

ABSTRACT
Through unknown mechanisms, the host cytosol restricts bacterial colonization; therefore, only professional cytosolic pathogens are adapted to colonize this host environment. Listeria monocytogenes is a Gram-positive intracellular pathogen that is highly adapted to colonize the cytosol of both phagocytic and nonphagocytic cells. To identify L. monocytogenes determinants of cytosolic survival, we designed and executed a novel screen to isolate L. monocytogenes mutants with cytosolic survival defects. Multiple mutants identified in the screen were defective for synthesis of menaquinone (MK), an essential molecule in the electron transport chain. Analysis of an extensive set of MK biosynthesis and respiratory chain mutants revealed that cellular respiration was not required for cytosolic survival of L. monocytogenes but that, instead, synthesis of 1,4-dihydroxy-2-naphthoate (DHNA), an MK biosynthesis intermediate, was essential. Recent discoveries showed that modulation of the central metabolism of both host and pathogen can influence the outcome of host-pathogen interactions. Our results identify a potentially novel function of the MK biosynthetic intermediate DHNA and specifically highlight how L. monocytogenes metabolic adaptations promote cytosolic survival and evasion of host immunity.
KEYWORDS: Listeria monocytogenes, cell autonomous defense, cytosolic pathogen, inflammasome, menaquinone, nutritional immunity, respiration
IMPORTANCE
Cytosolic bacterial pathogens, such as Listeria monocytogenes and Francisella tularensis, are exquisitely evolved to colonize the host cytosol in a variety of cell types. Establishing an intracellular niche shields these pathogens from effectors of humoral immunity, grants access to host nutrients, and is essential for pathogenesis. Through yet-to-be-defined mechanisms, the host cytosol restricts replication of non-cytosol-adapted bacteria, likely through a combination of cell autonomous defenses (CADs) and nutritional immunity. Utilizing a novel genetic screen, we identified determinants of L. monocytogenes cytosolic survival and virulence and identified a role for the synthesis of the menaquinone precursor 1,4-dihydroxy-2-naphthoate (DHNA) in cytosolic survival. Together, these data begin to elucidate adaptations that allow cytosolic pathogens to survive in their intracellular niches.
INTRODUCTION
Despite major advances in understanding how cells detect pathogens in the cytosol (1), little is known about the factors that protect the cytosol from invasion by bacterial pathogens. Likewise, despite a number of studies over the past two decades demonstrating that cells protect their cytosol from bacterial invasion (2–4), how cytosolic pathogens avoid these host defenses and utilize the cytosol as a replication niche remains largely unknown. Listeria monocytogenes is a deadly food-borne pathogen (5) that invades the cytosol of a wide variety of cell types and maintains its intracellular niche through coordinated expression of well-characterized virulence factors (6). Maintenance of an intracellular niche is essential for L. monocytogenes pathogenesis, and induction of host cell death highly attenuates bacterial virulence (7). We had previously identified a highly conserved protein of unknown function (YvcK) required for survival of L. monocytogenes in the macrophage cytosol (8). Bacterial killing in the cytosol resulted in the release of DNA from lysed bacteria, activation of the AIM2 inflammasome, and induction of a programmed cell death process known as pyroptosis (8–11). Pyroptosis attenuates infection by eliminating the replication niche of L. monocytogenes (12, 13); as such, cytosolic survival and avoidance of detection by the AIM2 inflammasome are critical for L. monocytogenes pathogenesis.
Although numerous determinants of L. monocytogenes cytosolic replication have been identified (14–16), few determinants of L. monocytogenes cytosolic survival are known (8, 17, 18). Thus, we designed and executed a novel genetic screen to identify L. monocytogenes mutants which lyse in the cytosol of macrophages. We identified mutations in genes regulating central metabolism and genes of unknown function critical for L. monocytogenes survival. Some genes were selectively required for survival in macrophages but not in other cell types, signifying cell type-specific cytosolic defenses. Unexpectedly, through an as-yet-undefined mechanism, a subset of mutants that lyse in the cytosol still avoided inflammasome activation. Despite this, all mutants identified in the screen were attenuated in a murine model of listeriosis. Next we investigated the function of menaquinone (MK) in cytosolic survival. We found that MK’s canonical functions in cellular respiration and the electron transport chain (ETC) were not critical for L. monocytogenes cytosolic survival. Instead, synthesis of the MK biosynthetic intermediate 1,4-dihydroxy-2-naphthoate (DHNA), but not of fully functional, isoprenylated menaquinone, was required for L. monocytogenes cytosolic survival. Taking the data together, our genetic screen uncovered factors required for L. monocytogenes survival in the host cytosol and evasion of the innate immune system and ultimately revealed a novel, ETC-independent function for DHNA. Additionally, these results add to the growing body of literature demonstrating that central metabolism plays a key role during host-pathogen interactions. Not only do host cells monitor and modulate their metabolism to sense and respond to pathogens (19), but cytosolic pathogens must also modulate their metabolism to avoid detection and/or killing by hosts.
RESULTS
Identification of genes required for cytosolic survival within macrophages.
To explicitly identify genes required for cytosolic survival of L. monocytogenes, we designed and executed a novel genetic screen to identify mutants of L. monocytogenes that lyse in the cytosol of host cells. Bacteriolysis of L. monocytogenes at the population level is indirectly measured through delivery of a luciferase-based reporter plasmid (pBHE573) (8) to the host cytosol during infection. Luciferase expression occurs only if the reporter translocates from the bacteria to the host cytosol since the luciferase gene is transcribed from a cytomegalovirus (CMV) promoter (see Fig. S1A in the supplemental material). We performed a nonsaturating screen using approximately 6,500 independent L. monocytogenes transposon mutants carrying pBHE573 and monitored for bacteriolysis (Fig. S1B). Type I interferon receptor-deficient immortalized macrophages (iIFNAR−/−) were used to avoid type I interferon-mediated suppression of translation (20), which would impact luciferase expression. Following secondary screening, we prioritized isolates which induced 2-fold or greater increases in luciferase expression compared to wild-type L. monocytogenes.
Screening for L. monocytogenes transposon mutants with intracellular survival defects in macrophages. (A) L. monocytogenes mutants carrying bacteriolysis reporter pBHE573 that can neither access the host cytosol nor avoid bacteriolysis in the host cytosol induce low levels of luciferase production in the host. However, L. monocytogenes mutants with survival defects transfer pBHE573 to the host cytosol and induce high levels of luciferase production. (B) Approximately 6,500 random transposon mutants were tested for increased bacteriolysis within macrophages. Data are normalized to wild-type levels of bacteriolysis. Download FIG S1, EPS file, 0.5 MB (545.5KB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Six unique isolates possessed single transposon insertions within the coding region of six different genes (see Table S1 in the supplemental material). nrdD encodes an anaerobic nucleotide reductase required for production of nucleotides during anaerobic growth of L. monocytogenes (21). pdhC encodes the E2 subunit of pyruvate dehydrogenase, which converts pyruvate to acetyl-coenzyme A (acetyl-CoA) while generating NADH (22). menF and menD encode enzymes for the first two dedicated steps in the biosynthesis of MK, a membrane-bound molecule which shuttles electrons between different complexes of the election transport chain (23). Finally, two genes of unknown function were identified in this genetic screen: lmo1602 and yvcJ. lmo1602 appears to be a sigma B-regulated gene associated with stress responses (24, 25), whereas yvcJ is cotranscribed with yvcK (26), a gene previously found to be required for cytosolic survival of L. monocytogenes (8).
Genes identified for intracellular survival of L. monocytogenes. Download TABLE S1, DOCX file, 0.05 MB (51KB, docx) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To exclude the possibility of secondary mutations, we transduced the transposon mutations into clean wild-type backgrounds and reexamined these strains for survival in macrophages (Fig. 1A). Holin-lysin, a L. monocytogenes strain expressing inducible bacteriophage holin and lysin proteins, and ΔyvcK mutants were used as positive bacteriolysis controls (8). Δhly mutants which cannot escape the host vacuole (27) were used as negative controls (8, 18). L. monocytogenes bacteriolysis mutants displayed bacteriolysis that was 4-fold to 8-fold higher than that seen with the wild-type strain, results similar to those obtained with ΔyvcK mutants (Fig. 1A). Importantly, while the mutants that we identified lysed more frequently than the wild-type strain, lysis of these strains was still a relatively rare event compared to lysis of the ~100% lysis control, holin-lysin. We also constructed markerless deletions and genetic complements for every gene identified in the genetic screen and tested them for survival in macrophages (Fig. S2A). With the exception of a ΔpdhC strain which we were unable to construct, the deletion strains reliably phenocopied their transposon mutant counterparts. Additionally, complementation of clean deletions and transposon mutants of the majority of mutants was successful, with the exception of ΔnrdD and ΔyvcJ mutants. Furthermore, to test whether mutants undergo complete bacteriolysis and not simply plasmid secretion, we created a chromosomal CMV-luciferase reporter and assayed L. monocytogenes mutants for intracellular survival. pdhC::Tn, menD::Tn, menF::Tn, and yvcJ::Tn mutants underwent complete bacteriolysis (Fig. S2B). The level of luciferase production from the chromosomal lysis reporter was significantly reduced compared to that seen in the plasmid-based assay, and as such, both the wild-type strain and the mutants demonstrated low levels of lysis that were below the limit of detection.
