

Abstract
Triple-negative breast cancer (TNBC) is more aggressive than other breast cancer subtypes. Collagen type IV alpha 2 (COL4A2), a major component of the basement membrane, dynamically influences a wide range of biological processes, including cancer pathogenesis and progression. This study evaluated the effects of COL4A2 siRNA delivered by lentiviral vector to TNBC cells. COL4A2 siRNA lenti-viral vector was constructed and transfected into MDA-MB-231 and MDA-MB-468 cells. The COL4A2 mRNA levels were quantified by RT-PCR. CCK8 assay was performed to evaluate cell proliferation and migration. Cell migration and invasion assays were carried out using Transwell. Cell apoptosis and cell cycle analyses were conducted using flow cytometric approach. We found that COL4A2 mRNA levels were significantly down-regulated in MDA-MB-231 and MDA-MB-468 cells after transfection with COL4A2 siRNA. Furthermore, cell migration and proliferation were significantly decreased and the cell cycle was arrested. Our results indicated that COL4A2 siRNA significantly suppresses the migration and proliferation of TNBC cells. Inhibition of COL4A2 may be a new target for the prevention and treatment of TNBC.
Keywords: triple-negative breast cancer (TNBC), COL4A2, small interfering RNA (siRNA), lentiviral vector
INTRODUCTION
Breast cancer consists of four molecular subtypes, including luminal A, luminal B, human epidermal growth factor receptor 2 (HER2)-enriched, and triple-negative breast cancer (TNBC) [1–3]. TNBC, which is defined by the negative expression of the estrogen receptor (ER), progesterone receptor (PR) and HER2 receptor, accounts for approximately 15% of all breast cancers [4]. Previous studies suggest that TNBC is more aggressive than other breast cancer subtypes due to a high probability of recurrence and metastasis, causing poor prognosis in patients [5–8]. Although major advances have been achieved in the treatment of ER-positive or HER2-amplified breast cancers, there are no clinically targeted therapies for TNBC, thus leaving cytotoxic chemotherapy as the only choice for systemic therapy [8].
Collagen type IV alpha 2 (COL4A2, MIM 120090), is the major structural component of basement membranes. The C-terminal portion of the protein, known as canstatin, is an inhibitor of angiogenesis and tumor growth [9]. COL4A2 forms heterotrimers together with COL4A1 (MIM120130), which is critical for the stability and functions of vascular basement membrane [10]. In the extracellular space, this kind of heterotrimers polymerize to form flexible sheets, that not only provide structural integrity but also participate in dynamic biological processes, including cell–matrix and cell–cell communication through interactions with growth factors and cell surface receptors [11–14]. Mutations in COL4A2 may contribute to intracerebral hemorrhage (ICH) [15], porencephaly and small-vessel disease with reduced penetrance and variable phenotype [16, 17]. Recently studies revealed that COL4A2 is involved in the pathogenesis and progression of esophageal cancer, lung cancer, and prostate cancer and other cancers.
Previously, it has been shown that COL4A2 is a diagnostic marker signature for esophageal cancer [18]. In addition, COL4A2 is significantly overexpressed in tissues obtained from brain metastases of lung cancer and melanoma patients [19]. Oktem G found a high level of COL4A2 when cancer stem cells (CSC) were maintained as serum-grown prostate CSC spheroids [20]. Furthermore, several studies suggested that COL4A2 may play a role in the pathogenesis of prostate cancer [21], epithelial ovarian cancer [22], colorectal cancer [23], and uterine leiomyoma [24]. A recent study performed in the breast cancer cell line MCF-7 revealed that the reduction of COL4A2 mRNA levels by miR-29b may contribute to the invasiveness of MCF-7 cells [25]. It has been postulated that extracellular matrix-related COL4A2 is associated with breast cancer and may be a potential biomarker for early diagnosis and therapy of breast cancer [26]. All of the aforementioned studies suggest that COL4A2 may play a role in the pathogenesis of cancer, notably breast cancer. However, it remains to be determined if COL4A2 is involved in the cell proliferation and migration of TNBC.
