

Abstract
Purpose
Enhanced regeneration of retinal ganglion cell (RGC) axons can be achieved by modification of numerous neuronal-intrinsic factors. However, axon growth initiation and the pathfinding behavior of these axons after traumatic injury remain poorly understood outside of acute injury paradigms, despite the clinical relevance of more chronic settings. We therefore examined RGC axon regeneration following therapeutic delivery that is postponed until 2 months after optic nerve crush injury.
Methods
Optic nerve regeneration was induced by virally mediated (adeno-associated virus) ciliary neurotrophic factor (AAV-CNTF) administered either immediately or 56 days after optic nerve crush in wild-type or Bax knockout (KO) mice. Retinal ganglion nerve axon regeneration was assessed 21 and 56 days after viral injection. Immunohistochemical analysis of RGC injury signals and extrinsic factors in the optic nerve were also examined at 5 and 56 days post crush.
Results
In addition to sustained expression of injury response proteins in surviving RGCs, we observe axon regrowth in wild-type and apoptosis-deficient Bax KO mice following AAV-CNTF treatment. Fewer instances of aberrant axon growth are seen, at least in the area near the lesion site, in animals given treatment 56 days after crush injury compared to the animals given treatment immediately after injury. We also find evidence of long distance growth into a visual target in Bax KO mice despite postponed initiation of this regenerative program.
Conclusions
These studies provide evidence against an intrinsic critical period for RGC axon regeneration or degradation of injury signals. Regeneration results from Bax KO mice imply highly sustained regenerative capacity in RGCs, highlighting the importance of long-lasting neuroprotective strategies as well as of RGC axon guidance research in chronically injured animals.
Keywords: axon regeneration, axon guidance, neuroprotection, ganglion cell, axon injury
Optic nerve injury triggers molecular events in retinal ganglion cells (RGCs), ultimately leading to axonal degeneration and apoptotic cell death. A small population of RGCs that survive injury do not spontaneously regenerate axons to long distances, causing permanent disconnection and loss of visual functions. Demise of RGCs and axonal degeneration occur within a few days following an intraorbital axotomy, and the majority of RGCs die within 2 weeks.1–3 At the molecular level, axonal injury causes upregulation of several stress-related and apoptosis-promoting proteins including activating transcription factor 3 (ATF3) and c-Jun.4–7 In an attempt to overcome injury, RGCs also increase expression of proteins known to promote cell survival (e.g., endoplasmic reticulum chaperones)8 and/or axon regeneration (e.g., STAT3). However, when death-promoting cascades outweigh these reparative efforts, RGCs undergo apoptosis.
In addition to changes that take place within the retina, axonal injury leads to drastic changes in the optic nerve, where distal axonal portions begin to undergo Wallerian degeneration 2 to 3 days following axotomy. Within several hours to days post injury, bloodborne macrophages infiltrate the lesion while astrocytes and oligodendrocytes in the lesion core undergo cell death. In the lesion periphery, astrocytes become reactive and release inflammatory and extracellular matrix proteins. Axonal and myelin debris are cleared away by both resident microglial cells and hematogenous macrophages.9–12
In mice, modulating the expression of certain genes within adult RGCs promote some RGC axons to elongate through the optic nerve and toward visual targets.13–18 Nonetheless, many axons induced to regenerate before or acutely after injury (i.e., 5–7 days post injury) are misguided in multiple regions, including the optic nerve, optic chiasm, and brain.19–21 Aberrant axon growth in the optic nerve is particularly prominent in the regions adjacent to crush site,20 indicating that the activated glial cells likely influence the path of regenerating axons. These observations raise a question of whether RGC axons that are stimulated to regenerate long after injury (when some myelin debris is cleared and astrocytic activation subsides) would exhibit distinct regrowth rates and pathfinding patterns.
Although the cellular and molecular changes that occur acutely (i.e., within hours to a week) after axotomy (e.g., crush injury) have been well characterized, changes that take place in RGCs and optic nerves of chronically injured animals (i.e., months or years after crush injury) are ill-defined. Furthermore, few studies utilize chronic optic nerve injury models, and it is currently unclear to what extent RGC axons regenerate and, importantly, find paths in the chronically injured optic pathway.
In this study, we examined (1) RGC survival, (2) expression of several injury-response proteins in RGCs, and (3) local cellular changes in the optic nerve in chronically injured animals (i.e., 56 days post crush), and assess axon regeneration and pathfinding in wild-type and apoptosis-deficient Bax knockout (KO) mice when regeneration-promoting stimulation via adeno-associated virus serotype 2 ciliary neurotrophic factor (AAV-CNTF) is given 56 days after crush injury.
Methods
Animals
All animal procedures were performed with the approval of Institutional Animal are and Use Committee at University of Miami and in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. C57BL/6N mice (male or female; Charles River Laboratories, Wilmington, MD, USA) were used in this study, except for Bcl2-associated X protein (BAX) KO experiments, in which Baxtm1Sjk mice (B6.129X1-Baxtm1Sjk/J, stock number 002994; Jackson Laboratory, Bar Harbor, ME, USA)22 were used. Mice were aged 5 to 6 weeks at the start of each experiment and were randomly assigned to different treatment groups. The investigator who analyzed the data was not blinded to the animal identity.
Intraorbital Optic Nerve Crush
Optic nerves were crushed intraorbitally with jeweler's forceps (Dumont no. 5; Roboz Surgical Instrument Co.; Gaithersburg, MD, USA) for 10 seconds approximately 1 mm behind the optic disc. Mice received a subcutaneous injection of buprenorphine (0.05 mg/kg; Bedford Laboratories, Bedford, OH, USA) as postoperative analgesic. Eye ointment containing atropine sulfate was applied preoperatively to protect the cornea during surgery.
Intravitreal Injection
Pulled glass micropipettes were inserted into the temporal eye posterior to the ora serrata, angled away from the lens. Approximately 2 μL vitreal fluid was withdrawn prior to injection of an equal volume of virus or cholera toxin β (CTB) subunit conjugated with Alexa 555 (C34776; ThermoFisher Scientific, Waltham, MA, USA). Adeno-associated virus serotype 2 (AAV) was administered either immediately or 56 days after crush. Cholera toxin β (2 μg/μl) was injected 2 to 3 days prior to death.