FIG 1 .
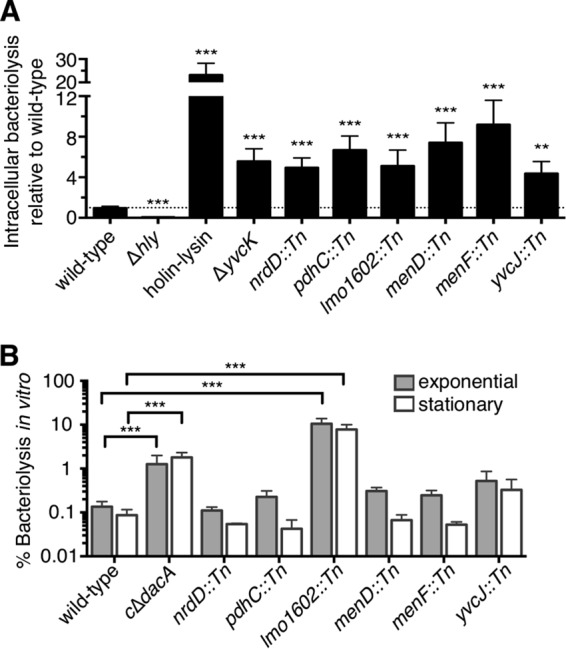
Identification of genes required for cytosolic survival of L. monocytogenes in macrophages. (A) Cleanly transduced L. monocytogenes bacteriolysis mutants (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages over a 6-h infection. All data are normalized to wild-type levels of bacteriolysis and presented as means ± standard errors of the mean (SEM) of results from five independent experiments. (B) Cleanly transduced L. monocytogenes bacteriolysis mutants were grown to the exponential or stationary phase in BHI media at 37°C and then examined for in vitro lysis. β-Galactosidase activities of supernatants are normalized to β-galactosidase activities of 100% lysed cultures, and data are presented as mean percent ± SEM of results from four independent experiments.
Bacteriolysis mutants identified in the genetic screen undergo complete bacteriolysis, inside macrophages. (A) Genetic complements of clean deletions and transposon mutants (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages using the bacteriolysis assay. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from six independent experiments. NT, not tested. (B) Bacteriolysis mutants carrying chromosomal bacteriolysis reporter pYL56 (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages 6 h postinfection. Ampicillin (Ap; 1 mg/ml) was added to the wild-type infection as a positive control. All data are presented as mean fluorescence ± SEM from six independent experiments. Download FIG S2, EPS file, 1.2 MB (1.3MB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Using an in vitro bacteriolysis assay (28), we also examined each mutant for bacteriolysis during exponential-phase and stationary-phase growth in broth culture (Fig. 1B). As a positive control, we used a conditional mutant of diadenylate cyclase (cΔdacA), a gene essential for growth of L. monocytogenes in nutrient-rich media and for survival in vitro (28, 29). The lmo1602::Tn mutant was the only mutant which significantly lysed in broth culture, suggesting that this gene is essential for survival in vitro. These data highlight the idea that other genes identified in our genetic screen are required for survival in the macrophage cytosolic environment but not during extracellular replication.
Salmonella enterica subsp. Typhimurium ΔsifA mutants escape to the host cytosol and replicate in epithelial cells but are killed upon entry into the macrophage cytosol, suggesting the presence of both cell type-specific cell autonomous defenses (CADs) and bacterial survival mechanisms (3). We next asked whether the cytosolic survival defects of our mutants were macrophage specific. Using the plasmid-based bacteriolysis assay, we assessed survival of each of our mutants in epithelial (Caco-2) cells and fibroblasts (BHKs) (Fig. S3A and B). Only the yvcJ::Tn mutant was significantly impaired for survival in all cell types, suggesting that YvcJ is generally required for cytosolic survival. nrdD::Tn, pdhC::Tn, menD::Tn, and menF::Tn mutants were significantly impaired for survival in macrophages and Caco-2 cells but not BHKs. Surprisingly, given the in vitro and macrophage bacteriolysis phenotypes, lmo1602::Tn mutants were not impaired for survival in either Caco-2 cells or BHKs. These observations of cell type-specific bacteriolysis are consistent with the idea of specific CADs and/or nutritional immunity in the cytosol of different cell types.
Bacteriolysis mutants differentially lyse in Caco-2 and BHKs. (A and B) Bacteriolysis mutants were tested for bacteriolysis in Caco-2 cells (A) or BHKs (B) (MOI of 64 and 100, respectively) using the bacteriolysis assay. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from six independent experiments. Download FIG S3, EPS file, 0.8 MB (869.2KB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes required for cytosolic survival are also required for virulence.
On the basis of the holin-lysin data that suggest that lysis of the mutants is a rare event, we asked whether cytosolic survival correlates with intracellular replication. We first assessed intracellular replication in wild-type C57BL/6 bone marrow-derived macrophages (BMDMs) (Fig. 2A and Fig. S4A). pdhC::Tn mutants are gradually cleared over time, whereas yvcJ::Tn mutants displayed partial replication defects in macrophages, similarly to previous observations for ΔyvcK mutants (8). Other mutants did not display replication defects within macrophages, suggesting that bacteriolysis is incomplete and that intracellular bacteriolysis does not strictly correlate with intracellular replication. Although MK mutants could not grow in minimal media lacking MK, MK-deficient mutants grown in MK-limited media displayed a small but reproducible invasion defect, potentially due to decreased internalization or decreased phagosomal survival. In addition, and consistent with previous results (30, 31), MK-starved MK-deficient mutants were impaired for intracellular replication (Fig. S4B), suggesting that carryover of MK from brain heart infusion can support intracellular replication over the course of infection. Furthermore, while individual bacteriolysis mutants displayed differential growth phenotypes in a variety of cell types, the ability to replicate intracellularly did not correlate with cell type-specific lysis (Fig. S4C to E).
FIG 2 .
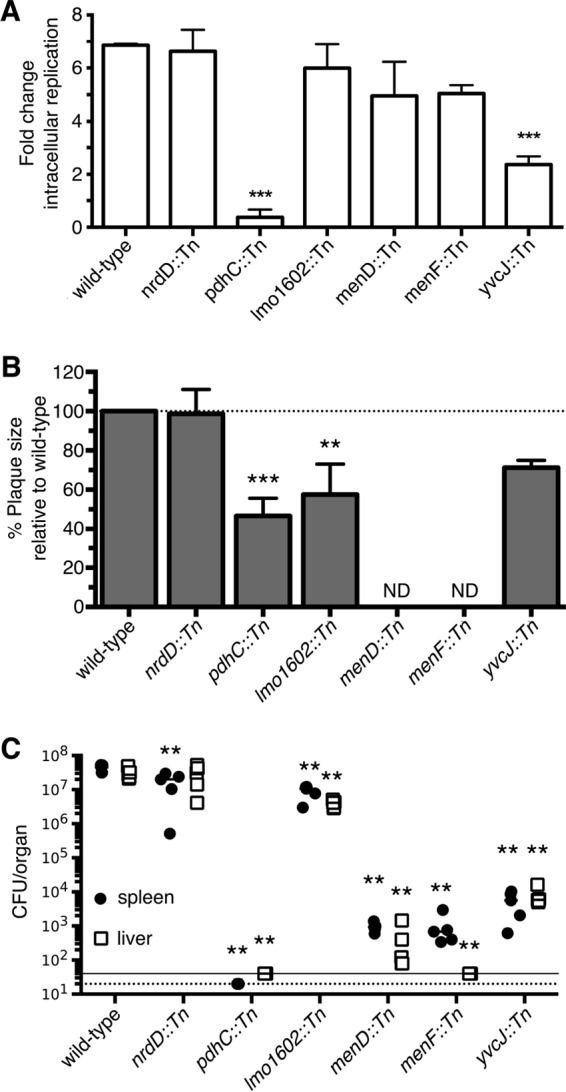
Genes required for cytosolic survival are also required for virulence. (A) Bacteriolysis mutants (MOI of 0.2) were grown in bone marrow-derived macrophages (BMDMs) and then enumerated for CFU at 2 and 5 h postinfection. Data are calculated as fold change between 2 and 5 h and representative of four independent experiments. (B) L. monocytogenes bacteriolysis mutants (MOI of 0.5) were examined for plaque formation in L2 fibroblasts 6 days postinfection. Data are normalized to wild-type plaque size and represent means ± SEM of results from three independent experiments. ND, not detected. (C) Bacterial burdens (CFU) from the spleen (●) and liver (□) were enumerated at 48 h postinfection. Data are representative of results from two biological replicates. The horizontal dotted and solid lines denote the limits of detection for the spleen and livers, respectively. Mann-Whitney statistical analysis was performed to measure statistical significance in comparison to wild-type results.