Previously, we reported that COL4A2 was significantly up-regulated in TNBC [27], suggesting that COL4A2 may play an important roles in the pathogenesis of TNBC. In the current study, we used siRNA to suppress COL4A2 expression and investigated how it affects the TNBC cell proliferation and migration.
RESULTS
Interference efficiency of siRNA on the expression of Col4A2 mRNA and protein
RNA interference was performed in both MDA-MB-231 and MDA-MB-468 cells. As shown in Figure 1A and 1B, RT-PCR results showed the levels of Col4A2 mRNA were significantly lower in the siRNA transfected cells compared to the negative control (NC) (p < 0.0001) and control groups (p < 0.0001). In addition, the interference efficiency was 82.1% and 74.9% in MDA-MB-231 and MDA-MB-468, respectively.
Figure 1. A and B, The relative expression of Col4A2 mRNA and protein in TNBC cells.
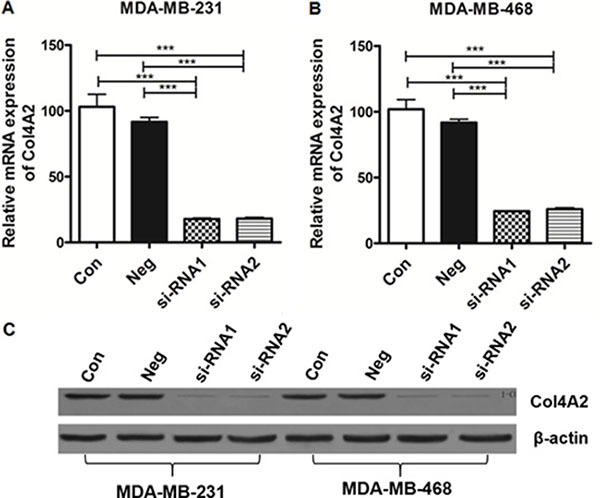
(A) MDA-MB-231; (B) MDA-MB-468; (C) Western blotting validated the interference efficiency.***p < 0.0001 (Con = non-transfected cells;Neg = cells transfected with control shRNA lentiviral vector).
Western blotting was used to further validate the interference efficiency. Figure 1C showed that the levels of Col4A2 protein were significantly lower in the siRNA transfected cells compared to NC (p < 0.0001) and control groups (p < 0.0001), and the interference efficiency was 91.3% and 89.4% in MDA-MB-231 and MDA-MB-468, respectively. These results indicated that lentivirus siRNA significantly downregulates the expression of Col4A2 mRNA and protein in TNBC cells.
siRNA inhibited the proliferation of MDA-MB-231 and MDA-MB-468 cells
The effect of siRNA on cell proliferation was evaluated by CCK8 assay. The results obtained from the CCK8 assay indicated that MDA-MB-231 and MSA-MB-468 cell proliferation was significantly decreased in the siRNA groups 72 h and 96 h after transfection, while there were no significant differences between the NC and control group after 24 h and 48 h after transfection (Figure 2). Together, the above results suggested that Col4A2 lentivirus siRNA significantly inhibits the proliferation of TNBC cells.
Figure 2. CCK8 assay on cell proliferation at 24, 48, 72, and 96 hours after transfection.
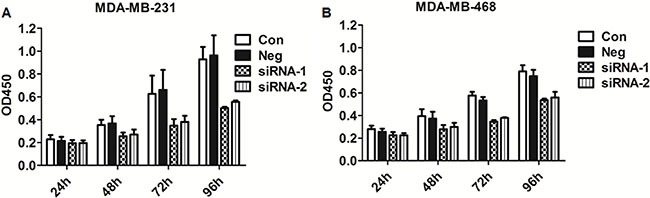
(A) MDA-MB-231; (B) MDA-MB-468. (Con = non-transfected cells; Neg = cells transfected with control shRNA lentiviral vector).
The effect of siRNA on cell migration of MDA-MB-231 and MDA-MB-468 cells
The effect of siRNA on cell migration was evaluated. Figure 3 indicates that MDA-MB-231 and MDA-MB-468 cell migration is significantly decreased 48 h after siRNA transfection. The relative migration rate of cells transfected with siRNA group was significantly lower compared to the control and NC groups. These results indicate that Col4A2 lentivirus siRNA significantly inhibits the migration of TNBC cells.