In this current study, we used AAV2-CNTF generated from our previous publication.21,23 Polymerase chain reaction-amplified CNTF was inserted into a Stratagene AAV plasmid (AAV-multiple cloning site [MCS] (Stratagene, La Jolla, CA, USA). We note that this plasmid contains a knockdown cassette of a SIBR containing anti-luciferase control shRNA. The region of the SIBR knockdown vector comprising the ubiquitin promoter, intronic sequences, knockdown cassette, and enhanced green fluorescent protein (EGFP) open reading frame (ORF) was cloned into the Stratagene AAV plasmid, from which the cytomegalovirus (CMV) promoter, intron, and MCS were removed. Polymerase chain reaction–amplified CNTF was then used to replace the EGFP ORF in the AAV-SIBR vector via standard restriction digest and ligation. This AAV-CNTF was used in the previous publication to compare axon regeneration rates in animals treated with AAV2-SIBR expressing shRNA against phosphatase and tensin homolog.21 The same AAV-CNTF is used in this study to promote axon regeneration and examine CNTF effects in animals following delayed treatment. Plasmid DNA encoding human CNTF was purchased from GE Dharmacon (Accession BC068030; Lafayette, CO, USA), and the ORF was amplified using a forward primer that incorporated both a 5′Kpn1 restriction site and the nerve growth factor signal peptide sequence (5′-GGTACCATGTCCATGTTGTTCTACACTCTGATCACAGCTTTTCTGATC; GGCATACAGGCGGCTTTCACAGAGCATTCACCGC-3′), and a reverse primer that incorporated 3′ BglII site (5′-AGATCTCTACATTTTCTTGTTGTTAGCAA-3′). All enzymes were purchased from New England Biolabs (Ipswith, MA, USA). Plasmids were then used to produce AAV2 (1–4 × 1013 particles per milliliter) at the University of Miami Viral Vector Core.
Tissue Preparation and Immunohistochemistry
Animals were perfused transcardially with PBS followed by 4% paraformaldehyde (PFA) in PBS, then tissues dissected and postfixed with 4% PFA in PBS for 1 hour at room temperature. For retina whole mount, the eye was bisected at the ora serrata before removal of lens and iris. The retina was gently separated from the sclera before four incomplete radial incisions were made to create a cloverleaf shape for mounting. For histologic sectioning, samples were cryoprotected by incubating in 30% sucrose in PBS for 48 hours. Optic nerves were cryosectioned to 12-μm thickness and retinae and brain tissue to 20 μm. Tissue sections were blocked in 5% normal goat serum and 0.3% Triton X-100 in PBS for 1 hour and incubated with primary antibodies diluted in blocking buffer overnight at 4°C, followed by 1-hour incubation with secondary antibodies at room temperature. Primary antibodies used were 1:800 dilution anti-βIII-tubulin (PRB-435P; Covance, Princeton, NJ, USA); 1:200 dilution dual leucine zipper kinase (DLK) (a gift from Syu-ichi Hirai of Wakayama Medical University School of Medicine); 1:200 dilution p-c-Jun (3270S; Cell Signaling Technology, Danvers, MA, USA); 1:200 dilution ATF3 (SC188; Santa Cruz Biotechnology, Dallas, TX, USA); 1:1600 dilution glial fibrillary acidic protein (GFAP) (ab4674; Abcam, Cambridge, MA, USA); 1:500 dilution CD11b (RM2800; ThermoFisher Scientific); and 1:500 dilution myelin basic protein (MBP) (MAB386; Merck Millipore, Billerica, MA, USA). All secondary antibodies (Cy2, Cy3 or Cy5, used at dilution of 1:400) were purchased from Jackson ImmunoResearch (West Grove, PA, USA).
Cell Survival and Axon Regeneration Quantification
To quantify RGCs, the number of β III tubulin immunoreactive cells in the ganglion cell layer of 8 to 12 nonconsecutive retina sections per animal (which included the central and peripheral retinal regions) was counted. To quantify regenerating axons, the crush site was identified by tissue morphology, CTB signal intensity, and/or GFAP immunosignal, and the number of CTB+ axons that projected various distances from the lesion was recorded. To quantify axon misguidance rates, the number of CTB+ axons whose orientations were over 30 degrees from the axis of the optic nerve were counted and reported as a percentage of regenerating axons at 1 mm from the lesion. At least three to four nonconsecutive sections were counted for each animal.
Western Blot
Mice were transcardially perfused with 1× PBS and optic nerves dissected and segmented with microscissors by length from optic disc (region spanning 1–3 mm for regions surrounding the lesion and 3–5 mm for distal nerve region). Optic nerve tissue was incubated in lysis buffer composed of 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS (Sigma-Aldrich Corp., St. Louis, MO, USA), and a protease inhibitor tablet, one tablet per 10 mL of buffer (Complete Mini EDTA-Free Protease Inhibitor Cocktail tablet; Roche, Basel, Switzerland). The samples were centrifuged at 16,000g for 10 minutes at 4°C, and the supernatants were stored at −80°C for no more than 1 week. The supernatants' protein concentration was determined using an assay kit (Pierce BCA Protein Assay Kit; ThermoFisher Scientific). Five micrograms of protein was loaded and separated in a 15% acrylamide-Bis gel (Bio-Rad Laboratories, Hercules, CA, USA). The protein was transferred onto membrane (Hybond-C Super membrane; Amersham Biosciences, Little Chalfont, UK) and blocked with 3% BSA in 0.1% Tween 20 in PBS (TPBS). The membranes were incubated with primary antibodies in 3% BSA in TPBS overnight at 4°C. The membranes were incubated in horseradish peroxidase–conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA) in 3% BSA in TPBS at a 1:1000 dilution for 1 hour at room temperature before the labeled proteins were detected using the ECL agent (Pierce, Rockford, IL, USA) following the supplier's manual. The intensity of each band was quantified using software (Image Studio Lite, Ver. 5.2; LI-COR Biosciences, Lincoln, NE, USA). Quantification was the mean obtained from at least 2 or 3 biological replicates. Primary antibodies used were 1:500 dilution GFAP (mAb 13-0300; ThermoFischer Scientific) and 1:1000 dilution MBP (mAb 78896; Cell Signaling).
Statistics
Data were analyzed using ANOVA and Bonferroni test within-groups comparison for axon regeneration quantification in Bax mice and Supplementary Figure S1, respectively; otherwise, Student's t-test was used. Significant differences required P values < 0.05. Values were displayed as mean + standard error of the mean.
