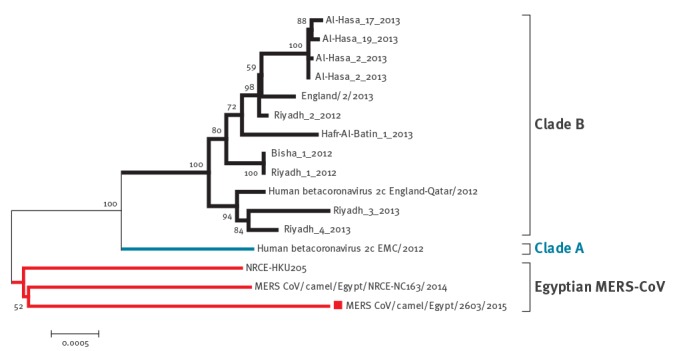
Figure 3.
Phylogenic analysis of a full MERS-CoV genome sequence retrieved from an imported dromedary camel from Sudan between August 2015 and January 2016
Representative viruses from the two major MERS-CoVs clades (A and B) are indicated and marked with vertical bar. Phylogenetic analysis was done using the neighbour-joining algorithm with the Kimura two-parameter model. The reliability of phylogenetic inference at each branch node was estimated by the bootstrap method with 1,000 replications. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Evolutionary analysis was conducted in MEGA 6.06. The virus sequenced for this study is marked by a red square.