Intracellular replication of bacteriolysis in different cell types. (A) Bacteriolysis mutants (MOI of 0.2) were examined for intracellular replication in BMDMs. This panel depicts the data from the experiment in Fig. 2A but displayed as CFU at 0.5, 2, 5, and 8 h postinfection. (B) ΔmenD and ΔmenF strains were grown in minimal media plus 25 ng/ml MK overnight. Infected BMDMs at a starting MOI of 0.2 were then enumerated for CFU at 2 and 5 h postinfection. MK (50 μg/ml) was added exogenously to infected cells to complement growth of MK-deficient strains. Data are calculated as fold change between 2 and 5 h and representative of results from three independent experiments. (C to E) Bacteriolysis mutants were grown in primary IFNAR−/− macrophages (MOI of 0.2) (C), Caco-2 cells (MOI of 5) (D), and BHKs (MOI of 5) (E) and then enumerated for CFU at 2 and 5 h postinfection. Data are calculated as fold change between 2 and 5 h and representative of results of two independent experiments. (B to E) The panels on the right depict the same data but graphed as CFU at 0.5, 2, 5, and 8 h postinfection. Download FIG S4, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Despite the lack of correlation between bacteriolysis and intracellular replication, we hypothesized that cytosolic survival was essential for virulence. To assess virulence ex vivo, we tested these mutants for plaque formation within L2 fibroblasts (32) (Fig. 2B). menD::Tn and menF::Tn mutants did not form visible plaques even after 6 days of infection. lmo1602::Tn and yvcJ::Tn mutants had intermediate virulence phenotypes, while nrdD::Tn mutants made plaques similarly to wild-type L. monocytogenes. Strikingly, pdhC::Tn mutants were capable of forming intermediate-sized plaques despite being cleared in macrophages, consistent with the pdhC::Tn mutant’s minimal bacteriolysis phenotype in fibroblasts (Fig. S3B).
Finally, we examined virulence in vivo using a murine acute infection model of listeriosis (Fig. 2C). Most mutants were significantly attenuated for virulence in both the spleen and the liver. Interestingly, nrdD::Tn mutants, which should be unable to grow anaerobically (21), were attenuated only moderately in the spleen and were not attenuated in the liver, suggesting that these particular niches for L. monocytogenes are not completely devoid of oxygen. The most attenuated strains were pdhC::Tn, menD::Tn, menF::Tn, and yvcJ::Tn mutants. Together, these data show that, independently of cell type-specific survival defects, genes required for cytosolic survival are essential for virulence in vivo.
Bacteriolysis mutants differentially activate the inflammasome.
Previous studies discovered that L. monocytogenes and Francisella species mutants hyperactivated the inflammasome as a consequence of bacteriolysis in the host cytosol (8, 10); hence, we hypothesized that our bacteriolysis mutants would also induce pyroptosis in macrophages. Holin-lysin and ΔyvcK strains were used as positive controls, since lysis of these strains triggers the AIM2 inflammasome (8). The Δhly strain induces undetectable levels of cell death because it cannot access the cytosol to be recognized by AIM2 (8). Infections with pdhC::Tn and yvcJ::Tn mutants induced significantly higher levels of caspase-1- and AIM2-dependent cell death in macrophages in comparison to wild-type results as expected (Fig. 3 and Fig. S5A). Like that seen with the ΔyvcK mutants (8), yvcJ::Tn mutant intracellular replication was partially caspase-1 dependent, as indicated by a moderate rescue in intracellular replication in caspase-1-deficient macrophages (Fig. S5B). Unlike that seen with the ΔyvcK and yvcJ::Tn mutants, pdhC::Tn mutant intracellular replication was not rescued in caspase-1-deficient macrophages, suggesting that the intracellular growth defect of these mutants is independent of induction of host cell death (Fig. S5C). Additionally, despite delivering plasmid and chromosomal DNA to the cytosol (Fig. 1A and Fig. S2C), infections with the other bacteriolysis mutants induced cell death at levels indistinguishable from those seen with the wild-type strain (Fig. 3). These mutants also did not induce caspase-3/caspase-7 (caspase-3/7)-dependent apoptosis in macrophages as an alternative to pyroptosis (Fig. S5D). In contrast to previous reports demonstrating a consistent link between bacteriolysis and inflammasome activation (8, 10), our data suggest that cytosolic bacteriolysis alone may be insufficient to trigger the inflammasome or that L. monocytogenes possesses additional strategies to avoid inflammasome activation.
FIG 3 .
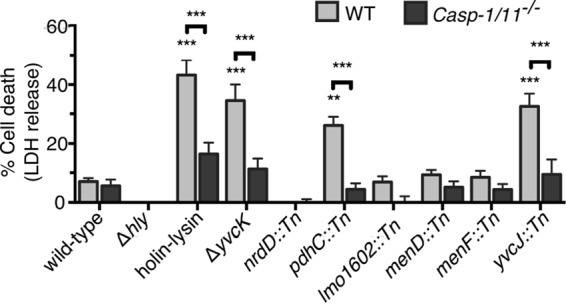
Genes required for cytosolic survival differentially activate the inflammasome. Lactate dehydrogenase assays were performed by infecting wild-type (WT) or caspase-1- and caspase-11-deficient (Casp-1 and Casp-11−/−) BMDMs with L. monocytogenes bacteriolysis mutants (MOI of 1). Data are presented as means ± SEM of results from three independent experiments. LDH, lactate dehydrogenase.
FIG 5 .

The electron transport chain plays a minor role in survival of L. monocytogenes within macrophages. (A) ΔmenD, cydA::Tn, qoxA::Tn, and atpH::Tn strains were grown in aerated BHI cultures at 37°C and monitored for OD600. Data are representative of results from three independent experiments. (B) ΔmenD, cydA::Tn, qoxA::Tn and atpH::Tn strains grown to exponential phase in BHI media at 37°C were examined for membrane potential generation. Data are normalized to the wild-type red/green fluorescence ratios and presented as mean percent ± SEM of results from three independent experiments. (C and E) ΔmenD, cydA::Tn, qoxA::Tn, and atpH::Tn strains (C) or ΔmenD, ΔcydAB, ΔqoxA, and ΔcydAB/ΔqoxA strains (E) (MOI of 0.5) were examined for plaque formation in L2 fibroblasts 6 days postinfection. Data are normalized to wild-type plaque size and represent means ± SEM of results from three independent experiments. ND, not detected. (D and F) ΔmenD, cydA::Tn, qoxA::Tn, and atpH::Tn strains (D) or ΔmenD, ΔcydAB, ΔqoxA, and ΔcydAB/ΔqoxA strains (F) (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages using the bacteriolysis assay. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from five independent experiments.
Bacteriolysis mutants do not induce apoptosis. (A) Lactate dehydrogenase assays were performed infecting wild-type (WT) and AIM2−/− BMDMs with bacteriolysis strains (MOI of 1). Data are presented ± SEM of results from two independent experiments. (B and C) yvcJ::Tn (B) and pdhC::Tn (C) strains (MOI of 0.2) were grown in BMDMs and then enumerated for CFU at 2 and 5 h postinfection. Data are calculated as fold change between 2 and 5 h and representative of results of two independent experiments. The panels on the right depict the same data but graphed as CFU at 0.5, 2, 5, and 8 h postinfection (D) iIFNAR−/− macrophages infected with bacteriolysis mutants at a MOI of 10 and measured for caspase-3/7 activation 6 h postinfection. Data are presented as means ± SEM of results from three independent experiments. NS, not significant. Download FIG S5, EPS file, 1.8 MB (1.8MB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Menaquinone biosynthesis is required for intracellular survival of L. monocytogenes.
In addition to the original 12 bacteriolysis mutants, we isolated a strain with multiple mutations in its chromosome. In this mutant, deletion of shikimate biosynthesis genes (aroED) was responsible for the bacteriolysis phenotype (data not shown) and led to MK auxotrophy since this pathway produces a precursor for MK (30) (see Fig. 6A). This, combined with our isolation of menD and menF mutants, prompted us to examine the role of MK in L. monocytogenes cytosolic survival. Consistent with our data from the transposon mutants as well as with previously published results (30, 31), the ΔmenD and ΔmenF strains can neither efficiently replicate aerobically nor generate a robust membrane potential (Fig. 4A to C). Additionally, these mutants were significantly attenuated for virulence and intracellular survival in macrophages (Fig. 4D and E).