Results
As previously understood, we found significantly elevated rates of RGC death after administration of optic nerve crush.1–3 Within 5 days of injury (Figs. 1A, 1D), 77.1% ± 4.3% of RGCs survived compared to uninjured contralateral retina (n = 5) as assessed by counting β III tubulin immunoreactive RGCs. In contrast, survival was only 4.0% ± 0.3% at 8 weeks, or 56 days post crush (dpc) (Figs. 1B, 1D; n = 5). Thus, the pool of living RGCs from which axon regeneration can be initiated is severely limited in a chronic injury paradigm.
Figure 1.
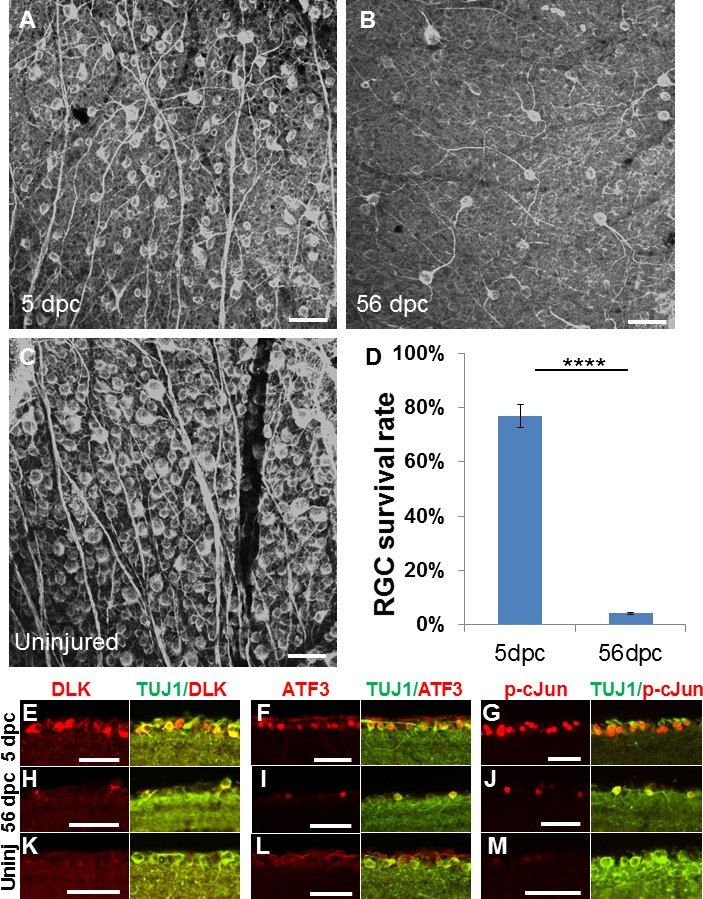
Axotomy-induced cell death and injury-signal proteins at acute and chronic time points. (A–B) β III tubulin immunoreactive (i.e., TUJ1 antibody immunostained) whole-mount retinae at 5 days (A) and 56 days (B) after optic nerve crush, and uninjured control retina (C). (D) Quantification of RGC survival, normalized to uninjured contralateral retina (n = 5/group, ****P = 0.0000026, Student's unpaired t-test). (E–M) Retinal section images of immunohistochemical analysis for DLK, ATF3, p-c-Jun signals in the ganglion cell layer of mice 5 days (E–G) and 56 days (H–I) after injury and without injury (K–M) (n = 5/group). Error bars: SEM. Scale bars: 50 μm.
We examined the presence of molecular injury signals in RGCs to better understand the possibility of axon regeneration at acute and chronic time points. Commonly used to indicate neuronal injury is DLK, also known as MAP3K12, which initiates multiple signaling cascades, including apoptosis.24,25 We also measured phosphorylated c-Jun (p-c-Jun) and ATF3, part of the CREB protein family involved in cellular stress response.7 We observed that nearly all surviving RGCs in both time points (5 and 56 days post crush) show visible immunoreactive signals of p-c-Jun, DLK, and ATF3 that are clearly higher in intensity compared to the weak background or no signal in control uninjured RGCs (Figs. 1E–M). These data indicate that RGCs retain awareness of axotomy long after it occurs.
Environmental factors have a major role in preventing or modulating patterns of axon regeneration through the injured central nervous system (CNS) tracts (reviewed by van Niekerk).26 We examined the expression of the astrocytic marker GFAP, along with cluster of differentiation marker 11b (CD11b) and MBP, which label immune cells and myelin, respectively, at 5 dpc and 56 dpc, in order to understand how the chronically injured environment after crush injury may affect axon outgrowth. We observed the well-characterized contraction of the astrocytic scar (GFAP-negative region) and the increased density of immune cells within (Figs. 2A, 2B). Glial fibrillary acidic protein–negative region at the lesion core is visibly smaller at 56 days post injury compared to 5 days post injury (Figs. 2A, 2B). Western blot of protein extracts isolated from different regions of the injured optic nerve shows no obvious difference in GFAP expression at 5 and 56 days post injury compared to uninjured optic nerve (Supplementary Fig. S1). There are decreases in MBP expression (Figs. 2C–E), especially near the lesion site (Supplementary Fig. S1), that indicates more thorough debris clearance at this later stage by hematogenous macrophages and/or resident microglia.
Figure 2.
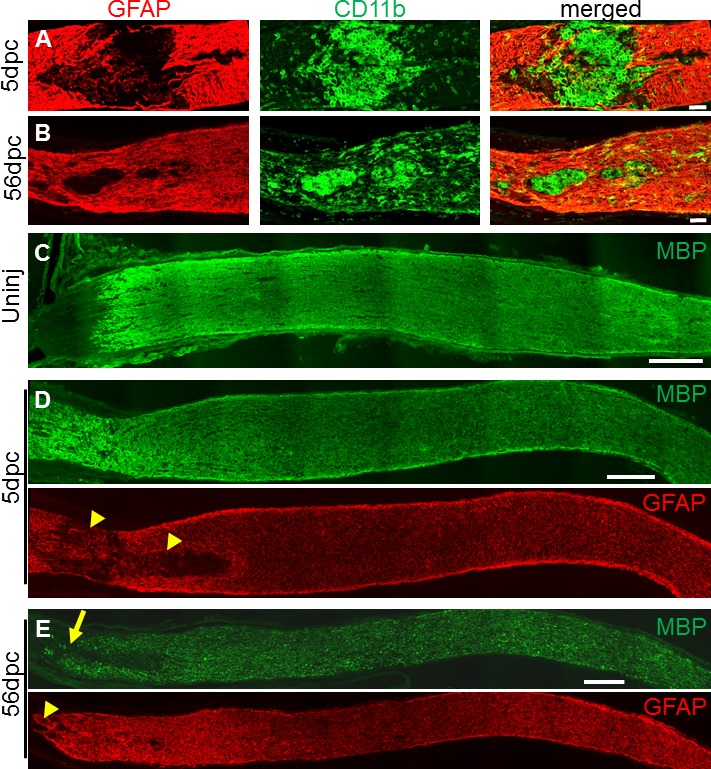
Changes in lesion and its penumbra within acutely and chronically injured optic nerves. (A–B) Glial fibrillary acidic protein and CD11b immunostained lesion sites at 5 days (A) and 56 days (B) after optic nerve crush (untreated). Note the contracted chronic lesion, defined by GFAP-negative area. (C–E) Myelin basic protein and GFAP immunostained optic nerve sections reveal large acute lesion (yellow arrowheads) and region with chronically decreased MBP expression (yellow arrow). Scale bars: 50 μm (A–B), 200 μm (C–E).