Figure 3. Migration of TNBC cells.
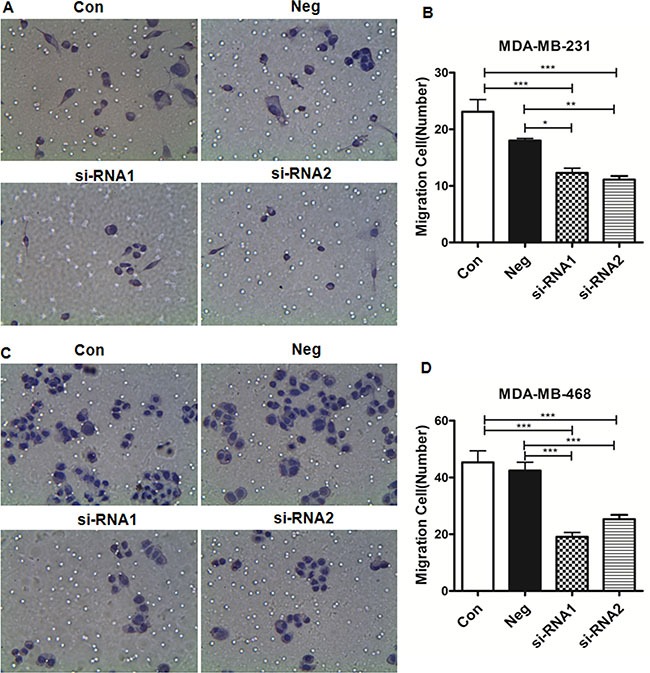
Light microscopic examination shows the migration of MDA-MB-231 (A) and MDA-MB-468 (C) cells; the relative migration rate of MDA-MB-231 (B) and MDA-MB-468 (D). Cells that migrated through the membrane were counted in five random fields for each group, and the relative migration rate= number of migrated cells/number of migrated cells in the control group).*P < 0.05; **P < 0.001; ***P < 0.0001. (Con = non-transfected cells; Neg = cells transfected with control shRNA lentiviral vector).
The effect of siRNA on apoptosis of MDA-MB-231 and MDA-MB-468 cells
As shown in Figure 4, in both MDA-MB-231 and MDA-MB-468 cells, after Col4A2 interference treatment, the cells undergoing early apoptosis were significantly increased in the siRNA1 and siRNA2 groups, compared with the NC and control groups. However, siRNA transfection did not significantly affect the number of cells undergoing late apoptosis (Figure 4). These results indicate that the Col4A2 lentivirus siRNA decreased TNBC cell growth by inducing apoptosis.
Figure 4. Col4A2 lentivirus siRNA induced apoptosis in TNBC cells.
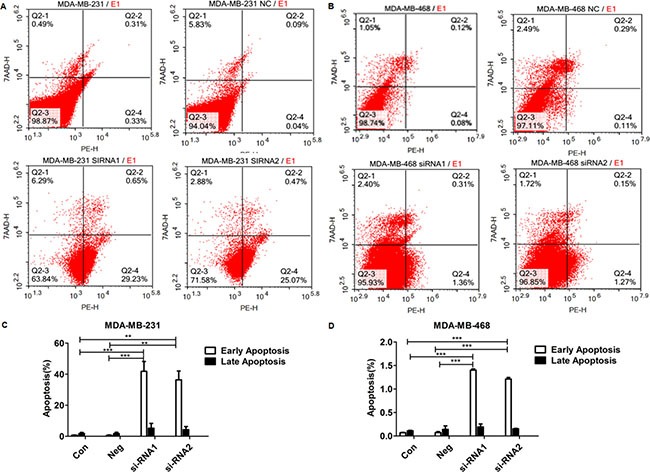
FACS results derived from MDA-MB-231 (A) and MDA-MB-468 (B) in three groups; Early and Late Apoptosis, (%)of MDA-MB-231 (C) and MDA-MB-468 (D). **P < 0.001; ***P < 0.0001. (Con = non-transfected cells; Neg = cells transfected with control shRNA lentiviral vector).
