

Abstract
A 32-year-old male presented with advanced renal failure and nephrotic proteinuria due to lambda light chain multiple myeloma. Renal biopsy showed a proliferative glomerulonephritis with isolated C3 deposits. Renal recovery was obtained after chemotherapy and autologous stem cell transplant. We review previously described cases of C3 glomerulopathy associated with monoclonal gammopathy.
Key words: C3 glomerulopathy, complement alternative pathway, monoclonal gammopathy
Introduction
Since its first description in 1971, the spectrum of glomerular diseases associated with deposition and precipitation of monoclonal immunoglobulin components has expanded.[1] These diseases can be classified into two categories.[2] The first category includes diseases with organized fibrillar (amyloidosis) and microtubular (cryoglobulinemia, immunotactoid glomerulonephritis [GN]) deposits. The second category is characterized by nonorganized electron-dense deposits, usually granular, located along the basement membranes in most tissues, mainly in the kidney and defines the monoclonal immunoglobulin deposition disease (MIDD). Furthermore, in some cases, nonorganized electron-dense deposits correspond to dense deposit disease (DDD) and C3 GN (C3GN), now classified under the heading of C3 glomerulopathies and differ from classic MIDD by the presence of isolated deposits of C3 on immunofluorescence.[3,4,5,6] Hypocomplementemia is common in these latter cases.
Rare cases of C3 glomerulopathies have been reported in association with plasma cell disorders.[7,8] We report the case of a patient with monoclonal gammopathy and glomerular disease, characterized by isolated C3 deposits without monoclonal immunoglobulin deposits. Unlike other rare reported cases, the evolution in this case was favorable. To our knowledge, this is the second observation of a case where a complete renal remission was obtained after controlling hematologic disease by autologous stem cell (ASC) transplant.[9] The role of monoclonal component in the constitution of kidney damage is discussed.
Case Report
A 32-year-old male was admitted to our department for acute renal failure. This patient reported diffuse bone pain and asthenia from a few weeks ago. Clinical examination revealed no abnormalities. Urine dipstick revealed 3+ protein and 2+ blood.
The serum creatinine was 4.5 mg/dl and 24- urine protein 10 g/day. Blood count revealed normochromic normocytic anemia at 8.2 g/dl of hemoglobin. The rest of biological data showed hypoalbuminemia at 3.0 g/dl and hypercalcemia at 13.3 mg/dl. Tests for serum rheumatoid factor, hepatitis (B and C), and HIV infection were negative. Antinuclear, anti-double-stranded deoxyribonucleic acid and M2 antimitochondrial antibodies were not detected. The serum protein electrophoresis test identified a thin monoclonal band in the beta 2 globulin position. The immunofixation confirmed the presence of monoclonal lambda light chains in serum and urine. The free lambda light chains were 6330 mg/L (kappa at 331 mg/L and kappa/lambda ratio at 19). The bone marrow aspiration showed atypical plasma cells >90% of marrow cells with some normal hematopoietic elements. X-rays revealed a patchy distribution of skeletal lesions. Cryoglobulinemia was negative. The dosage of the C3 complement fraction was low (0.55 g/L) with normal levels for the C4 fraction. Exploration of specific complement alternative pathway (CAP) proteins has not been performed.
The patient was diagnosed with a lambda light chain multiple myeloma associated with renal impairment. Common causes of renal failure were excluded and a renal biopsy was performed and showed the aspect of a membranoproliferative GN with isolated C3 deposits, which confirmed the diagnosis of C3 glomerulopathy. The light microscopic appearance of glomerular lesions was characterized by a mesangial proliferation with neutrophilic leukocyte afflux in glomerular tufts and deposited in the mesangium and glomerular capillary walls [Figure 1]. Moderate chronic lesions, including global glomerular sclerosis, tubular atrophy, interstitial fibrosis, and arteriosclerosis, were also present. Immunofluorescence showed diffuse and intense staining for C3 along the glomerular, tubular, and Bowman's capsule basement membranes, as well as mesangial rings [Figure 2]. No significant staining was observed with anti-kappa, lambda, C4, C1q, and immunoglobulins (A, G, and M) conjugates. Unfortunately, electronic microscopy was not performed to establish the distinction between DDD and C3GN.
Figure 1.