To measure the ability of chronically injured RGCs to regenerate their axon, we intravitreally treated surviving RGCs with AAV expressing the secretable form of CNTF.21 In this modified, chronic paradigm (Fig. 3A), AAV-CNTF was delivered at 56 dpc, and axon regeneration proceeded for 21 days afterward. Of note, we and others have previously shown that AAV-mediated protein expression (e.g., transgene under CMV or uBC promoter) in RGCs starts as early as 5 days after intravitreal injection (unpublished observation for AAV with uBC promoter).17 As indicated by the CTB signal, we observed small numbers of regenerate axons 1 mm beyond the lesion site in five of five animals examined (Fig. 3B, yellow arrows; quantified in 3D). In contrast, we did not see CTB-positive axons at this distance following AAV-GFP treatment in five control animals (Fig. 3C). Immediate AAV-CNTF treatment (time frame shown in Fig. 3E) results in much higher rates of axon regeneration (Figs. 3F), at least in part due to the approximately 20-fold larger pool of surviving RGCs (Fig. 1D). In these animals, the average numbers of regenerating axons per optic nerve section at 1 and 2 mm distal to the lesion site were 20 (±6.9 SEM) and 6 (±3.1 SEM), respectively (n = 3 mice). Thus, despite 8 weeks elapsing between administration of nerve injury and therapeutic treatment, some surviving RGCs were nevertheless capable of initiating a regenerative response.
Figure 3.
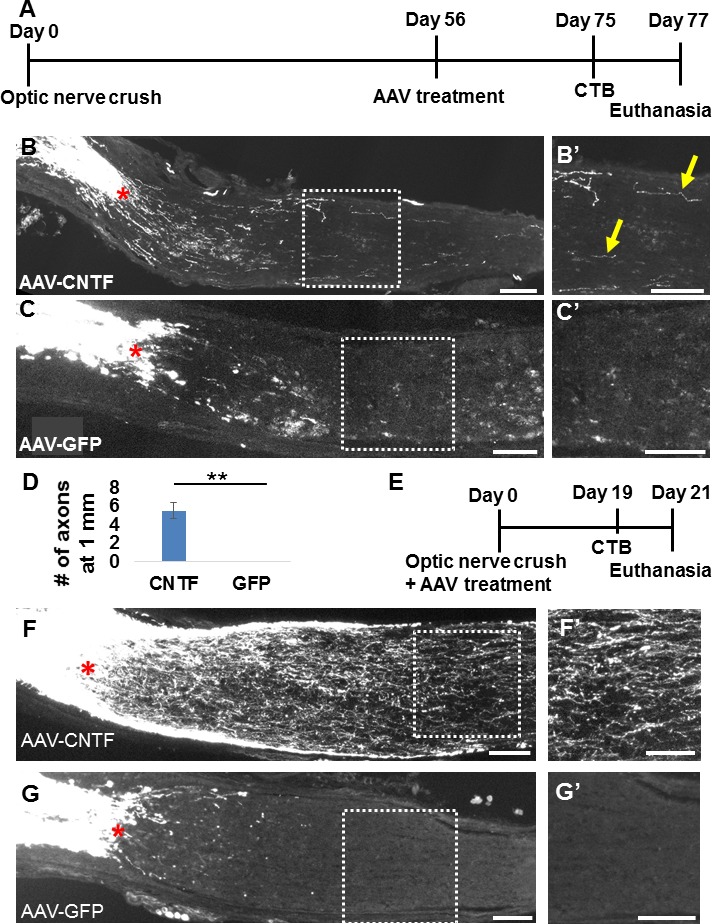
Adeno-associated virus--ciliary neurotrophic factor treatment in chronically injured mice promotes RGC axon regeneration beyond the injury site. (A) Time course of chronic experiments. (B–C) Sectioned optic nerves with CTB-traced RGC axons in AAV-CNTF–treated (B) and control AAV-GFP–treated (C) mice 11 weeks after injury and 3 weeks after intravitreal viral administration. (B′, C′) High-magnification micrographs of the boxed regions in (B) and (C) (i.e., ∼1 mm distal to the injury site). Regenerating RGC axons (yellow arrows) were found >1 mm from the lesion in all AAV-CNTF–treated mice, and never after AAV-GFP (n = 5/group). (D) Quantification of axon regeneration rates at 1 mm from lesion (**P = 0.0029, Student's unpaired t-test). (E) Time course of acute experiments. In comparison, immediate treatment (F) with AAV-CNTF (F), but not AAV-GFP (G) induces significantly greater axon regeneration. Scale bars: 100 μm.
Since these few surviving RGCs still have the capacity to regenerate axons long after injury, we sought to examine whether a larger pool of RGCs could be triggered to regenerate their axons at any point after an injury as long as they are protected from cell death. To examine this, we repeated the chronic treatment paradigm (Fig. 3A) in apoptosis-deficient Bax KO mice, which undergo little appreciable cell death following axotomy or during neuronal pruning stages in development.27–33 We observed immunohistochemically that the number of β III tubulin immunoreactive RGCs does not decrease at 5 dpc or 56 dpc (Figs. 4A–C), compared to uninjured contralateral retinae (n = 3–4/group). Elevated RGC survival notwithstanding, Bax KO mice did not regenerate axons (i.e., to 1 mm distal to the lesion) following AAV-GFP treatment given immediately after injury (Fig. 4D). However, intravitreal injection of AAV-CNTF given 56 days after crush injury promoted strong axon regeneration within 3 weeks (Fig. 4F), despite the large span of time between injury and treatment. Similarly high rates of axon regeneration were seen in Bax KO mice receiving an immediate AAV-CNTF treatment (Fig. 4E, quantified in 4G), pointing toward a time-independent limit on regenerative response, at least at 3 weeks post treatment. Furthermore, with comparable numbers of regenerating axons afforded by Bax KO mice, we observed that axon misguidance may be less common at chronic stages (Figs. 4E′, 4F′), with significantly fewer axons turning at 1 mm from lesion site in chronic (2.7% ± 2.3%) than in acute (14.7% ± 2.1%) conditions, expressed as percentage of total regenerating axons (P = 0.0029) (see Methods). Nevertheless, at this 3-week posttreatment time point, regenerating axons have had insufficient time to elongate long distances into the brain to reinnervate visual targets.
