirin to be administered clinically, despite updated guidelines and numerous studies that indicate high‐dose aspirin has no additional benefit but a reduced safety profile as compared with low‐dose aspirin.5, 21, 22, 23, 24, 25 In past years, substantial effort has been made to identify the genetic and nongenetic determinants of aspirin response, and although these investigations have increased our understanding, identification of bona fide aspirin response candidate polymorphisms have remained elusive. Multiple factors contribute to lack of genetic replication, including study design, participant population, clinical indication, and several other confounding variables. Furthermore, the ways in which aspirin response is defined (e.g., inhibition of platelet aggregation vs. rates of cardiovascular outcomes) as well as the use of multiple platelet function tests has made it difficult to compare results across investigations. Given that aspirin irreversibly acetylates COX resulting in the inhibition of thromboxane A2 formation from AA, traditional tests of on‐aspirin platelet function include stimulation of platelets with AA as well as assessment of urinary 11‐dehydrothromboxane B2, a stable metabolite of thromboxane A2. However, an increasing body of evidence suggests that evaluation of non‐COX‐dependent platelet pathways complement traditional aspirin response approaches and can aid in the identification of factors that lead to variation in response to this drug.4, 5, 21, 26
PEAR1 was identified in 2005 as a type 1 transmembrane receptor that is highly expressed in platelets and endothelial cells, among other cell types.7 In platelets, PEAR1 acts as a contact receptor that, upon ligand binding, becomes phosphorylated in an Src family kinase‐dependent manner, leading to downstream PI3K and AKT signaling, ultimately resulting in the stabilization of platelet aggregates through secondary activation of glycoprotein αIIbβ3.6, 7 Continual refinement of the biological processes that are influenced by PEAR1 in platelets and other cell types are consistent with the results of genetic investigations of PEAR1 (see below) and allow for better understanding of PEAR1‐dependent changes in platelet aggregation before as well as after antiplatelet treatment.
To date, multiple investigations have convincingly shown that genetic variation in PEAR1 significantly influences agonist‐induced platelet aggregation at baseline9, 10, 11, 12, 13, 27 as well as after treatment with aspirin,9, 28 prasugrel,29 and dual antiplatelet therapy with aspirin and clopidogrel.4, 30 Although several polymorphisms that influence platelet aggregation have been identified in this gene, rs12041331 seems to be the most highly and consistently associated of these variants.4, 9, 10, 11, 13 The rs12041331 polymorphism results in G to A substitution in intron 1 and has been previously implicated in reducing PEAR1 expression.9 In a genome‐wide meta‐analysis of basal platelet function in over 4,500 European and African American participants, Johnson et al.13 showed that PEAR1 rs12041331 was among the strongest determinants of basal ADP‐induced and epinephrine‐induced platelet aggregation (P = 3.8 × 10−16 and 4.9 × 10−19, respectively) but did not report associations with collagen or collagen lag time. Similarly, evaluation of Caucasian Americans of the GeneSTAR cohort revealed that PEAR1 rs12041331 was among the strongest determinants of platelet aggregation pre‐aspirin administration, accounted for ∼15% of the total phenotypic variation in platelet function, and sequencing of the full PEAR1 gene in 104 individuals confirmed that rs12041331 accounted most strongly for the relation between PEAR1 and platelet aggregation.11 Surprisingly, however, a different pattern of association emerged post‐aspirin administration (81 mg/day for 14 days), in which significant associations were observed with collagen‐induced and epinephrine‐induced platelet aggregation but not when ADP was used as a platelet agonist.9 To complicate matters further, in African Americans of the GeneSTAR cohort, in which the minor allele frequency of this variant is substantially higher compared with Caucasian participants (0.37 vs. 0.09, respectively), rs12041331 was significantly associated with ADP‐induced, collagen‐induced, and epinephrine‐induced platelet aggregation pre‐aspirin and post‐aspirin exposure. In our own genome‐wide association studies of dual antiplatelet therapy response (aspirin and clopidogrel) in 565 healthy Amish participants of the PAPI study, PEAR1 rs12041331 was the strongest modifier of collagen‐stimulated platelet aggregation (P = 7.7 × 10−9).4 We also observed that African American and Caucasian patients with non‐emergent percutaneous coronary intervention who carried the minor allele of rs12041331 had reduced 1‐year survival (hazard ratio = 3.97; P = 0.04 and hazard ratio = 2.62; P = 0.06, respectively) and that Caucasian patients with stable coronary artery disease on aspirin experienced higher rates of myocardial infarction compared with noncarriers (odds ratio = 2.03; P = 0.05). Interestingly, rs12041331 was not associated with myocardial infarction in patients with stable coronary artery disease who were not treated with aspirin.4