FIG 6 .

MK biosynthesis intermediates are required for cytosolic survival of L. monocytogenes. (A) Major intermediates and enzymes present in the MK biosynthesis pathway of L. monocytogenes according to the KEGG database (http://www.genome.jp/kegg/). Specific men mutants examined in this study are highlighted in red. (B and E) MK biosynthesis mutants (B) or ΔmenB, ΔmenA, and ΔmenB/ΔmenA strains (E) (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages using the bacteriolysis assay. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from four to six independent experiments. (C) The menD::Tn, menB::Tn, ΔmenA, and menG::Tn strains were grown to exponential phase in BHI media at 37°C of stained and measured for membrane potential generation. Data are normalized to wild-type red/green fluorescence ratios and presented as mean percent ± SEM of results from three independent experiments. (D and F) The menB::Tn, ΔmenA, and menG::Tn strains (D) or ΔmenB, ΔmenA, and ΔmenB/ΔmenA strains (MOI of 0.5) were examined for plaque formation in L2 fibroblasts at 6 days postinfection. Data are normalized to wild-type plaque size and represent means ± SEM of results from three independent experiments.
FIG 4 .

ΔmenD and ΔmenF strains are deficient for generation of the membrane potential and intracellular survival. (A and B) Genetic (A) or chemical (B) complementation of ΔmenD and ΔmenF strains grown in aerated cultures in BHI media at 37°C and monitored at OD600. Data are representative of results from three independent experiments. (C) ΔmenD and ΔmenF cultures grown to exponential phase in BHI media at 37°C were examined for membrane potential generation. Data are normalized to the wild-type red/green fluorescence ratios and represented as mean percent ± SEM of results from three independent experiments. MFI, median fluorescence intensity; PMF, proton motive force. (D) ΔmenD and ΔmenF strains (MOI of 0.5) were examined for plaque formation in L2 fibroblasts 6 days postinfection. Data are normalized to wild-type plaque size and represent mean percent ± SEM of results from three independent experiments. ND, not detected. (E) ΔmenD and ΔmenF strains (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages over 6 h. All data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from four independent experiments.
To understand what host defenses may induce intracellular bacteriolysis, we examined these mutants for sensitivity to a variety of stresses. MK auxotrophs were not sensitive to cell wall- or cell membrane-targeting antimicrobials (Fig. S6A and B). Although sensitivity to these antimicrobials could be masked by trace amounts of MK in the brain heart infusion (BHI) media, MK auxotrophs were still hypersensitive to oxidative stress in BHI cultures (Fig. S6C). We next examined cellular reactive oxygen species (ROS) production in macrophages infected with the wild-type strain or the ΔmenD mutant but did not detect increases in ROS production during infection (Fig. S6D). Though ROS scavenging with N-acetylcysteine (NAC) did partially lower ROS production in menadione-induced macrophages (positive control), NAC treatment did not rescue MK mutants from bacteriolysis, suggesting that cytosolic ROS is not responsible for the intracellular survival defects of MK-deficient mutants, though additional evidence may be required (Fig. S6D and E). These data leave open the possibility that another unknown cytosolic stress(es) or host defense may be responsible for lysis of MK-deficient L. monocytogenes in macrophages.
MK-deficient strains are sensitive to ROS in vitro but not ex vivo. (A to C) The menD::Tn and menF::Tn strains were tested for sensitivity to various concentrations of ceftriaxone (CRO), ampicillin (AMP), bacitracin (BAC), and daptomycin (DAP) (A); lysozyme and LL-37 (B); and hydrogen peroxide (C) in vitro to calculate MICs. Data represent median MICs of results from three or more independent experiments. (D) iIFNAR−/− macrophages were treated with 100 µM menadione for 1 h or infected with the wild-type strain or the ΔmenD strain at an MOI of 10 for 6 h. Cells were treated with or without 1mM N-acetylcysteine (NAC) throughout the assay and were then examined for ROS production. (E) The ΔmenD strain (MOI of 10) was examined for bacteriolysis in iIFNAR−/− macrophages 6 h postinfection. NAC (1 mM) was added to infections to scavenge ROS. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from four independent experiments. NS, not significant. Download FIG S6, EPS file, 0.7 MB (713.9KB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Loss of the electron transport chain does not fully account for MK mutant survival defects.
Loss of MK biosynthesis in L. monocytogenes disrupts the electron transport chain (ETC) and prevents L. monocytogenes from producing a robust membrane potential (Fig. 4C). To investigate whether the ETC is required for intracellular survival of L. monocytogenes, we characterized transposon mutants with mutations in cydA, qoxA, and atpH (genes encoding ETC components cytochrome bd oxidase, cytochrome aa3 oxidase, and ATP synthase, respectively) (33). Similarly to MK-deficient mutants, the cydA::Tn mutant was deficient for generating a membrane potential and displayed in vitro aerobic growth defects (Fig. 5A and B). qoxA::Tn mutants were partially defective with respect to their ability to generate a membrane potential, whereas atpH::Tn mutants produced a robust membrane potential similar to that seen with the wild-type strain and grew to wild-type levels in vitro. No visible growth of the atpH::Tn mutant was observed under anoxic conditions, likely because ATP synthase is essential for generating a membrane potential during fermentative growth (34) (Fig. S7A). Interestingly, cydA::Tn mutants did not have a growth defect within macrophages but made significantly smaller plaques (Fig. 5C and Fig. S7B). In contrast, qoxA::Tn and atpH::Tn mutants also replicated in macrophages but formed plaques similar to those seen with the wild-type strain, suggesting that neither the cytochrome aa3 oxidase nor the ATP synthase of L. monocytogenes is important for virulence ex vivo (Fig. 5C and Fig. S7B). The atpH::Tn mutant displayed an initial invasion defect in macrophages (Fig. S7B), though this was likely be due to poorer growth in statically grown overnight cultures. Next we examined ETC complex mutants for intracellular survival. cydA::Tn mutants, but not qoxA::Tn or atpH::Tn mutants, lysed in the cytosol of macrophages, albeit not to ΔmenD levels (Fig. 5D), suggesting that the ETC or ability to generate a membrane potential may contribute, at least partially, to L. monocytogenes intracellular survival.
Electron transport chain mutations are not impaired for intracellular replication. (A) The atpH::Tn strain was grown on BHI plates under oxic and anoxic conditions. (B) cydA::Tn, qoxA::Tn, and atpH::Tn strains (MOI of 0.2) were grown in BMDMs and enumerated for CFU at 2 and 5 h postinfection. Data are calculated as fold change between 2 and 5 h and representative of results from three independent experiments. The panel on the right depicts the same data but graphed as CFU at 0.5, 2, 5, and 8 h postinfection. Download FIG S7, EPS file, 1 MB (1MB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Since cydA::Tn mutants did not phenocopy ΔmenD mutants for intracellular survival, we hypothesized that disruption of both cytochrome oxidases might be necessary to completely abolish membrane potential; hence, we generated a double cytochrome oxidase mutant, the ΔcydAB/ΔqoxA strain. This mutant was more attenuated for virulence than either single cytochrome oxidase mutant (Fig. 5E), suggesting at least partial redundancy of the two cytochrome oxidases in L. monocytogenes. Unexpectedly, the double ΔcydAB/ΔqoxA mutants lysed at the same level as single ΔcydAB mutants (Fig. 5F). Although the cytochrome oxidase mutants exhibited moderate intracellular survival defects, our data suggest that noncanonical functions of MK, independently of MK-dependent functions in the ETC, protect L. monocytogenes from cytosolic bacteriolysis.
Biosynthesis of DHNA is required for cytosolic survival of L. monocytogenes in macrophages.
The results described above led us to reexamine the role of MK biosynthesis in intracellular survival of L. monocytogenes. MK is synthesized from chorismate, the end product of the shikimate biosynthesis pathway, by eight well-characterized enzymes encoded by the men genes (23) (Fig. 6A). Following conversion of o-succinylbenzoyl-CoA (OSB-CoA) to 1,4-dihydroxy-2-naphthoyl-CoA (DHNA-CoA) by MenB, an unknown DHNA-CoA thioesterase is predicted to convert DHNA-CoA to 1,4-dihydroxy-2-naphthoate (DHNA) (35). MenA and MenG are responsible for the final two steps in MK biosynthesis by addition of the polyprenyl side chain and methylation of the naphthoquinone ring, respectively (Fig. 6A).