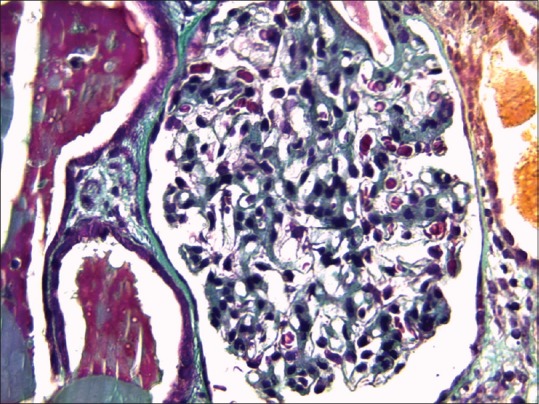
Glomerular lesions with mesangial proliferation and neutrophilic leukocyte afflux in glomerular tufts (light microscopy, Masson trichrome stain, ×100)
Figure 2.
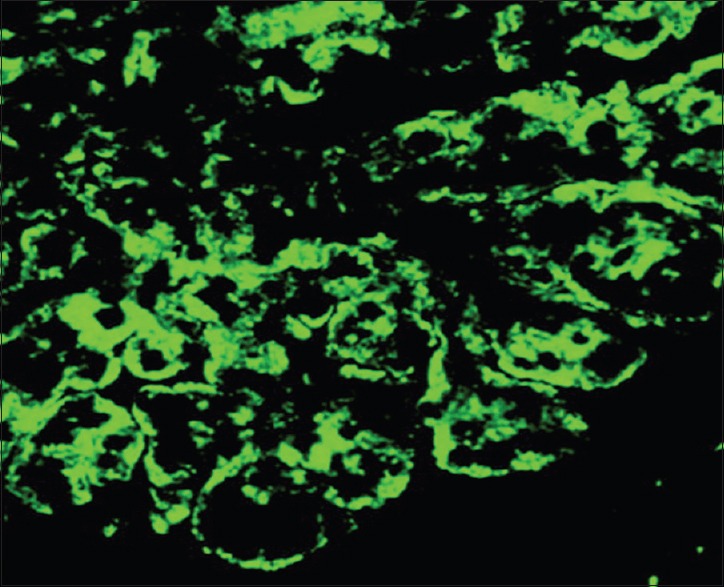
Intense staining for C3 along the glomerular basement membranes and mesangial rings (immunofluorescence, ×100)
The patient received hydration with alkalinization which allowed his renal function to improve (creatinine 30 mg/L at the time of his discharge) and his serum calcium to normalize. Chemotherapy consisted of cyclophosphamide (500 mg on days 1, 8, and 15 per month), dexamethasone (40 mg from day 1 to day 4 each week during the first 2 months, then from day 1 to day 4 each month), and thalidomide (100 mg daily for 1 month followed by 200 mg/day). This treatment was extended through 9 months after which the patient was referred for an ASC transplant.
The clinical course was favorable with an improved renal function, remission of the nephrotic syndrome, and complete hematologic remission over 1 year follow-up after ASC transplant. Serum C3 complement levels returned to normal at 6 months after the initiation of treatment.
Discussion
The combination of C3 glomerulopathy and monoclonal gammopathy has rarely been described before.[10] Prevalence of monoclonal gammopathy appears to be higher in patients with C3 glomerulopathy compared to the general population, which does not favor a simple coincidence.[11,12] Nasr et al. report a plasma cell disorder in 4 of 32 patients with DDD.[13] Sethi et al. found that among 14 patients with DDD, ten (71.4%) had a serum monoclonal immunoglobulin G.[8]
Several hypotheses have been advanced to explain how kidney damages in C3 glomerulopathy can be linked to monoclonal gammopathy. The role of monoclonal immunoglobulin in the systemic activation of CAP, leading to glomerular proliferative lesions and subsequent C3 deposits, is questionable.
Bridoux et al. reported six cases of similar patients with C3GN and evidence of monoclonal gammopathy.[14] All patients had nephrotic proteinuria with a variable degree of renal failure. Most patients had low serum C3 levels. No genetic abnormality was found in all tested patients, so a genetic predisposition in favor of activation of the CAP does not seem necessary to provoke kidney lesions. In one of the six patients, anti-factor H autoantibodies were detected without any immunoglobulin deposit. However, a control biopsy performed 4 years later showed a colocalization of C3 deposits and lambda light chain, suggesting that the isolated C3 deposition might be the initial step, which precedes the secondary deposit of immunoglobulin.