Figure 4.
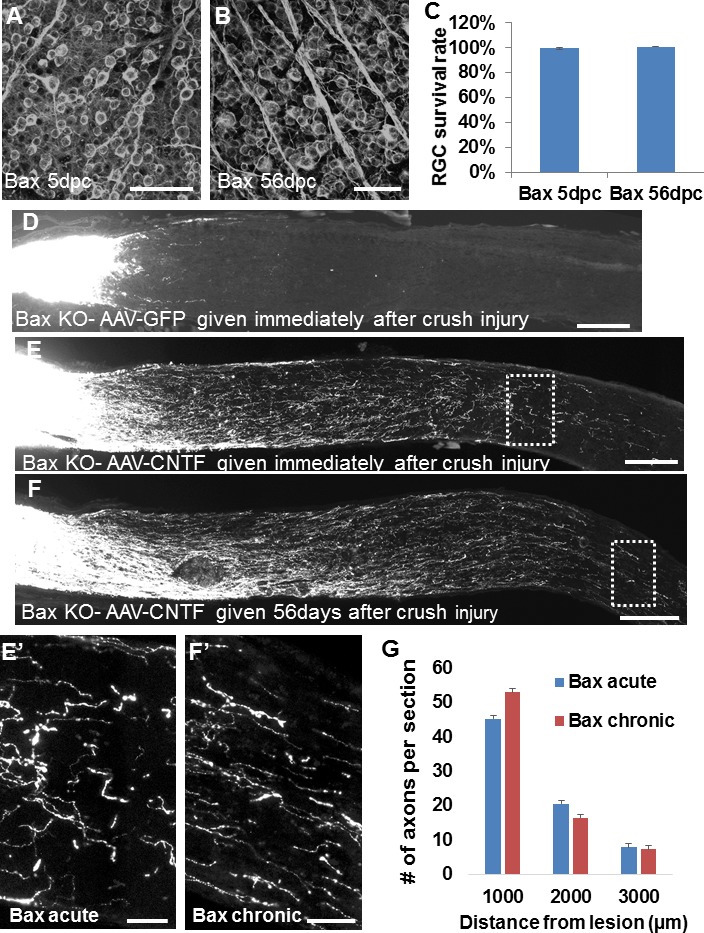
Axon regeneration in chronically injured, apoptosis-deficient Bax−/− mice. (A–B) β III tubulin immunoreactive (i.e., TUJ1 antibody immunostained) whole-mount retinae in injured Bax KO mice show no significant decrease in RGC density up to 56 days after optic nerve crush. (C) Quantification of RGC survival, normalized to uninjured contralateral retinae (P = 0.261 by Student's t-test; n = 4/group). (D) Sectioned optic nerves in Bax KO mice 3 weeks after intravitreal AAV-GFP administration, which was given immediately after injury. (E–F) Bax KO mice 3 weeks after intravitreal AAV-CNTF administration, which was given either immediately (E) or 56 days (F) after crush. High-magnification micrographs of distal boxed regions in (E) and (F) are shown in (E′) and (F′). (G) Quantification of axon regeneration rates at 1000, 2000, and 3000 μm from lesion shows no significant difference between chronic and acute treatment (P = 0.533 by 2-way ANOVA; n = 3/group). Error bars: SEM. Scale bars: 50 μm (A–B, E′, F′), 200 μm (D–F).
We also examined myelin and the glial scar in Bax KO mice after optic nerve crush. As seen in Supplementary Figures S2 and S3, there are GFAP-negative areas in the lesion site of Bax KO animals at 21 days after injury (Supplementary Fig. S3), indicating that the infiltration of astrocytic processes into the lesion site is comparable to (or at least is not more robust than) the wild-type animals (Supplementary Fig. S2). We also observed that in the lesion site (i.e., GFAP-negative area; regions indicated by the white arrows in Supplementary Figs. S2, S3), more intense MBP immunoreactivity is seen in Bax KO mice than in wild-type animals.
With poorly understood environmental modifications occurring at chronic stages of optic nerve injury, it is unclear whether the axons of neuroprotected RGCs might be capable of reinnervating visual targets. For this reason, we looked for regenerate axons in the brains of Bax KO mice at 8 weeks after AAV-CNTF treatment (i.e., 16 weeks after crush, the (time frame shown in Fig. 5A). We observed CTB-positive RGC axons in the distal optic nerve, optic chiasm, optic tracts, and hypothalamic regions of the brain (Figs. 5B–D). Axons regenerated into ipsi- and contralateral optic tracts as in an acute injury paradigm, described in our earlier work,21 as well as into the contralateral optic nerve. We also observed axons that ascend dorsally near the third ventricle and, in each of the four mice examined, in the suprachiasmatic nucleus (SCN) (Fig. 5D). In Bax KO animals without AAV-CNTF injection (i.e., control animals; n = 3), no regenerating axons were found in the optic chiasm or in the SCN (Fig. 5E). In the Bax KO mice with AAV-CNTF, regenerate axons were not found in more distal visual targets (i.e., lateral geniculate nucleus and superior colliculus; data not shown), as in acutely injured wild-type and Bax KO mice. Nonetheless, these data demonstrate a degree of proper visual target recognition and reinnervation, even at chronic timing.
Figure 5.