Cell cycle of MDA-MB-231 and MDA-MB-468 cells
As shown in Figure 5, after Col4A2 interference transfection, there was no significant difference in the number of cells in the G0/G1 phase compared to the NC and control groups. In contrast, a higher percentage of cells in the G2 phase and a lower percentage of cells in the S phase were detected in the siRNA group compared to the NC and control groups. These results indicate that transfection with Col4A2 lentivirus siRNA arrested the TNBC cell cycle in the G2 phase.
Figure 5. Cell cycle in each group.
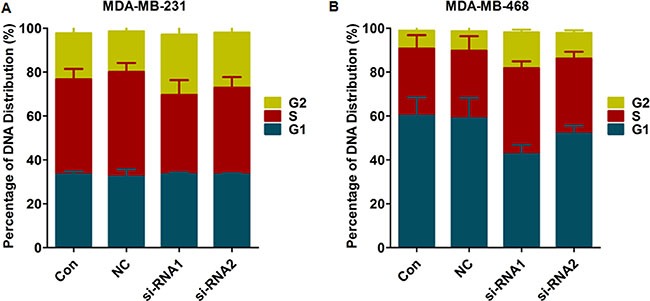
(A) MDA-MB-231; (B) MDA-MB-468. (Con = non-transfected cells; Neg = cells transfected with control shRNA lentiviral vector).
DISCUSSION
TNBC is a highly invasive clinical subtype of breast cancer characterized by lack of effective targeted therapies, aggressive phenotype and poor prognosis. Therefore, it is important to identify new therapeutic targets for TNBCs. In our previous study, gene expression analysis was performed by microarray to compare gene expression profiling of TNBC to that of non-TNBC. Our previous results demonstrated that Col4A2 was the most significant up-regulated gene in the integrated analysis, and the high expression of Col4A2 in TNBC was further confirmed by qRT-PCR in MDA-MB-231 cells. Protein expression analysis showed that Col4A2 protein was up-regulated. Jak-STAT signaling pathway was found to be highly active for differentially expressed genes including Col4A2 in cancer [27]. It was suggested that the JAK/STAT was one of the TNBC-specific regulatory feedback programs, and its critical signaling nodes could be used as the therapeutic target for the treatment of TNBC [28].
In the present study, we constructed the COL4A2 siRNA lenti-viral vector and transfected it into MDA-MB-231 cells and MDA-MB-468 cells. Col4A2 mRNA and protein expressions were effectively down-regulated in MDA-MB-231 and MDA-MB-468 cells after siRNA lentivirus transfection. The Col4A2 siRNA-transduced cells showed inhibition in cell proliferation and migration in MDA-MB-231 and MDA-MB-468 cells, indicating the migration and proliferation of TNBC cells were regulated by Col4A2 expression. Some classical pathways such as Wnt signaling pathway, PI3K-Akt signaling pathway, Jak-STAT signaling pathway, MAPK signaling pathway, ErbB signaling pathway [29] and some others together may regulate tumor growth and metastasis based on integrated analysis. These results may provide us new insight for the prevention and treatment of TNBC [27]. Apoptosis is an active process of cellular self-destruction, not only functions to limit viral replication by eliminating infected cells but also contributes to limit viral dissemination. After Col4A2 interference treatment in both MDA-MB-231 and MDA-MB-468 cells, the cells undergoing early apoptosis were significantly increased in the siRNA1 and siRNA2 groups, compared to the NC and control groups. However, siRNA did not significantly affect the number of cells undergoing late apoptosis. These results indicate that the Col4A2 lentivirus siRNA decreased the proliferation of TNBC cells by inducing apoptosis. In the future, we might be able to induce programmed cell death through the use of a targeted Col4A2 inhibitor, in order to achieve the goal of treating TNBC. As shown in Figure 5, a higher percentage of cells in the G2 phase and a lower percentage of cells in the S phase were detected in the siRNA group compared to the NC and control groups. These results indicate that transfection with Col4A2 lentivirus siRNA arrested the TNBC cell cycle in the G2 phase to suppress DNA synthesis.