Although several retrospective studies have independently shown that PEAR1 rs12041331 influences platelet aggregation and cardiovascular outcomes, others have not.31 In addition, differences in participant populations (e.g., healthy patients vs. patients with cardiovascular disease), study design (e.g., presence vs. absence of antiplatelet agents), as well as utilized platelet function tests (i.e., the agonist used to stimulate platelet aggregation) have made it challenging to definitively determine the platelet activation pathways influenced by PEAR1. To better define the influence of PEAR1 rs12041331 on platelet aggregation pathways, we, for the first time, prospectively evaluated this polymorphism before and after aspirin administration (81, 162, and 324 mg/day) using multiple platelet agonists (i.e., ADP, collagen, epinephrine, and AA). Prior to aspirin administration, we showed a strong relationship between PEAR1 genotype and ADP‐stimulated platelet aggregation but not with collagen‐induced platelet aggregation, consistent with the results observed in nontreated patients in both the Framingham Heart Study and GeneSTAR cohorts.9, 13 Interestingly, after aspirin treatment, PEAR1 rs12041331 substantially influenced collagen‐induced platelet aggregation but not ADP‐induced platelet aggregation, suggesting a potential shift in the platelet pathways impacted by PEAR1 in the presence of aspirin. Although we found that on‐aspirin epinephrine‐induced platelet aggregation was altered by PEAR1 genotype, consistent with prior reports, we failed to observe such a relationship prior to aspirin administration. This latter observation is in contrast to previous investigations; however, it should be noted that we did observe a non‐significant reduction in platelet aggregation in rs12041331 minor allele homozygotes after stimulation with epinephrine prior to aspirin treatment (maximal platelet aggregation = 33.6, 64.0, and 60.8 for PEAR1 rs12041331 genotypes A/A, G/A, and G/G, respectively). Importantly, we did not observe evidence of association between AA‐induced platelet aggregation and urinary 11‐dehydrothromboxane B2 levels with rs12041331 pre‐aspirin or post‐aspirin administration. In other words, assessment of non‐COX‐dependent biological pathways, in addition to canonical pathways, provided useful insights regarding the determinants of variable aspirin response, consistent with previous investigations.4, 5, 9, 21, 26, 28, 30
Given the prior observation that aspirin‐treated patients with coronary artery disease who carried the minor allele of PEAR1 rs12041331 experienced higher rates of myocardial infarction compared with nontreated carriers,4 we, for the first time, prospectively investigated the potential interaction between therapeutically used aspirin doses and PEAR1 genotype on agonist‐induced platelet aggregation. However, we did not observe any such interaction, nor did we observe any impact of aspirin dose on degree of inhibition of platelet aggregation in the entire cohort overall, consistent with recent US guidelines that favor low‐dose aspirin antiplatelet therapy.25
Although it is increasingly clear that genetic variation in the PEAR1 gene substantially impacts agonist‐induced platelet aggregation, it is still unclear by what mechanism they may influence cardiovascular event rates. For example, the minor allele of rs12041331 leads to reduced on‐treatment platelet aggregation yet higher rates of thrombotic outcomes.4 It has been speculated that the additive effect of aspirin therapy in addition to reduced platelet aggregation in carriers of the rs12041331 minor allele may lead to unstable thrombi and subsequent plaque rupture; however, no evidence exists to support this hypothesis. Alternatively, it is possible that reduced platelet aggregation in minor allele carriers is unrelated to the increased cardiovascular event risk. PEAR1 expression in the endothelium is sixfold higher than expression in platelets.7 Vandenbriele et al.32 recently showed that PEAR1 is a negative regulator of neoangiogenesis and wound healing.32 Furthermore, Fisch et al.16 showed through meta‐analysis of publically available microarray data that PEAR1 expression is highly correlated with several genes and phenotypes that are integral to endothelial function and that PEAR1 rs12041331 is significantly associated with flow‐mediated dilation of the brachial artery, a widely used noninvasive measure of endothelial function. Additional mechanistic studies both in the endothelium and in platelets will be required to clarify the role of PEAR1 in cardiovascular disease risk in both the presence and absence of aspirin.