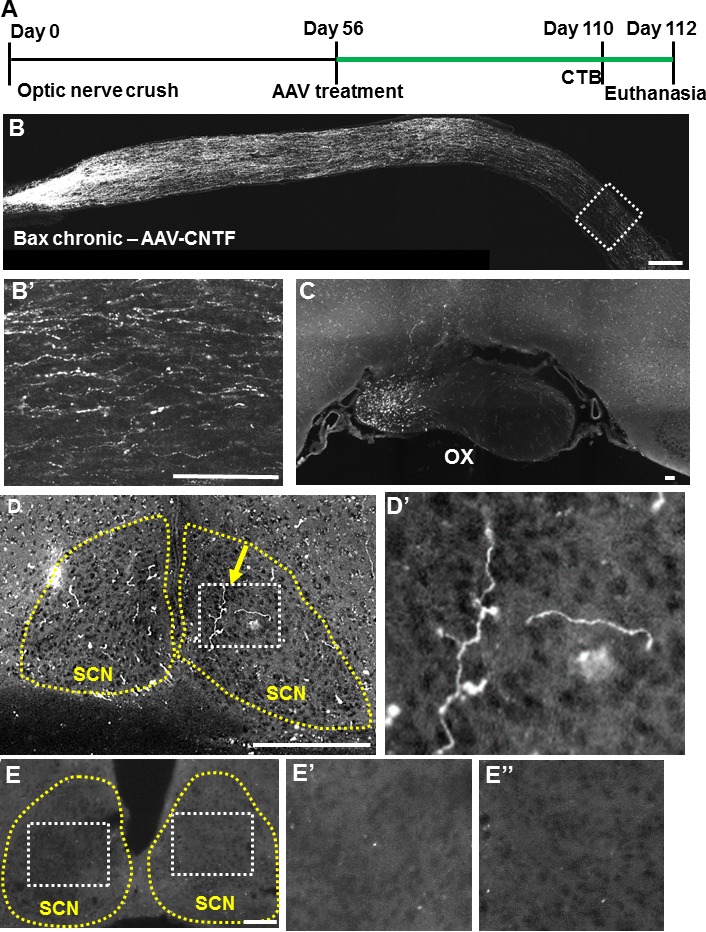
Visual target in the brain is reinnervated after postponed AAV-CNTF treatment in Bax−/− mice. (A) Time course of chronic experiment, with longer duration for regeneration (highlighted in green). (B) Sectioned optic nerve with CTB-traced RGC axons 16 weeks after injury and 8 weeks after intravitreal viral administration. High-magnification micrograph of distal region shown in (B′). (C–D) Coronal brain sections showing regenerating axons in the optic chiasm (C) and suprachiasmatic nucleus (SCN) (D), including at least one branched axon (yellow arrow) inside the visual target SCN (yellow outline). (D′) High magnification of the white boxed area in (D). (E) Representative coronal brain section of a control animal (i.e., Bax KO without AAV-CNTF) showing absence of regenerating axons. (E′, E′′) High magnification of the white boxed area in (E). OX, optic chiasm; SCN, suprachiasmatic nucleus. Scale bars: 150 μm.
Discussion
This study demonstrates RGCs' remarkable regenerative potential long after optic nerve injury. Few studies have examined RGC axon regeneration in a postinjury treatment paradigm,17,18,21 and as far as we know, none with treatment given beyond 10 days after injury. More work has been done in the spinal cord to address chronic stages of central nerve injury, and in this context, studies have shown that neurons (e.g., dorsal root ganglion neurons, cerebrospinal neurons, rubrospinal neurons, etc.) may be induced to regenerate an axon (with varying degrees of success) at 4 weeks, and even up to a year, after experimental injury.34–41 These findings have not been reproduced in RGCs, likely a consequence of their widespread axotomy-induced death due to proximity of injury to soma. We provide evidence against an intrinsic critical period for axon regeneration in wild-type and Bax KO mice by showing significantly enhanced growth rates following delayed AAV-CNTF treatment. Interestingly, there are similar rates of axon regeneration in Bax KO mice, whether treatment is provided acutely or chronically. However, any comparison of regenerative capacity between Bax KO and wild-type mice must take into account significant phenotypic differences, including approximately double the RGC number prior to injury,28–30 enhanced survival of proximal axonal segments,31,32 and muted macrophage and microglial activation.33 It is also worth noting that approximately twice as many regenerating RGC axons were found in Bax KO animals compared to the WT counterparts. This is in line with the previous reports that showed that approximately twice as many RGC axons regenerate in Bcl-2 transgenic mice (in which apoptosis is also blocked) compared to wild-type mice.42 Since approximately 20% to 30% of RGCs survive (∼10,000 RGCs) with AAV-CNTF treatment at 21 days post crush (data not shown), approximately 0.2% of surviving RGCs may be regenerating axons at least to 1 mm distal to lesion in the wild-type mice. On the other hand, given that almost all RGCs survive in Bax KO mice (i.e., ∼80,000 RGCs),28 approximately 0.05% of RGCs are regenerating axons. Thus, although more axons regenerate in Bax KO mice, perhaps due to higher cell number, it seems that a smaller portion of RGCs is actually capable of regenerating axons compared to wild-type. In this regard, it is worth noting that MBP level is higher in the lesion site of Bax KO mice compared to wild-type animals 3 weeks after injury. This could be due to (1) slower myelin clearance, (2) less oligodendrocyte death in the lesion area, or (3) more rapid regeneration of myelin in Bax KO mice. We do not know which of these possibilities contribute to the higher presence of MBP in the lesion site in the KO mice. Nonetheless, these results indicate that the smaller portion of RGCs able to regenerate in Bax KO mice despite the higher cell number could in part be explained by the intense level of myelin (i.e., often referred to as a growth inhibitor) in the lesion site in the KO mice. While the cause of regenerative failure in the majority of RGCs remains to be elucidated, this finding highlights the resilient ability of surviving RGCs to regenerate axons long after injury.
It is likely that the sustained presence of some injury signals may be required for the observed chronic capacity to regenerate axons; however, the relationship between these markers, cell survival, and the regenerative response is not clear. While our findings confirm previous reports of the increased expression of injury signals following injury,4,7 it is less clear how, for example, DLK and c-Jun somehow regulate both apoptotic and regenerative cellular pathways. Perhaps, as some suggest, axotomy primes CNS neurons for these disparate responses, and that cell death is triggered by insufficient axon regeneration.25 It is similarly unclear how cells with chronically elevated prodeath phosphorylated c-Jun43–45 fail to undergo apoptosis. Despite high levels of DLK, ATF3, and c-Jun expression in nearly all RGCs, we find only a small number of regenerating axons compared to surviving RGCs in both wild-type and Bax KO mice. Recent work has identified melanopsin-expressing intrinsically photosensitive RGCs (ipRGCs) as comprising the majority of axotomy-resistant cells,46 though these cells do not appear to possess regenerative capacity, at least in the peripheral nerve graft paradigm,47 leaving the identity of long-surviving, axon-regenerating RGCs a mystery. Possibly using transgenic mice that label ipRGCs, we may examine whether the surviving and regenerating RGCs are, in fact, ipRGCs and whether their axons reach the SCN.