In conclusion, we showed that Col4A2 can restrain both MDA-MB-231 and MDA-MB-468 cells proliferation, migration, cell cycle, and ultimately lead to apoptosis. The data indicated that Col4A2 was significantly correlated with the proliferation and invasive potential of TNBC. Moreover, the result of Col4A2 inhibition can suppressed cell proliferation and invasion in MDA-MB-231 cells and MDA-MB-468 cells which may help to understand the pathogenesis of TNBC. Inhibition of Col4A2 might represent a new target for the prevention and treatment of TNBC. Further in vivo and in vitro experiments are needed to uncover the biological function of the differentially regulated genes in the pathogenesis of TNBC. A future challenge will be to determine the mechanisms of Col4A2 in regulating breast cancer incidence.
MATERIALS AND METHODS
Construction of Col4A2 RNAi lentiviral vector
Specific primers for Col4A2-si-RNA, as well as negative controls (NC), were designed based on the gene sequence in GenBank (Gene ID: 1284), which can be found in Table 1. The pGLV3/H1/GFP+Puro vector was used for selecting clones. All the procedures were performed according to the manufacturer's instructions.
Table 1. Primers of Col4A2 si-RNA and negative controls.
| Primers(5′–3′) | |
|---|---|
| Si-1 F | GATCCGGGACCCAACGGGATTCCATTTCAAGAGAATGGAATCCCGTTGGGTCCCTTTTTTG |
| Si-1 R | AATTCAAAAAAGGGACCCAACGGGATTCCATTCTCTTGAAATGGAATCCCGTTGGGTCCCG |
| Si-2 F | GATCCGGGAATGCAGATGTACAGAATTCAAGAGATTCTGTACATCTGCATTCCCTTTTTTG |
| Si-2R | AATTCAAAAAAGGGAATGCAGATGTACAGAATCTCTTGAATTCTGTACATCTGCATTCC CG |
| shNC F | GATCCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAACTTTTTTG |
| shNC R | AATTCAAAAAAGTTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAACG |
“F”: Forward; “R”: Reverse.
Lentivirus packaging and titer measurement
Human embryonic kidney 293T cells (Beijing Dingguo Changsheng Biotech Co., Ltd., Beijing, China) were selected for packaging and titer measurement of the lenti-virus. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Carlsbad, USA), supplemented with 10% heat-inactivated fetal bovine serum, 100 mg/L streptomycin, and 100 U/mL penicillin (Gibco) under 5% CO2, at 37°C in a humidified incubator. When cells were grown to approximately 70–80% confluence, they were transfected with NC (control shRNA lenti-viral vectors) and Col4A2 RNAi lentiviral vectors using Lipofectamine2000 (Invitrogen, Carlsbad, USA). After 48h, the viruses were harvested and concentrated, followed by titer detection. A serial dilutions of the lentivirus from 10−1 to 10−4 were prepared by diluting the concentrated lentivirus. 4 days later, fluorescent cells were counted in the last two concentrations, then divided by their corresponding n dilution factors to obtain the original virus titer.
Transfection
In this study, the effects of down-regulated COL4A2 expression were investigated in two types of human breast cancer cell lines, MDA-MB-231and MDA-MB-468. When cells were grown to approximately 90% confluence, polybrene was added according to the manufacturer's instructions. Then different vector was added according to the MOI ( multiplicity of infection) value and the virus titer, and incubated overnight under 5% CO2, at 37°C. Both cell types were divided into three subgroups: Con-transfected with empty lenti-virus vector, NC group-transfected with NC vector ands iRNA group-transfected with the Col4A2 RNAi lenti-viral vector.
Real-time PCR
Total RNA was extracted from the cultured MDA-MB-231and MDA-MB-468 cells and the concentration of extracted RNA was estimated by optical density measurement (A260/A280 ratio) with a Q5000 Spectrophotometer (Quawell, Sunnyvale, USA). Real-time PCR amplification was carried out using the SYBR Green-based PCR Master Mix (Applied Biosystems/Life Technologies, Carlsbad, CA). The ABI PRISM 7500 system (ABI, Grand Island, NY, USA) was used for all reactions in a total volume of 25 μL.