In the previous reported cases, isolated glomerular C3 deposits were associated with monoclonal gammopathy of unknown significance, rather than a high-mass myeloma.[4,6,8] This finding can suggest that prolonged CAP activation by the monoclonal immunoglobulin is required for the development of glomerular lesions. The role of monoclonal immunoglobulin in the activation of CAP has been demonstrated in recent observations. Meri et al. in a similar case showed that purified monoclonal light chains can interact with factor H and activated the CAP.[15] Jokiranta et al. also isolated monoclonal light chains from a patient with DDD and demonstrated the ability of these autoantibodies to activate the CAP.[16]
Other factors could be implicated in the pathogenesis. Recently, Zand et al. suggested that elevated cytokines in multiple myeloma such as interleukin 6 could explain an excessive activation of CAP by an elevated activity of complement factor I.[17] Indeed, transcriptional activity of factor I is enhanced in the presence of these cytokines.[18]
Decreased serum C3 and a normal C4 may help distinguish C3 glomerulopathies from other types of GN. The main differential diagnosis is postinfectious GN which can produce a similar clinical syndrome. As poststreptococcal GN, C3 glomerulopathies may also be preceded by upper respiratory tract infection.[19] In our case, several features help distinguish C3 glomerulopathy from postinfectious GN. Depression of C3 persisted for several months and became normal only after hematologic remission, whereas low C3 is usually transient in postinfectious GN. On the other hand, in immunofluorescence microscopy, immunoglobulins were absent which indicate that it was not an immune complex-mediated disease.
In addition to serum C3 and C4, special diagnostic tests should be obtained in patients with C3 glomerulopathy (if possible) since the results can help to determine a specific therapeutic approach (C3 nephritic factor (C3NeF), serum factor H, complement factor H-related protein gene mutations, serum factor B, serum factor I, membrane cofactor protein (CD46), and soluble C5b-9).
C3NeF, an autoantibody that stabilizes the C3 convertase, is found frequently in patients with C3 glomerulopathy.[20] In this case, C3NeF was undetectable; however, unfortunately, its determination was not performed until several months after treatment beginning when the patient was already in remission of his hematological disease.
The improvement of renal disease and complement abnormalities were observed after complete hematologic remission. Plasma exchange, fresh frozen plasma infusions, or immunosuppressive therapy has not been required to maintain normal levels of C3. These arguments are not in favor of a genetic anomaly as a possible pathogenic cause. An acquired disorder (for example, the suggested role of the monoclonal immunoglobulin in CAP excessive activation) rather than genetic seems more likely, but genetic testing for an inherited disorder was not performed.
The peculiarity of our case is the favorable renal outcome after the hematological remission. Indeed, the outcome of the most reported cases was poor with frequent progression to end-stage renal disease.[1,6,16] Improvement in renal function, reduction of proteinuria, and normalization of C3 level in parallel with the control of the hematologic disease are a strong argument for a probable role of monoclonal gammopathy in the pathogenesis of this case. However, it is possible that the improvement in proteinuria following treatment is attributable to switching off the production of lambda light chains, thus preventing their overflow into the urine, rather than the regression of glomerular lesions. Overall, we considered that the renal damage in our case was reversible, and a posttreatment biopsy was not performed.
Conclusion
Monoclonal gammopathy should be considered in the patients with C3 glomerulopathy. An excessive activation of CAP by monoclonal immunoglobulin remains the most plausible reported explanation. However, additional studies are needed to prove this hypothesis. Renal remission could be subject to an effective treatment of plasma cell dyscrasias.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We acknowledge Dr. Hanan Oubenyahya for his invaluable help in drafting this case report.