We would like to acknowledge some limitations of our investigation. Given the moderate sample size of our cohort, it is possible that we may have been statistically underpowered for some of our association analyses. However, the effect sizes of PEAR1 rs12041331 on platelet aggregation in this study are similar to the effect sizes observed in previous investigations.4, 9 In addition, our study design evaluated PEAR1 genotype in the context of acute aspirin administration. Given the methods used in this investigation (i.e., ex vivo platelet aggregation) and average lifespan of platelets (7–10 days), we do not believe that platelet aggregation results would differ considerably between short‐term and long‐term aspirin exposure. However, if the relationship between PEAR1 genotype and platelet aggregation is significantly impacted by PEAR1 activity in the endothelium, where it is highly expressed, it is possible that chronic aspirin exposure might influence our findings. Finally, we conducted our investigation in healthy Amish participants. Although our results are consistent with previously conducted retrospective analyses in white populations, these results may not be generalizable to participants of different race/ethnicities or to patients with cardiovascular disease. Additional evaluation of the influence of PEAR1 rs12041331 genotype in these groups will be needed to confirm our findings.
In summary, we prospectively investigated the impact of a recently identified aspirin response polymorphism pre‐aspirin and post‐aspirin administration using several important platelet agonists. Although no difference in platelet function was observed using traditional tests of aspirin response (i.e., AA‐induced platelet aggregation and circulating levels of urinary thromboxane B2), PEAR1 genotype significantly impacted non‐COX‐dependent platelet aggregation pathways. Furthermore, exposure to aspirin led to a shift in the platelet aggregation pathways influenced by rs12041331, whereby ADP‐stimulated platelet aggregation was affected prior to aspirin administration yet collagen‐induced and epinephrine‐induced platelet aggregation were impacted post‐aspirin exposure. Continual investigation will be necessary to better understand the dynamics of PEAR1‐mediated platelet aggregation in vivo as well as the implications of these findings on adverse event rates in aspirin‐treated patients with cardiovascular disease.
Author Contributions
J.P.L., J.D.B., and R.B.H. wrote the manuscript. J.P.L. designed the research. R.B.H., S.N., S.S., M.M., P.D., M.D., and K.T. performed the research. J.P.L., J.D.B., L.M.Y.‐A., M.A.P., and B.D.M. analyzed the data. L.M.Y.‐A. and J.O. contributed new reagents/analytical tools.
Conflicts of Interest
J.P.L. grant support from the National Institutes of Health to study the pharmacogenomics of antiplatelet therapy. L.M.Y.‐A. is an employee of GlaxoSmithKline PLC.
Supporting information
Supplementary Table S1. Mean maximal platelet aggregation (%) during washout periods
Supplementary Table S2. Mean maximal platelet aggregation (%) by aspirin dose
Acknowledgments
We gratefully acknowledge the Amish community, our Amish liaisons, and field workers for their extraordinary cooperation. We would also like to acknowledge Alan R. Shuldiner for his role in developing the PAPI study as well as insights in the development of this investigation. This study was supported by the National Institutes of Health grants NIH K23 GM102678, U01 GM074518, U01 HL105198, the Mid‐Atlantic Nutrition and Obesity Center (P30 DK072488), and the University of Maryland General Clinical Research Center (M01 RR 16500).
References
- 1. Serebruany, V.L. , Steinhubl, S.R. , Berger, P.B. , Malinin, A.I. , Bhatt, D.L. & Topol, E.J. Variability in platelet responsiveness to clopidogrel among 544 individuals. J. Am. Coll. Cardiol. 45, 246–251 (2005). [DOI] [PubMed] [Google Scholar]
- 2. Mega, J.L. et al Cytochrome p‐450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 360, 354–362 (2009). [DOI] [PubMed] [Google Scholar]
- 3. Shuldiner, A.R. et al Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 302, 849–857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lewis, J.P. et al Genetic variation in PEAR1 is associated with platelet aggregation and cardiovascular outcomes. Circ. Cardiovasc. Genet. 6, 184–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Faraday, N. et al Heritability of platelet responsiveness to aspirin in activation pathways directly and indirectly related to cyclooxygenase‐1. Circulation 115, 2490–2496 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Kauskot, A. , Di Michele, M. , Loyen, S. , Freson, K. , Verhamme, P. & Hoylaerts, M.F. , A novel mechanism of sustained platelet αIIbβ3 activation via PEAR1. Blood 119, 4056–4065 (2012). [DOI] [PubMed] [Google Scholar]
- 7. Nanda, N. et al Platelet endothelial aggregation receptor 1 (PEAR1), a novel epidermal growth factor repeat‐containing transmembrane receptor, participates in platelet contact‐induced activation. J. Biol. Chem. 280, 24680–24689 (2005). [DOI] [PubMed] [Google Scholar]
- 8. Kauskot, A. et al PEAR1 attenuates megakaryopoiesis via control of the PI3K/PTEN pathway. Blood 121, 5208–5217 (2013). [DOI] [PubMed] [Google Scholar]