Western blotting
Cells were harvested and total protein was isolated using RIPA buffer (50 mM Tris-HCl at pH 7.4, 1 mM EDTA, 150 mM NaCl, 1% NP-40 and 0.5% SDS) supplemented with proteinase cocktail (Roche, Switzerland), and heated for 5 min at 100°C. Pierce BCA Protein Assay Kit (Thermo, USA) was used to determine protein concentration. Equal amount of denatured protein samples were loaded and separated by 12% SDS-polyacrylamide gels, and then transferred onto polyvinylidene difluoride membranes (PALL). After blocking with 5% non-fat milk power in Tris-buffered saline/0.05% Tween 20 (TBST), the membrane was incubated with a specific primary antibody, followed by the horseradixh peroxidase-conjugated secondary antibody. Proteins were visualized using ECL reagents (Pierce).
CCK8 assay
After transfection for 72 hours, MDA-MB-231 and MDA-MB-468 cells were incubated for 0, 24, 48, and 72 hours in 96-wells plate with 2000 cells/well. Then 20 μL CCK8 (Dojindo Molecular Technologies, Inc.,Xiongben, Japan) was added to each well for 1 hour, followed by light absorbance measurement at a wavelength of 450 nm.
Transwell assay
After incubating for 72 hours, MDA-MB-231 and MDA-MB-468 cells were seeded in the upper chambers of 6-well transwell with 25,000 cells/well. The chambers were then incubated for 48 hours at 37°C in a humidified incubator with 5% CO2. Subsequently, cells in the lower chamber was stained with hematoxylin and photographed.
Flow cytometry analyses
For cell cycle analysis, MDA-MB-231 and MDA-MB-468 cells were seeded in the 6-well plates with 500,000 cells/well, and then incubated at 37°C in a humidified incubator with 5% CO2. After transfection for 72 hours, the cells were collected, washed with Dulbecco's phosphate buffered saline (DPBS; Genview, El Monte,USA), fixed in 70% ethanol, and incubated overnight at −20°C. Ethanol was removed by centrifugation. The cell pellets were washed with DPBS, followed by incubation with 100 μL propidium iodide (PI, Sigma, St.Louis, USA) solution for 5–10 minutes in the dark at 37°C. The cells were analyzed using a flow cytometer (Beckman Coulter, Brea, USA).
For apoptosis analysis, cells were stained using the Annexin V-FITC Apoptosis Detection Kit (ShanghaiGenechem Biotech Co., Ltd.) and PI staining. Cells were harvested 72 hours after transfection, binding buffer (5 μL Annexin V/FITC) was added according to the manufacturer's instructions. After 15 minutes, PI staining was performed in the dark at room temperature, followed by incubation in the dark at room temperature for another 15 minutes. Apoptosis was then detected by flow cytometry.
Statistical analysis
Data are shown as the means ± standard deviation. The statistical significance of differences between groups was assessed via one-way analysis followed by Student's t-tests of comparison. p-values less than 0.05 were considered to be statistically significant. Statistical analyses were conducted using the GraphPad Prism 4.0 software.
ACKNOWLEDGMENTS AND FUNDING
This work were supported by the National Natural Science Foundation of China (81372583 and 81402370), The Scientific and Technology Foundation of Guangdong Province (2015B090904007), The Natural Science Foundation of Guangdong Province (2015A030313829), the Science and Technology Foundation of Shenzhen (CXZZ20150430092951135, KQTD20140630100658078, and JCYJ20150422150029094), Shenzhen city science and technology innovation international cooperation projects 2014 (NO.GJHZ20140414170821180) ,the Natural Science Foundation of SZU (201573), and the Key Laboratory Project of Shenzhen (ZDSY20130329101130496).