References
- 1.Glenner GG, Terry W, Harada M, Isersky C, Page D. Amyloid fibril proteins: Proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150–1. doi: 10.1126/science.172.3988.1150. [DOI] [PubMed] [Google Scholar]
- 2.Touchard G. Ultrastructural pattern and classification of renal monoclonal immunoglobulin deposits. In: Touchard G, Aucouturier P, Hermine O, Ronco P, editors. Monoclonal Gammopathies and the Kidney. Dordrecht: Kluwer Academic Publishers; 2003. pp. 95–117. [Google Scholar]
- 3.Bridoux F, Binaut R, Zanetta G, Mougenot B, Goujon JM, Vanhille P, et al. Glomerulopathy with non-organized and non-Randall type monoclonal immunoglobulin deposits: A rare entity. J Am Soc Nephrol. 2001;12:94. [Google Scholar]
- 4.Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: A distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65:85–96. doi: 10.1111/j.1523-1755.2004.00365.x. [DOI] [PubMed] [Google Scholar]
- 5.Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20:2055–64. doi: 10.1681/ASN.2009010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sethi S, Zand L, Leung N, Smith RJ, Jevremonic D, Herrmann SS, et al. Membranoproliferative glomerulonephritis secondary to monoclonal gammopathy. Clin J Am Soc Nephrol. 2010;5:770–82. doi: 10.2215/CJN.06760909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sepandj F, Trillo A. Dense deposit disease in association with monoclonal gammopathy of unknown significance. Nephrol Dial Transplant. 1996;11:2309–12. doi: 10.1093/oxfordjournals.ndt.a027156. [DOI] [PubMed] [Google Scholar]
- 8.Sethi S, Sukov WR, Zhang Y, Fervenza FC, Lager DJ, Miller DV, et al. Dense deposit disease associated with monoclonal gammopathy of undetermined significance. Am J Kidney Dis. 2010;56:977–82. doi: 10.1053/j.ajkd.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen CA, Marcussen N, Gregersen JW. Recovery of renal function succeeding stem cell transplant: A case of C3 glomerulonephritis secondary to monoclonal gammopathy. Clin Kidney J. 2013;6:639–42. doi: 10.1093/ckj/sft124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): An update. J Am Soc Nephrol. 2005;16:1392–403. doi: 10.1681/ASN.2005010078. [DOI] [PubMed] [Google Scholar]
- 11.Saleun JP, Vicariot M, Deroff P, Morin JF. Monoclonal gammopathies in the adult population of Finistère, France. J Clin Pathol. 1982;35:63–8. doi: 10.1136/jcp.35.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362–9. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 13.Nasr SH, Valeri AM, Appel GB, Sherwinter J, Stokes MB, Said SM, et al. Dense deposit disease: Clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol. 2009;4:22–32. doi: 10.2215/CJN.03480708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bridoux F, Desport E, Frémeaux-Bacchi V, Chong CF, Gombert JM, Lacombe C, et al. Glomerulonephritis with isolated C3 deposits and monoclonal gammopathy: A fortuitous association? Clin J Am Soc Nephrol. 2011;6:2165–74. doi: 10.2215/CJN.06180710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meri S, Koistinen V, Miettinen A, Törnroth T, Seppälä IJ. Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis. J Exp Med. 1992;175:939–50. doi: 10.1084/jem.175.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jokiranta TS, Solomon A, Pangburn MK, Zipfel PF, Meri S. Nephritogenic lambda light chain dimer: A unique human miniautoantibody against complement factor H. J Immunol. 1999;163:4590–6. [PubMed] [Google Scholar]
- 17.Zand L, Kattah A, Fervenza FC, Smith RJ, Nasr SH, Zhang Y, et al. C3 glomerulonephritis associated with monoclonal gammopathy: A case series. Am J Kidney Dis. 2013;62:506–14. doi: 10.1053/j.ajkd.2013.02.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minta JO, Fung M, Paramaswara B. Transcriptional and post-transcriptional regulation of complement factor I (CFI) gene expression in Hep G2 cells by interleukin-6. Biochim Biophys Acta. 1998;1442:286–95. doi: 10.1016/s0167-4781(98)00189-4. [DOI] [PubMed] [Google Scholar]
- 19.Bennett WM, Fassett RG, Walker RG, Fairley KF, d'Apice AJ, Kincaid-Smith P. Mesangiocapillary glomerulonephritis type II (dense-deposit disease): Clinical features of progressive disease. Am J Kidney Dis. 1989;13:469–76. doi: 10.1016/s0272-6386(89)80004-6. [DOI] [PubMed] [Google Scholar]
- 20.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–64. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]