To investigate how MK biosynthesis drives intracellular survival of L. monocytogenes in macrophages, we obtained transposon mutants (from an unpublished genetic screen that identified small-colony variants) with mutations of every gene encoding enzymes in the MK biosynthesis pathway, with the exception of menH and the unknown thioesterase gene. Mutations in genes menF through menB, which are responsible for the first six steps in MK biosynthesis, led to survival defects of L. monocytogenes in macrophages, confirming the findings of our screen (Fig. 6B). Remarkably, the loss of DHNA polyprenyltransferase (MenA) did not lead to intracellular bacteriolysis of L. monocytogenes. The menG::Tn mutant, with a disruption in demethylmenaquinone (DMK) methyltransferase (MenG), displayed only a minor bacteriolysis phenotype, reminiscent of the ΔcydAB mutant results. These data suggest that synthesis of DHNA, but not menaquinone, is required for intracellular survival of L. monocytogenes. Additionally, all of the MK biosynthesis mutants, including the ΔmenA mutant, were sensitive to oxidative stress in vitro (Fig. S6C), further suggesting that cytosolic ROS is not responsible for bacteriolysis of these mutants. Importantly, mutations in menB, menA, or menG abolish the ability of L. monocytogenes to generate a membrane potential (Fig. 6C), highlighting, moreover, that the ETC/membrane potential plays a minor (if any) role in intracellular survival. Curiously, these data also demonstrate that, unlike Staphylococcus aureus (36), L. monocytogenes cannot use demethylmenaquinone (DMK) in the electron transport since the menG::Tn strain, which should accumulate DMK, does not respire (Fig. 6C). ΔmenA and menG::Tn mutants were impaired for plaque formation similarly to the ΔcydAB mutant, though not to the extent seen with other men mutants, further suggesting that, in addition to the function of menaquinone for respiration and generation of a membrane potential, biosynthesis of the DHNA is critical for L. monocytogenes virulence (Fig. 6D). Finally, to pinpoint the exact step of MK biosynthesis critical for cytosolic survival, we generated a double deletion strain (mutant ΔmenA/ΔmenB) and tested it for bacteriolysis in macrophages. ΔmenA/ΔmenB mutants phenocopied a single ΔmenB mutant, lysing significantly in macrophages and being drastically attenuated for virulence in a plaquing assay (Fig. 6E and F). Taken together, our data support the model that DHNA, or a derivative of it, is a critical virulence determinant independently of its role in the synthesis of MK for the ETC. Additionally, synthesis of DHNA, through a yet-to-be-defined mechanism, prevents cytosolic lysis of L. monocytogenes in macrophages.
DISCUSSION
How cytosolic pathogens avoid killing in the highly restrictive host cytosol is unknown. Using a novel genetic screen, we identified factors required for cytosolic survival of L. monocytogenes inside the host. Our approach uncovered genes involved in central metabolism and genes of unknown function required for the cytosolic survival and, ultimately, virulence of L. monocytogenes (see Table S1 in the supplemental material). Our data suggest that L. monocytogenes may have distinct strategies to overcome different nutritional limitations, stresses, and/or active CADs in different cytosolic environments (Fig. 1A; see also Fig. S3A and B in the supplemental material). Finally, we demonstrated that synthesis of the MK biosynthesis intermediate DHNA, but not of full-length isoprenylated MK, is essential for L. monocytogenes cytosolic survival in macrophages (Fig. 6B and E). Our findings identify some critical metabolic pathways for survival and immune evasion in the host cytosol. Finally, these findings are consistent with (and add to) recent studies demonstrating the intricate connection between pathogen and host metabolism in the context of pathogenesis and innate immune regulation.
Although the minimal amount of DNA required to trigger the AIM2 inflammasome is unknown, studies of Francisella have shown that inflammasome components can be recruited to singly lysed bacteria (9). One of the most striking findings from our screen was the observation that, despite a wealth of data suggesting that DNA in the cytosol is sufficient to activate the AIM2 inflammasome (8, 9, 37, 38), a subset of our mutants which lyse and release both plasmid and chromosomal DNA into the cytosol of macrophages do not hyperactivate the inflammasome (Fig. 3). MK-deficient mutants lyse at frequencies similar to those seen with pdhC::Tn, ΔyvcK and yvcJ::Tn mutants but avoid activation of the AIM2 inflammasome. It is possible that during infection with MK mutants, changes in host metabolism activate host cell nucleases that modify or degrade bacterial DNA such that inflammasome signals are destroyed. Alternatively, it is possible that the response to infection with MK mutants leads to changes in signaling or cellular redox balance that inactivate either the AIM2 receptor or downstream signaling components, including caspase-1 itself (19, 39). Finally, although less likely given the ability of sterile transfected DNA to activate the AIM2 inflammasome, it is possible that DNA alone is not sufficient for AIM2 activation during infection and that non-respiring L. monocytogenes mutants lack the metabolic signals required to activate the inflammasome. Bacterial metabolism and regulation of innate immune signaling pathways appear to be intimately linked (19, 40–42). For example, S. Typhimurium metabolic mutants induce mitochondrial reactive oxygen production, resulting in NALP3-inflammasome activation (42). How MK-deficient mutants avoid inflammasome activation despite the release of cytosolic DNA is the subject of ongoing studies.
Previous studies performed with vacuolar pathogens that mislocalize to the cytosol highlighted differences between immune cells and nonimmune cells with respect to the ability to restrict pathogens in the cytosol. S. enterica ΔsifA mutants, which access the cytosol due to loss of vacuole integrity (43), appear to be killed in macrophages but not HeLa cells (3). Legionella pneumophila ΔsdhA mutants, which similarly mislocalize to the host cytosol (4), also display cell-specific survival defects (44) and are ultimately killed in the cytosol, resulting in activation of the AIM2 inflammasome (45). Our genetic screen also identified mutants with differential survival defects in different cell types and suggested that, similarly to the phagosomal killing capacity results, professional phagocytes such as macrophages may have more-potent cytosolic CADs (Fig. 1B). This may translate directly to virulence, as evidenced by the results seen with pdhC::Tn mutants, which were slowly cleared from macrophages but were capable of forming plaques in fibroblasts consistent with their improved survival in the cytosol of these cells (Fig. 2A and B). L. monocytogenes and other cytosolic pathogens likely employ various strategies to counter CADs in different cell types.
To kill cytosolic invaders, macrophages may utilize antimicrobial effectors such as ubiquicidin (46), interferon-inducible guanylate binding proteins (GBPs) (47), lysozyme (18), or autophagy (48). Unlike the results previously determined with Francisella novicida (47), recent data from our laboratory suggest that GBPs do not contribute to bacteriolysis of L. monocytogenes in the cytosol (49). Although the host factors which restrict cytosolic pathogens are unknown, a recent study found that S. enterica ΔsifA mutants experience oxidative and/or nitrosative stress in the macrophage cytosol (50), although it is unclear whether this stress originates from the host, from the bacterium, or from both. Our findings suggest that, despite increased sensitivity to ROS, cytosolic ROS is unlikely to be responsible for survival of L. monocytogenes MK-deficient mutants (Fig. S6C to E). Finally, in addition to identifying novel targets for therapeutic intervention, L. monocytogenes mutants susceptible to killing in the host cytosol act as tools to identify the host pathways responsible for killing these mutants.
Consistent with previous studies, L. monocytogenes MK-deficient mutants were highly attenuated for virulence ex vivo and in vivo (30, 31) (Fig. 2C and Fig. 4D and E); however, the function of MK itself, MK biosynthetic intermediates, or simply MK-dependent respiration in virulence has not been fully explored. Previous reports have found that MK biosynthesis genes and the cytochrome bd oxidase are upregulated during infection ex vivo (51) and in vivo (52), emphasizing the importance of MK for virulence. Here we uncovered a novel requirement for the MK intermediate DHNA in protecting L. monocytogenes from intracytosolic bacteriolysis (Fig. 6B and E). This phenotype was not due to disruption of the electron transport chain or loss of ATP generation through oxidative phosphorylation (Fig. 5D and F) but rather to the fact that DHNA synthesis is crucial for virulence and intracellular survival (Fig. 6B). Unfortunately, since the enzymatic steps at this stage in the pathway are incompletely understood (Fig. 6A) (35), we cannot determine whether DHNA or DHNA-CoA is the relevant molecule. Inexplicably, cytochrome bd oxidase and menG::Tn mutants displayed intermediate bacteriolysis phenotypes (Fig. 5D and F and Fig. 6B). Our favored hypothesis is that mutants with an incomplete ETC have depleted pools of available DHNA. This could be due either to increased funneling of DHNA into MK biosynthesis caused by incomplete electron transfer or, alternatively, to a transcriptional/posttranslation negative-feedback loop that inhibits DHNA synthesis in the absence of a productive ETC.