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Peppercorn J, Perou CM, Carey LA. Molecular subtypes in breast cancer evaluation and management: divide and conquer. Cancer investigation. 2008;26:1–10. doi: 10.1080/07357900701784238. [DOI] [PubMed] [Google Scholar]
- 2.Donepudi MS, Kondapalli K, Amos SJ, Venkanteshan P. Breast cancer statistics and markers. Journal of cancer research and therapeutics. 2014;10:506–511. doi: 10.4103/0973-1482.137927. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 4.Park YH, Jung HH, Ahn JS, Im YH. Statin induces inhibition of triple negative breast cancer (TNBC) cells via PI3K pathway. Biochemical and biophysical research communications. 2013;439:275–279. doi: 10.1016/j.bbrc.2013.08.043. [DOI] [PubMed] [Google Scholar]
- 5.Penault-Llorca F, Viale G. Pathological and molecular diagnosis of triple-negative breast cancer: a clinical perspective. Annals of oncology. 2012;23:vi19–22. doi: 10.1093/annonc/mds190. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy N, Mitchell G, Bilous M, Wilcken N, Lindeman GJ. Triple-negative breast cancer: making the most of a misnomer. Asia-Pacific journal of clinical oncology. 2012;8:145–155. doi: 10.1111/j.1743-7563.2012.01533.x. [DOI] [PubMed] [Google Scholar]
- 7.Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Annals of oncology. 2012;23:2223–2234. doi: 10.1093/annonc/mds067. [DOI] [PubMed] [Google Scholar]
- 8.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. The New England journal of medicine. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 9.Turner AW, Nikpay M, Silva A, Lau P, Martinuk A, Linseman TA, Soubeyrand S, McPherson R. Functional interaction between COL4A1/COL4A2 and SMAD3 risk loci for coronary artery disease. Atherosclerosis. 2015;242:543–552. doi: 10.1016/j.atherosclerosis.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Jeanne M, Jorgensen J, Gould DB. Molecular and Genetic Analyses of Collagen Type IV Mutant Mouse Models of Spontaneous Intracerebral Hemorrhage Identify Mechanisms for Stroke Prevention. Circulation. 2015;131:1555–1565. doi: 10.1161/CIRCULATIONAHA.114.013395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkin JD, San Antonio JD, Pedchenko V, Hudson B, Jensen ST, Savige J. Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Human mutation. 2011;32:127–143. doi: 10.1002/humu.21401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bunt S, Hooley C, Hu N, Scahill C, Weavers H, Skaer H. Hemocyte-secreted type IV collagen enhances BMP signaling to guide renal tubule morphogenesis in Drosophila. Developmental cell. 2010;19:296–306. doi: 10.1016/j.devcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Harris RE, Bayston LJ, Ashe HL. Type IV collagens regulate BMP signalling in Drosophila. Nature. 2008;455:72–77. doi: 10.1038/nature07214. [DOI] [PubMed] [Google Scholar]
- 14.Paralkar VM, Vukicevic S, Reddi AH. Transforming growth factor beta type 1 binds to collagen IV of basement membrane matrix: implications for development. Developmental biology. 1991;143:303–308. doi: 10.1016/0012-1606(91)90081-d. [DOI] [PubMed] [Google Scholar]
- 15.Jeanne M, Labelle-Dumais C, Jorgensen J, Kauffman WB, Mancini GM, Favor J, Valant V, Greenberg SM, Rosand J, Gould DB. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. American journal of human genetics. 2012;90:91–101. doi: 10.1016/j.ajhg.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoneda Y, Haginoya K, Arai H, Yamaoka S, Tsurusaki Y, Doi H, Miyake N, Yokochi K, Osaka H, Kato M, Matsumoto N, Saitsu H. De novo and inherited mutations in COL4A2, encoding the type IV collagen alpha2 chain cause porencephaly. American journal of human genetics. 2012;90:86–90. doi: 10.1016/j.ajhg.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verbeek E, Meuwissen ME, Verheijen FW, Govaert PP, Licht DJ, Kuo DS, Poulton CJ, Schot R, Lequin MH, Dudink J, Halley DJ, de Coo RI, den Hollander JC, Oegema R, Gould DB, Mancini GM. COL4A2 mutation associated with familial porencephaly and small-vessel disease. European journal of human genetics. 2012;20:844–851. doi: 10.1038/ejhg.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warnecke-Eberz U, Metzger R, Holscher AH, Drebber U, Bollschweiler E. Diagnostic marker signature for esophageal cancer from transcriptome analysis. Tumour biology. 2015 doi: 10.1007/s13277-015-4400-4. [DOI] [PubMed] [Google Scholar]