In addition to initiating axon regrowth, misguidance of axons en route to visual centers in the brain poses a major problem in the recovery of visual function. It is poorly understood what, if any, axon guidance systems are in place in the adult CNS or how they might be altered over time. Our comparison of axon growth patterns in chronic and acute settings reveals fewer instances of aberrant axon growth, at least near the crush site; this more faithful and linear growth toward the brain could be due to enhanced permissiveness in the chronic nerve environment. For example, as macrophage infiltration proceeds, more efficient clearing of axonal and myelin debris (albeit incomplete at 56 dpc) may decrease axon meandering. It is also possible that decreased extracellular matrix protein deposition (e.g., chondroitin sulfate proteoglycans) in chronically injured animals may make scar tissue less inhibitory to RGC axon growth. Similarly, various growth factors and cytokines that affect axons' guidance in the injured optic nerve (e.g., brain-derived neurotrophic factor) may be downregulated as inflammation subsides, resulting in more linear growth of axons chronically.
Reactive astrocytes (at least those that fill an injury site) have been shown to facilitate axon regeneration.48 At chronic stages, astrocytes around the lesion fill the lesion site, at least partly, potentially providing more permissive astrocytic bridges48 for RGC axons to extend long after injury. In contrast, several studies have shown that reactive astrocytes can also inhibit axon growth.49 Increase in GFAP expression is a hallmark feature of reactive astrocytes. In Western blot analysis, we did not observe differences in GFAP expression in both acutely and chronically injured nerve compared to uninjured nerve. The lack of increase in GFAP expression acutely after injury (5 days post injury) could be due to localized increase in GFAP expression close to the lesion and our tissue sampling method, in which we collect the entire first half of optic nerve. Overall, understanding the interaction between regenerating axons and extracellular- and membrane-based elements in the injured milieu in order to promote proper axon pathfinding through the optic nerve and retinogeniculate and retinocollicular optic tracts will be of paramount importance in reestablishing topographically functional termination patterns.
In conclusion, we demonstrated sustained regenerative potential following long-delayed treatment with AAV-CNTF and compared axon regeneration rates and patterns between acutely and chronically treated wild-type and Bax KO mice. Our findings highlight the importance of chronic neuroregenerative and neuroprotective strategies and of elucidating mechanisms of axon pathfinding.
Supplementary Material
Acknowledgments
The authors thank Murray Blackmore for the CNTF vector, Syu-ichi Hirai of Wakayama Medical University School of Medicine for sharing the DLK antibody, and Pingping Jia at the University of Miami Viral Vector Core.
Supported by grants from NEI, 1R01EY022961-01 (KKP) and W81XWH-12-1-0319 (KKP), and from the Ziegler Foundation (KKP) and the Pew Charitable Trust (KKP).
Disclosure: B.J. Yungher, None; M. Ribeiro, None; K.K. Park, None
References
- 1. Berkelaar M,, Clarke DB,, Wang Y,, Bray GM,, Aguayo AJ. Axotomy ganglion results in delayed death and apoptosis of retinal cells in adult rats. J Neurosci. 1994; 14: 4368–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galindo-Romero C,, Avilés-Trigueros M,, Jiménez-López M,, et al. Axotomy-induced retinal ganglion cell death in adult mice: quantitative and topographic time course analyses. Exp Eye Res. 2011; 92: 377–387. [DOI] [PubMed] [Google Scholar]
- 3. Chauhan BC,, Stevens KT,, Levesque JM,, et al. Longitudinal in vivo imaging of retinal ganglion cells and retinal thickness changes following optic nerve injury in mice. PLoS One. 2012; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herdegen T,, Brecht S,, Mayer B,, et al. Long-lasting expression of JUN and KROX transcription factors and nitric oxide synthase in intrinsic neurons of the rat brain following axotomy. J Neurosci. 1993; 13: 4130–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koistinaho J,, Hicks KJ,, Sagar SM. Long-term induction of c-jun mRNA and Jun protein in rabbit retinal ganglion cells following axotomy or colchicine treatment. J Neurosci Res. 1993; 34: 250–255. [DOI] [PubMed] [Google Scholar]
- 6. Robinson GA. Immediate early gene expression in axotomized and regenerating retinal ganglion cells of the adult rat. Brain Res Mol Brain Res. 1994; 24: 43–54. [DOI] [PubMed] [Google Scholar]
- 7. Takeda M,, Kato H,, Takamiya A,, Yoshida A,, Kiyama H. Injury-specific expression of activating transcription factor-3 in retinal ganglion cells and its colocalized expression with phosphorylated c-Jun. Invest Ophthalmol Vis Sci. 2000; 41: 2412–2421. [PubMed] [Google Scholar]
- 8. Hu Y,, Park KK,, Yang L,, et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012; 73: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stoll G,, Trapp BD,, Griffin JW. Macrophage function during Wallerian degeneration of rat optic nerve: clearance of degenerating myelin and Ia expression. J Neurosci. 1989; 9: 2327–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blaugrund E,, Duvdevani R,, Lavie V,, Solomon A,, Schwartz M. Disappearance of astrocytes and invasion of macrophages following crush injury of adult rodent optic nerves: implications for regeneration. Exp Neurol. 1992; 118: 105–115. [DOI] [PubMed] [Google Scholar]
- 11. Ohlsson M,, Bellander B-M,, Langmoen I,, Svensson M. Complement activation following optic nerve crush in the adult rat. J Neurotrauma. 2003; 20: 895–904. [DOI] [PubMed] [Google Scholar]
- 12. Napoli I,, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009; 158: 1030–1038. [DOI] [PubMed] [Google Scholar]
- 13. Leaver SG,, Cui Q,, Plant GW,, et al. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006; 13: 1328–1341. [DOI] [PubMed] [Google Scholar]
- 14. Park KK,, Liu K,, Hu Y,, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008; 322: 963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moore DL,, Blackmore MG,, Hu Y,, et al. KLF family members regulate intrinsic axon regeneration ability. Science. 2009; 326: 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith PD,, Sun F,, Park KK,, et al. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009; 64: 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun F,, Park KK,, Belin S,, et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011; 480: 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hellström M,, Pollett M,, Harvey AR. Post-injury delivery of rAAV2-CNTF combined with short-term pharmacotherapy is neuroprotective and promotes extensive axonal regeneration after optic nerve trauma. J Neurotrauma. 2011; 28: 2475–2483. [DOI] [PubMed] [Google Scholar]