Apart from MK’s role in shuttling electrons between complexes in the electron transport chain, MK and its biosynthetic intermediates may play greater physiological roles in bacteria than previously appreciated. Shewanella oneidensis utilizes a derivative of DHNA to degrade carbon tetrachloride (53). In S. aureus, MK potentiates spermine and heme toxicity independently of MK’s function in the electron transport chain. Small-colony variants with mutations specifically in the MK biosynthesis pathway lead to increased resistance to spermine and heme (36, 54). Given that MK, DMK, and DHNA share naphthoquinone rings with similar redox characteristics, we surmise that these molecules may act as cofactors in chemical reactions crucial to the survival of L. monocytogenes in macrophages. It is also possible that L. monocytogenes uses DHNA as a substrate for synthesis of noncanonical MK (55) or another yet-to-be-characterized molecule. Alternatively, it has been proposed that two-component systems, such as ArcAB in Escherichia coli and SrrAB in Staphylococcus aureus, may sense changes in the redox state of MK as signals for environmental changes (56, 57). We posit that MK, DMK, and/or DHNA may be used as a sensor(s) of cytosolic stress. In the absence of these molecules, L. monocytogenes may not be able to adapt to this specific host environment.
Although decades of studies have focused on the host-pathogen interactions between macrophages and vacuolar pathogens, very little is known about how cytosolic pathogens survive in their primary niche and how the cell protects this environment from invasion by pathogens. Consistent with this study, previous work demonstrates that cytosolic survival of Francisella spp. relies on specific metabolic adaptations during invasion of the host cytosol (10), though the same metabolic pathways were not identified in this study. This outcome was expected since L. monocytogenes and Francisella spp. likely evolved mechanisms for cytosolic survival of the same host defense through convergent evolution. Here, we show that the MK biosynthetic intermediate DHNA promotes intracellular survival of L. monocytogenes in macrophages. These findings expand upon a series of recent studies demonstrating that central metabolism is at the nexus of host pathogen interactions. Host cells monitor and modulate their central metabolism both to sense and to defend against infection, whereas pathogens must modulate their metabolism both to make use of the available nutrients and to evade detection and survive in the face of host defenses. Finally, in addition to identification of a variety of new virulence determinants for L. monocytogenes, the mutants identified in this screen provide the tools necessary to identify host restriction factors that prevent cytosolic colonization by non-cytosol-adapted pathogens.
MATERIALS AND METHODS
Animals and cell cultures.
C57BL/6 mice were purchased from NCI. Casp-1/-11−/− and AIM2−/− mice were a kind gift from Russell Vance (University of California, Berkeley [UC Berkeley]) and Doug McNeel (University of Wisconsin—Madison [UW—Madison]), respectively. All experiments involving animals were performed according to protocols approved by the Animal Use and Care Committee of the University of Wisconsin—Madison.
iIFNAR−/− macrophages and BHK and L2 cells were all kind gifts from Daniel Portnoy (UC Berkeley). Caco-2 cells were purchased from ATCC. Bone marrow-derived macrophages (BMDM) were prepared from 6-to-8-week-old mice as previously described (58).
Bacterial strains, plasmid construction, and growth conditions in vitro.
L. monocytogenes 10403s was used as the wild-type strain. L. monocytogenes strains were grown at 37°C or 30°C in brain heart infusion (BHI), Luria broth (LB), or minimal defined media (min) supplemented with glucose as the sole carbon source (59). Escherichia coli strains were grown in Luria broth (LB) at 37°C. Antibiotics were used at a concentration of 100 µg/ml carbenicillin, 10 µg/ml chloramphenicol, 2 µg/ml erythromycin, or 30 µg/ml kanamycin when appropriate. For growth of MK-deficient strains in MK-limiting minimal media, MK (Sigma; V9378) was supplemented at 25 ng/ml. For anaerobic growth, bacteria were cultivated on BHI plates placed anaerobic jars carrying a GasPak EZ anaerobe container system (BD; 260678).
Vectors were shuttled into L. monocytogenes by the use of E. coli strain S17 or strain S10 through conjugation (60). In-frame deletions of genes in L. monocytogenes were performed by allelic exchange (61) using suicide plasmid pksv7-oriT as previously described (62). Integrative vector pIMK2 (63) was used for constitutive expression of L. monocytogenes genes. Transductions between L. monocytogenes strains were performed using U153 bacteriophage (64).
Generation of transposon libraries and screening for bacteriolysis mutants.
The Himar1 mariner transposon system was used to construct a random transposon library in wild-type L. monocytogenes carrying bacteriolysis reporter pBHE573 (8) as outlined in references 65 and 66. To perform the genetic screen, random mutant colonies from the Himar1 bacteriolysis library were isolated and grown in wells from a 96-well plate overnight in BHI media at 30°C. In parallel, iIFNAR−/− macrophages were seeded in opaque 96-well plates at 1 × 105 cells per well and then allowed to incubate at 37°C in a 5% CO2 atmosphere for approximately 16 h. iIFNAR−/− macrophages were infected at an estimated multiplicity of infection (MOI) of 10. At 1 h postinfection, the culture medium was exchanged for media containing 50 µg/ml gentamycin. At 6 h postinfection, the culture medium was replaced with TNT buffer to lyse cells. Luciferase reagent was added and measured for luciferase activity using a luminometer (BioTek Synergy HT).
Intracellular bacteriolysis assay.
For standard bacteriolysis assays, unprimed iIFNAR−/− macrophages, Caco-2 (epithelial) cells, or BHK (fibroblast) cells were plated at 5 × 105 cells per well in a 24-well plate and then infected with L. monocytogenes strains carrying the plasmid bacteriolysis reporter (pBHE573) or chromosomal bacteriolysis reporter (pYL56) at an MOI of 10, 64, or 100, respectively. The higher MOIs used in the Caco-2 and BHK cells were used to gain comparable levels of luciferase production similar to those in macrophage experiments. At 1 h postinfection, cultures were treated with 50 µg/ml gentamicin. At 6 h postinfection (iIFNAR−/− macrophages and BHK cells) or 8 h postinfection (Caco-2 cells), cells were assayed for luciferase activity as previously described (8). If indicated, infected cells were treated with 1 mg/ml ampicillin at 2 h postinfection.
In vitro bacteriolysis assay.
Measurements of bacteriolysis in vitro were performed as previously described (28) with slight modifications. Cultures of L. monocytogenes carrying the lacZ gene encoding a transposon (Tn917-LTV3) were grown to mid-logarithmic or early stationary phase in BHI medium. Half of the culture was subjected to centrifugation to separate bacteria from the supernatant. The other half of the culture was subjected to complete bacteriolysis by addition of SDS to reach a concentration of 0.1% and to bead beating performed with 0.1-mm-diameter silicon beads for 10 min at 2,000 rpm. One hundred microliters of culture supernatant or 100 µl of serially diluted lysate (standard curve) was monitored for β-galactosidase activity by combining the supernatant or the lysate with 100 µl of 200 µM MUG (4-methylumbelliferyl β-d-galactopyranoside; Invitrogen) (excitation/emission [ex/em] wavelengths = 360 nm/449 nm) resuspended in Z buffer (0.1 M phosphate, 0.01 M KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7.0). The rate of β-galactosidase activity in the culture supernatant was compared to the standard curve to calculate percent bacteriolysis in the broth culture.
Lactate dehydrogenase release.
BMDMs pretreated for 16 h with 100 ng/ml Pam3CSK4 (Invitrogen) were infected with L. monocytogenes at an MOI of 1. At 30 min postinfection, the medium was replaced with media containing 50 µg/ml gentamicin. At 6 h postinfection, supernatants were removed and measured for lactate dehydrogenase activity as previously described (67) using a BioTek Synergy HT spectrophotometer.
Caspase-3/7 activity assay.
iIFNAR−/− macrophages were seeded into white 96-well plates and infected with L. monocytogenes at an MOI of 10. Staurosporine (AG Scientific) (5 µM) was added to uninfected macrophages at 30 min postinfection to induce caspase-3/7 activation. At 6 h postinfection, caspase-3/7 activation was measured using a caspase-Glo 3/7 assay kit (Promega) according to the manufacturer’s instructions.
Intracellular growth curves.
Wild-type or IFNAR−/− BMDMs, Caco-2 cells, and BHK cells were infected with L. monocytogenes strains (at a multiplicity of infection [MOI] of 0.2 for the BMDMs and an MOI of 5 for the other cell types) and enumerated for CFU at various time points as previously described (58).
L2 plaque assay.
Plaque assays were conducted using a L2 fibroblast cell line as previously described (32) with minor modifications for visualization and quantification of plaques. L2 fibroblasts were seeded at 1.2 × 106 per 35-mm-diameter dish and infected at an MOI of 0.5 to obtain approximately 30 PFU per dish. At 4 to 6 days postinfection, cells were stained with 0.3% crystal violet for 10 min and washed twice with deionized water. Stained wells were imaged and areas of plaque formation were measured on Fiji image analysis software (68).
Acute virulence assay.