- 19.Ilhan-Mutlu A, Siehs C, Berghoff AS, Ricken G, Widhalm G, Wagner L, Preusser M. Expression profiling of angiogenesis-related genes in brain metastases of lung cancer and melanoma. Tumour biology. 2016;37:1173–1182. doi: 10.1007/s13277-015-3790-7. [DOI] [PubMed] [Google Scholar]
- 20.Oktem G, Sercan O, Guven U, Uslu R, Uysal A, Goksel G, Ayla S, Bilir A. Cancer stem cell differentiation: TGFbeta1 and versican may trigger molecules for the organization of tumor spheroids. Oncology reports. 2014;32:641–649. doi: 10.3892/or.2014.3252. [DOI] [PubMed] [Google Scholar]
- 21.Zhu J, Pan C, Jiang J, Deng M, Gao H, Men B, McClelland M, Mercola D, Zhong WD, Jia Z. Six stroma-based RNA markers diagnostic for prostate cancer in European-Americans validated at the RNA and protein levels in patients in China. Oncotarget. 2015;6:16757–16765. doi: 10.18632/oncotarget.4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown CW, Brodsky AS, Freiman RN. Notch3 overexpression promotes anoikis resistance in epithelial ovarian cancer via upregulation of COL4A2. Molecular cancer research. 2015;13:78–85. doi: 10.1158/1541-7786.MCR-14-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell SM, Ross JP, Drew HR, Ho T, Brown GS, Saunders NF, Duesing KR, Buckley MJ, Dunne R, Beetson I, Rand KN, McEvoy A, Thomas ML, Baker RT, Wattchow DA, Young GP, Lockett TJ, Pedersen SK, Lapointe LC, Molloy PL. A panel of genes methylated with high frequency in colorectal cancer. BMC cancer. 2014;14:54. doi: 10.1186/1471-2407-14-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maekawa R, Sato S, Yamagata Y, Asada H, Tamura I, Lee L, Okada M, Tamura H, Takaki E, Nakai A, Sugino N. Genome-wide DNA methylation analysis reveals a potential mechanism for the pathogenesis and development of uterine leiomyomas. PloS one. 2013;8:e66632. doi: 10.1371/journal.pone.0066632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Gao C, Zhuang JL, Ding C, Wang Y. A combined approach identifies three mRNAs that are down-regulated by microRNA-29b and promote invasion ability in the breast cancer cell line MCF-7. Journal of cancer research and clinical oncology. 2012;138:2127–2136. doi: 10.1007/s00432-012-1288-x. [DOI] [PubMed] [Google Scholar]
- 26.Huan JL, Gao X, Xing L, Qin XJ, Qian HX, Zhou Q, Zhu L. Screening for key genes associated with invasive ductal carcinoma of the breast via microarray data analysis. Genetics and molecular research. 2014;13:7919–7925. doi: 10.4238/2014.September.29.5. [DOI] [PubMed] [Google Scholar]
- 27.He J, Yang J, Chen W, Wu H, Yuan Z, Wang K, Li G, Sun J, Yu L. Molecular Features of Triple Negative Breast Cancer: Microarray Evidence and Further Integrated Analysis. PloS one. 2015;10:e0129842. doi: 10.1371/journal.pone.0129842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poage GM, Hartman ZC, Brown PH. Revealing targeted therapeutic opportunities in triple-negative breast cancers: a new strategy. Cell cycle. 2013;12:2705–2706. doi: 10.4161/cc.25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin S, Mutvei AP, Chivukula IV, Andersson ER, Ramskold D, Sandberg R, Lee KL, Kronqvist P, Mamaeva V, Ostling P, Mpindi JP, Kallioniemi O, Screpanti I, Poellinger L, Sahlgren C, Lendahl U. Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKalpha/IKKbeta. Oncogene. 2013;32:4892–4902. doi: 10.1038/onc.2012.517. [DOI] [PMC free article] [PubMed] [Google Scholar]