- 19. Pernet V,, Joly S,, Dalkara D,, et al. Long-distance axonal regeneration induced by CNTF gene transfer is impaired by axonal misguidance in the injured adult optic nerve. Neurobiol Dis. 2013; 51: 202–213. [DOI] [PubMed] [Google Scholar]
- 20. Luo X,, Salgueiro Y,, Beckerman SR,, Lemmon VP,, Tsoulfas P,, Park KK. Three-dimensional evaluation of retinal ganglion cell axon regeneration and pathfinding in whole mouse tissue after injury. Exp Neurol. 2013; 247: 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yungher BJ,, Luo X,, Salgueiro Y,, Blackmore MG,, Park KK. Viral vector-based improvement of optic nerve regeneration: characterization of individual axons' growth patterns and synaptogenesis in a visual target. Gene Ther. 2015; 22: 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knudson CM,, Tung KS,, Tourtellotte WG,, Brown G,, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995; 270: 96–99. [DOI] [PubMed] [Google Scholar]
- 23. Luo X,, Ribeiro M,, Bray ER,, et al. Enhanced transcriptional activity and mitochondrial localization of STAT3 co-induce axon regrowth in the adult central nervous system. Cell Rep. 2016; 15: 398–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiong X,, Wang X,, Ewanek R,, Bhat P,, Diantonio A,, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010; 191: 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watkins TA,, Wang B,, Huntwork-Rodriguez S,, et al. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013; 110: 4039–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Niekerk E,, Tuszynski MH,, Lu P,, Dulin JN. Molecular and cellular mechanisms of axonal regeneration after spinal cord injury. Mol Cell Proteomics. 2016; 15: 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deckwerth TL,, Elliott JL,, Knudson CM,, Johnson EM, Jr,, Snider WD,, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996; 17: 401–411. [DOI] [PubMed] [Google Scholar]
- 28. Mosinger Ogilvie J, Deckwerth TL, Knudson CM, Korsmeyer SJ. Suppression of developmental retinal cell death but not of photoreceptor degeneration in Bax-deficient mice. Invest Ophthalmol Vis Sci. 1998; 39: 1713–1720. [PubMed] [Google Scholar]
- 29. Li Y,, Schlamp CL,, Poulsen KP,, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000; 71: 209–213. [DOI] [PubMed] [Google Scholar]
- 30. Libby RT,, Li Y,, Savinova OV,, et al. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005; 1: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Howell GR,, Libby RT,, Jakobs TC,, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007; 179: 1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nickells RW,, Howell GR,, Soto I,, John SWM. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012; 35: 153–179. [DOI] [PubMed] [Google Scholar]
- 33. Mac Nair CE,, Schlamp CL,, Montgomery AD,, Shestopalov VI,, Nickells RW. Retinal glial responses to optic nerve crush are attenuated in Bax-deficient mice and modulated by purinergic signaling pathways. J Neuroinflammation. 2016; 13: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Houle JD. Demonstration of the potential for chronically injured neurons to regenerate axons into intraspinal peripheral nerve grafts. Exp Neurol. 1991; 113: 1–9. [DOI] [PubMed] [Google Scholar]
- 35. Ye JH,, Houle JD. Treatment of the chronically injured spinal cord with neurotrophic factors can promote axonal regeneration from supraspinal neurons. Exp Neurol. 1997; 143: 70–81. [DOI] [PubMed] [Google Scholar]
- 36. von Meyenburg J,, Brösamle C,, Metz GA,, Schwab ME. Regeneration and sprouting of chronically injured corticospinal tract fibers in adult rats promoted by NT-3 and the mAb IN-1, which neutralizes myelin-associated neurite growth inhibitors. Exp Neurol. 1998; 154: 583–594. [DOI] [PubMed] [Google Scholar]
- 37. Jin Y,, Tessler A,, Fischer I,, Houle JD. Fibroblasts genetically modified to produce BDNF support regrowth of chronically injured serotonergic axons. Neurorehabil Neural Repair. 2000; 14: 311–317. [DOI] [PubMed] [Google Scholar]
- 38. Kwon BK,, Liu J,, Messerer C,, et al. Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc Natl Acad Sci U S A. 2002; 99: 3246–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gonzenbach RR,, Zoerner B,, Schnell L,, Weinmann O,, Mir AK,, Schwab ME. Delayed anti-Nogo-A antibody application after spinal cord injury shows progressive loss of responsiveness. J Neurotrauma. 2012; 29: 567–578. [DOI] [PubMed] [Google Scholar]
- 40. Wang Z,, Reynolds A,, Kirry A,, Nienhaus C,, Blackmore MG. Overexpression of Sox11 promotes corticospinal tract regeneration after spinal injury while interfering with functional recovery. J Neurosci. 2015; 35: 3139–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Du K,, Zheng S,, Zhang Q,, et al. PTEN deletion promotes regrowth of corticospinal tract axons 1 year after spinal cord injury. J Neurosci. 2015; 35: 9754–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leaver SG,, Cui Q,, Bernard O,, Harvey AR. Cooperative effects of bcl-2 and AAV-mediated expression of CNTF on retinal ganglion cell survival and axonal regeneration in adult transgenic mice. Eur J Neurosci. 2006; 24: 3323–3332. [DOI] [PubMed] [Google Scholar]
- 43. Ham J,, Babij C,, Whitfield J,, et al. A c-jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995; 14: 927–939. [DOI] [PubMed] [Google Scholar]
- 44. Yoshida K,, Behrens A,, Le-Niculescu H,, et al. Amino-terminal phosphorylation of c-Jun regulates apoptosis in the retinal ganglion cells by optic nerve transection. Invest Ophthalmol Vis Sci. 2002; 43: 1631–1635. [PubMed] [Google Scholar]
- 45. Fernandes K,, Harder JM,, Kim J,, Libby RT. Jun regulates early transcriptional responses to axonal injury in retinal ganglion cells. Exp Eye Res. 2013; 112: 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. De Sevilla Müller LP,, Sargoy A,, Rodriguez AR,, Brecha NC. Melanopsin ganglion cells are the most resistant retinal ganglion cell type to axonal injury in the rat retina. PLoS One. 2014; 9: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson G,, Madison RD. Axotomized mouse retinal ganglion cells containing melanopsin show enhanced survival, but not enhanced axon regrowth into a peripheral nerve graft. Vision Res. 2004; 44: 2667–2674. [DOI] [PubMed] [Google Scholar]
- 48. Zukor K,, Belin S,, Wang C,, Keelan N,, Wang X,, He Z. Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J Neurosci. 2013; 33: 15350–15361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005; 5: 400–407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.