- 9. Faraday, N. et al Identification of a specific intronic PEAR1 gene variant associated with greater platelet aggregability and protein expression. Blood 118, 3367–3375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qayyum, R. et al Genome‐wide association study of platelet aggregation in African Americans. BMC Genet. 16, 58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim, Y. et al Targeted deep resequencing identifies coding variants in the PEAR1 gene that play a role in platelet aggregation. PLoS One 8, e64179 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herrera‐Galeano, J.E. et al A novel variant in the platelet endothelial aggregation receptor‐1 gene is associated with increased platelet aggregability. Arterioscler. Thromb. Vasc. Biol. 28, 1484–1490 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson, A.D. et al Genome‐wide meta‐analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat. Genet. 42, 608–613 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitchell, B.D. et al The genetic response to short‐term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am. Heart J. 155, 823–828 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Machlus, K.R. & Italiano, J.E. Jr. ,The incredible journey: from megakaryocyte development to platelet formation. J. Cell. Biol. 201, 785–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fisch, A.S. et al Genetic variation in the platelet endothelial aggregation receptor 1 gene results in endothelial dysfunction. PLoS One 10, e0138795 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Albert, J.S. et al Null mutation in hormone‐sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 370, 2307–2315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lewis, J.P. et al The functional G143E variant of carboxylesterase 1 is associated with increased clopidogrel active metabolite levels and greater clopidogrel response. Pharmacogenet. Genomics 23, 1–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis, J.P. et al The CYP2C19*17 variant is not independently associated with clopidogrel response. J. Thromb. Haemost. 11, 1640–1646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pollin, T.I. et al A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 322, 1702–1705 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mathias, R. et al A combined genome‐wide linkage and association approach to find susceptibility loci for platelet function phenotypes in European American and African American families with coronary artery disease. BMC Med. Genomics 3, 22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hall, H.M. et al Contemporary patterns of discharge aspirin dosing after acute myocardial infarction in the United States: results from the National Cardiovascular Data Registry (NCDR). Circ. Cardiovasc. Qual. Outcomes 7, 701–707 (2014). [DOI] [PubMed] [Google Scholar]
- 23. Peters, R.J. et al Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the clopidogrel in unstable angina to prevent recurrent events (CURE) study. Circulation 108, 1682–1687 (2003). [DOI] [PubMed] [Google Scholar]
- 24. Yu, J. et al Safety and efficacy of high‐ versus low‐dose aspirin after primary percutaneous coronary intervention in ST‐segment elevation myocardial infarction: the HORIZONS‐AMI (Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction) Trial. JACC Cardiovasc. Interv. 5, 1231–1238 (2012). [DOI] [PubMed] [Google Scholar]
- 25. O'Gara, P.T. et al 2013 ACCF/AHA guideline for the management of ST‐elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 127, 529–555 (2013). [DOI] [PubMed] [Google Scholar]
- 26. Voora, D. et al Aspirin exposure reveals novel genes associated with platelet function and cardiovascular events. J. Am. Coll. Cardiol. 62, 1267–1276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eicher, J.D. , Xue, L. , Ben‐Shlomo, Y. , Beswick, A.D. & Johnson, A.D. , Replication and hematological characterization of human platelet reactivity genetic associations in men from the Caerphilly Prospective Study (CaPS). J. Thromb. Thrombolysis 41, 343–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Würtz, M. , Nissen, P.H. , Grove, E.L. , Kristensen, S.D. & Hvas, A.M. , Genetic determinants of on‐aspirin platelet reactivity: focus on the influence of PEAR1. PLoS One 9, e111816 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiang, Q. , Cui, Y. , Zhao, X. & Zhao, N. , Identification of PEAR1 SNPs and their influences on the variation in prasugrel pharmacodynamics. Pharmacogenomics 14, 1179–1189 (2013). [DOI] [PubMed] [Google Scholar]
- 30. Yao, Y. et al Association of PEAR1 genetic variants with platelet reactivity in response to dual antiplatelet therapy with aspirin and clopidogrel in the Chinese patient population after percutaneous coronary intervention. Thromb. Res. 141, 28–34 (2016). [DOI] [PubMed] [Google Scholar]
- 31. Voora, D. , Horton, J. , Shah, S.H. , Shaw, L.K. & Newby, L.K. , Polymorphisms associated with in vitro aspirin resistance are not associated with clinical outcomes in patients with coronary artery disease who report regular aspirin use. Am. Heart J. 162, 166–172 (2011). [DOI] [PubMed] [Google Scholar]
- 32. Vandenbriele, C. et al Platelet endothelial aggregation receptor‐1: a novel modifier of neoangiogenesis. Cardiovasc. Res. 108, 124–138 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Mean maximal platelet aggregation (%) during washout periods
Supplementary Table S2. Mean maximal platelet aggregation (%) by aspirin dose