Female C57BL/6 mice (6 to 8 weeks of age) were injected intravenously with 1 × 105 CFU (50% lethal dose [LD50] = 1 for wild-type 10403s L. monocytogenes) of logarithmically growing L. monocytogenes (optical density at 600 nm [OD600] = 0.5). At 48 h postinfection, spleens and livers were harvested, homogenized, and enumerated for CFU as previously described (12).
Measuring bacterial membrane potential.
L. monocytogenes strains were grown to mid-late-logarithmic phase in BHI media at 37°C. Bacteria were diluted in phosphate-buffered saline (PBS) to a concentration of 106/ml and transferred to a fluorescence-activated cell sorter (FACS) tube. Next, cells were stained with 3 mM membrane potential indicator dye DIO2(3) (3,3′-diethyloxacarbocyanine iodide) (Sigma; 320684) and/or 500 µM proton ionophore CCCP (carbonyl cyanide 3-chlorophenylhydrazone) (Sigma; C2759) for 30 min. Samples were analyzed on a BD LSR-II flow cytometer, and data analysis was performed using FlowJo software. Samples were gated for bacteria using forward and side scatter, and then individual bacteria were measured for ratiometric fluorescence analysis using comparisons between red mean fluorescence intensity and green mean fluorescence intensity.
MIC measurements.
L. monocytogenes strains were grown in aerated BHI cultures with various concentrations of ceftriaxone (Sigma; C5793), ampicillin (Sigma; A0166), bacitracin (Fisher Bioreagent; BP29501), daptomycin (Merck), lysozyme (Sigma; L6876), LL-37 (AnaSpec; AS61302), or hydrogen peroxide. MICs were determined as the median concentration of antimicrobial required to prevent replication over 12 h as determined by OD600 in a BioTek Eon or BioTek Synergy HT plate reader.
Intracellular ROS measurements.
iIFNAR−/− macrophages were grown in 35 mm-diameter dishes at a density of 2 × 106 per dish. Cells were infected with L. monocytogenes at an MOI of 10 for 6 h or treated for 1 h with 100 µM menadione (positive control). N-acetylcysteine (NAC) (1 mM) was added to cell cultures throughout treatment to scavenge ROS. Treated cells were stained with 2.5 µM CellROX DeepRed (Molecular Probes; C10422) for 30 min at 37°C. Cells were then washed twice in FACS buffer, fixed with 3.7% formaldehyde for 15 min, and washed again prior to analysis on a BD LSR-II flow cytometer.
Statistical analysis.
Statistical significance analysis (GraphPad Prism 6.0h) was performed using one-way or two-way analysis of variance (ANOVA) with the Bonferroni posttest unless otherwise indicated in the figure legends. Bacteriolysis assay data were log transformed prior to statistical analysis (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001).
Strains and plasmids used in this study. Download TABLE S2, DOCX file, 0.04 MB (43.8KB, docx) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download TABLE S3, DOCX file, 0.1 MB (139.2KB, docx) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
We thank Dan Portnoy for the atpH::Tn, qoxA::Tn, and cydA::Tn strains, Darren Higgins for the ΔmenD strain, and Andrew A. Mehle the BHK cell line.
This work was funded by the National Institutes of Health (T32AI055397 [G.Y.C.], U54AI57153 [subaward to J.-D.S.], and R01CA188034 [J.-D.S.]). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Chen GY, McDougal CE, D’Antonio MA, Portman JL, Sauer J-D. 2017. A genetic screen reveals that synthesis of 1,4-dihydroxy-2-naphthoate (DHNA), but not full-length menaquinone, is required for Listeria monocytogenes cytosolic survival. mBio 8:e00119-17. https://doi.org/10.1128/mBio.00119-17.
REFERENCES
- 1.Broz P, Monack DM. 2013. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol 13:551–565. doi: 10.1038/nri3479. [DOI] [PubMed] [Google Scholar]
- 2.Goetz M, Bubert A, Wang G, Chico-Calero I, Vazquez-Boland JA, Beck M, Slaghuis J, Szalay AA, Goebel W. 2001. Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc Natl Acad Sci U S A 98:12221–12226. doi: 10.1073/pnas.211106398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beuzón CR, Salcedo SP, Holden DW. 2002. Growth and killing of a Salmonella enterica serovar Typhimurium sifA mutant strain in the cytosol of different host cell lines. Microbiology 148:2705–2715. doi: 10.1099/00221287-148-9-2705. [DOI] [PubMed] [Google Scholar]
- 4.Creasey EA, Isberg RR. 2012. The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc Natl Acad Sci U S A 109:3481–3486. doi: 10.1073/pnas.1121286109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lecuit M. 2007. Human listeriosis and animal models. Microbes Infect 9:1216–1225. doi: 10.1016/j.micinf.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Freitag NE, Port GC, Miner MD. 2009. Listeria monocytogenes—from saprophyte to intracellular pathogen. Nat Rev Microbiol 7:623–628. doi: 10.1038/nrmicro2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glomski IJ, Decatur AL, Portnoy DA. 2003. Listeria monocytogenes mutants that fail to compartmentalize listeriolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect Immun 71:6754–6765. doi: 10.1128/IAI.71.12.6754-6765.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. 2010. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7:412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. 2010. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A 107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng K, Broz P, Jones J, Joubert LM, Monack D. 2011. Elevated AIM2‐mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell Microbiol 13:1586–1600. doi: 10.1111/j.1462-5822.2011.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsuchiya K, Hara H, Kawamura I, Nomura T, Yamamoto T, Daim S, Dewamitta SR, Shen Y, Fang R, Mitsuyama M. 2010. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J Immunol 185:1186–1195. doi: 10.4049/jimmunol.1001058. [DOI] [PubMed] [Google Scholar]
- 12.Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, Portnoy DA. 2011. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci U S A 108:12419–12424. doi: 10.1073/pnas.1019041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warren SE, Duong H, Mao DP, Armstrong A, Rajan J, Miao EA, Aderem A. 2011. Generation of a Listeria vaccine strain by enhanced caspase-1 activation. Eur J Immunol 41:1934–1940. doi: 10.1002/eji.201041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Riordan M, Moors MA, Portnoy DA. 2003. Listeria intracellular growth and virulence require host-derived lipoic acid. Science 302:462–464. doi: 10.1126/science.1088170. [DOI] [PubMed] [Google Scholar]
- 15.Joseph B, Przybilla K, Stühler C, Schauer K, Slaghuis J, Fuchs TM, Goebel W. 2006. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol 188:556–568. doi: 10.1128/JB.188.2.556-568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chico-Calero I, Suárez M, González-Zorn B, Scortti M, Slaghuis J, Goebel W, Vázquez-Boland JA; European Listeria Genome Consortium . 2002. Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in listeria. Proc Natl Acad Sci U S A 99:431–436. doi: 10.1073/pnas.012363899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pensinger DA, Aliota MT, Schaenzer AJ, Boldon KM, Ansari IU, Vincent WJB, Knight B, Reniere ML, Striker R, Sauer JD. 2014. Selective pharmacologic inhibition of a PASTA kinase increases Listeria monocytogenes susceptibility to β-lactam antibiotics. Antimicrob Agents Chemother 58:4486–4494. doi: 10.1128/AAC.02396-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rae CS, Geissler A, Adamson PC, Portnoy DA. 2011. Mutations of the Listeria monocytogenes peptidoglycan N-deacetylase and O-acetylase result in enhanced lysozyme sensitivity, bacteriolysis, and hyperinduction of innate immune pathways. Infect Immun 79:3596–3606. doi: 10.1128/IAI.00077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM, Weerapana E, Bogyo M. 2016. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 5:e13663. doi: 10.7554/eLife.13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ofer A, Kreft J, Logan DT, Cohen G, Borovok I, Aharonowitz Y. 2011. Implications of the inability of Listeria monocytogenes EGD-e to grow anaerobically due to a deletion in the class III NrdD ribonucleotide reductase for its use as a model laboratory strain. J Bacteriol 193:2931–2940. doi: 10.1128/JB.01405-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neveling U, Bringer-Meyer S, Sahm H. 1998. Gene and subunit organization of bacterial pyruvate dehydrogenase complexes. Biochim Biophys Acta 1385:367–372. doi: 10.1016/S0167-4838(98)00080-6. [DOI] [PubMed] [Google Scholar]
- 23.Meganathan R. 2001. Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): a perspective on enzymatic mechanisms. Vitam Horm 61:173–218. doi: 10.1016/S0083-6729(01)61006-9. [DOI] [PubMed] [Google Scholar]
- 24.Kazmierczak MJ, Mithoe SC, Boor KJ, Wiedmann M. 2003. Listeria monocytogenes σB regulates stress response and virulence functions. J Bacteriol 185:5722–5734. doi: 10.1128/JB.185.19.5722-5734.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milohanic E, Glaser P, Coppée JY, Frangeul L, Vega Y, Vázquez-Boland JA, Kunst F, Cossart P, Buchrieser C. 2003. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol Microbiol 47:1613–1625. doi: 10.1046/j.1365-2958.2003.03413.x. [DOI] [PubMed] [Google Scholar]
- 26.Wurtzel O, Sesto N, Mellin JR, Karunker I, Edelheit S, Bécavin C, Archambaud C, Cossart P, Sorek R. 2012. Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol Syst Biol 8:583. doi: 10.1038/msb.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camilli A, Paynton CR, Portnoy DA. 1989. Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc Natl Acad Sci U S A 86:5522–5526. doi: 10.1073/pnas.86.14.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witte CE, Whiteley AT, Burke TP, Sauer JD, Portnoy DA, Woodward JJ. 2013. Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. mBio 4:e00282-13. doi: 10.1128/mBio.00282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whiteley AT, Pollock AJ, Portnoy DA. 2015. The PAMP c-di-AMP Is essential for Listeria monocytogenes growth in rich but not minimal media due to a toxic increase in (p)ppGpp. Cell Host Microbe 17:788–798. doi: 10.1016/j.chom.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stritzker J, Janda J, Schoen C, Taupp M, Pilgrim S, Gentschev I, Schreier P, Geginat G, Goebel W. 2004. Growth, virulence, and immunogenicity of Listeria monocytogenes aro mutants. Infect Immun 72:5622–5629. doi: 10.1128/IAI.72.10.5622-5629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perry KJ, Higgins DE. 2013. A differential fluorescence-based genetic screen identifies Listeria monocytogenes determinants required for intracellular replication. J Bacteriol 195:3331–3340. doi: 10.1128/JB.00210-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun AN, Camilli A, Portnoy DA. 1990. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun 58:3770–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couvé E, de Daruvar A, Dehoux P, Domann E, Domínguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, García-del Portillo F, Garrido P, Gautier L, Goebel W, Gómez-López N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, de Pablos B, Pérez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, et al. . 2001. Comparative genomics of Listeria species. Science 294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida M, Muneyuki E, Hisabori T. 2001. ATP synthase—a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol 2:669–677. doi: 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- 35.Chen M, Ma X, Chen X, Jiang M, Song H, Guo Z. 2013. Identification of a hotdog fold thioesterase involved in the biosynthesis of menaquinone in Escherichia coli. J Bacteriol 195:2768–2775. doi: 10.1128/JB.00141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakeman CA, Hammer ND, Stauff DL, Attia AS, Anzaldi LL, Dikalov SI, Calcutt MW, Skaar EP. 2012. Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus. Mol Microbiol 86:1376–1392. doi: 10.1111/mmi.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. 2009. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM. 2013. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β Nat Immunol 14:52–60. doi: 10.1038/ni.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaudet RG, Sintsova A, Buckwalter CM, Leung N, Cochrane A, Li J, Cox AD, Moffat J, Gray-Owen SD. 2015. Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science 348:1251–1255. doi: 10.1126/science.aaa4921. [DOI] [PubMed] [Google Scholar]
- 41.Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wynosky-Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D, Zwack EE, Riblett AM, Hu B, Strowig T, Flavell RA, Jones RG, Freedman BD, Brodsky IE. 2014. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J Exp Med 211:653–668. doi: 10.1084/jem.20130627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beuzón CR, Méresse S, Unsworth KE, Ruíz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19:3235–3249. doi: 10.1093/emboj/19.13.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. 2006. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci U S A 103:18745–18750. doi: 10.1073/pnas.0609012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ge J, Gong YN, Xu Y, Shao F. 2012. Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc Natl Acad Sci U S A 109:6193–6198. doi: 10.1073/pnas.1117490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hiemstra PS, van den Barselaar MT, Roest M, Nibbering PH, van Furth R. 1999. Ubiquicidin, a novel murine microbicidal protein present in the cytosolic fraction of macrophages. J Leukoc Biol 66:423–428. [DOI] [PubMed] [Google Scholar]
- 47.Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Rühl S, Dussurgey S, Dick MS, Kistner A, Rigard M, Degrandi D, Pfeffer K, Yamamoto M, Henry T, Broz P. 2015. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol 16:476–484. doi: 10.1038/ni.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knodler LA, Celli J. 2011. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol 13:1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pensinger DA, Boldon KM, Chen GY, Vincent WJB, Sherman K, Xiong M, Schaenzer AJ, Forster ER, Coers J, Striker R, Sauer JD. 2016. The Listeria monocytogenes PASTA kinase PrkA and its substrate YvcK are required for cell wall homeostasis, metabolism, and virulence. PLoS Pathog 12:e1006001. doi: 10.1371/journal.ppat.1006001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Heijden J, Bosman ES, Reynolds LA, Finlay BB. 2015. Direct measurement of oxidative and nitrosative stress dynamics in Salmonella inside macrophages. Proc Natl Acad Sci U S A 112:560–565. doi: 10.1073/pnas.1414569112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chatterjee SS, Hossain H, Otten S, Kuenne C, Kuchmina K, Machata S, Domann E, Chakraborty T, Hain T. 2006. Intracellular gene expression profile of Listeria monocytogenes. Infect Immun 74:1323–1338. doi: 10.1128/IAI.74.2.1323-1338.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camejo A, Buchrieser C, Couvé E, Carvalho F, Reis O, Ferreira P, Sousa S, Cossart P, Cabanes D. 2009. In vivo transcriptional profiling of Listeria monocytogenes and mutagenesis identify new virulence factors involved in infection. PLoS Pathog 5:e1000449. doi: 10.1371/journal.ppat.1000449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward MJ, Fu QS, Rhoads KR, Yeung CHJ, Spormann AM, Criddle CS. 2004. A derivative of the menaquinone precursor 1,4-dihydroxy-2-naphthoate is involved in the reductive transformation of carbon tetrachloride by aerobically grown Shewanella oneidensis MR-1. Appl Microbiol Biotechnol 63:571–577. doi: 10.1007/s00253-003-1407-3. [DOI] [PubMed] [Google Scholar]
- 54.Joshi GS, Spontak JS, Klapper DG, Richardson AR. 2011. Arginine catabolic mobile element encoded speG abrogates the unique hypersensitivity of Staphylococcus aureus to exogenous polyamines. Mol Microbiol 82:9–20. doi: 10.1111/j.1365-2958.2011.07809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holsclaw CM, Sogi KM, Gilmore SA, Schelle MW, Leavell MD, Bertozzi CR, Leary JA. 2008. Structural characterization of a novel sulfated menaquinone produced by stf3 from Mycobacterium tuberculosis. ACS Chem Biol 3:619–624. doi: 10.1021/cb800145r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bekker M, Alexeeva S, Laan W, Sawers G, Teixeira de Mattos JT, Hellingwerf K. 2010. The ArcBA two-component system of Escherichia coli is regulated by the redox state of both the ubiquinone and the menaquinone pool. J Bacteriol 192:746–754. doi: 10.1128/JB.01156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kinkel TL, Roux CM, Dunman PM, Fang FC. 2013. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. mBio 4:e00696doi: 10.1128/mBio.00696-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones S, Portnoy DA. 1994. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun 62:5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phan-Thanh L, Gormon T. 1997. A chemically defined minimal medium for the optimal culture of Listeria. Int J Food Microbiol 35:91–95. doi: 10.1016/S0168-1605(96)01205-6. [DOI] [PubMed] [Google Scholar]
- 60.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 61.Camilli A, Tilney LG, Portnoy DA. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol 8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lauer P, Hanson B, Lemmens EE, Liu W, Luckett WS, Leong ML, Allen HE, Skoble J, Bahjat KS, Freitag NE, Brockstedt DG, Dubensky TW. 2008. Constitutive activation of the PrfA regulon enhances the potency of vaccines based on live-attenuated and killed but metabolically active Listeria monocytogenes strains. Infect Immun 76:3742–3753. doi: 10.1128/IAI.00390-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monk IR, Gahan CGM, Hill C. 2008. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl Environ Microbiol 74:3921–3934. doi: 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hodgson DA. 2000. Generalized transduction of serotype 1/2 and serotype 4b strains of Listeria monocytogenes. Mol Microbiol 35:312–323. doi: 10.1046/j.1365-2958.2000.01643.x. [DOI] [PubMed] [Google Scholar]
- 65.Cao M, Bitar AP, Marquis H. 2007. A mariner-based transposition system for Listeria monocytogenes. Appl Environ Microbiol 73:2758–2761. doi: 10.1128/AEM.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zemansky J, Kline BC, Woodward JJ, Leber JH, Marquis H, Portnoy DA. 2009. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J Bacteriol 191:3950–3964. doi: 10.1128/JB.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Decker T, Lohmann-Matthes ML. 1988. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods 115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- 68.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Screening for L. monocytogenes transposon mutants with intracellular survival defects in macrophages. (A) L. monocytogenes mutants carrying bacteriolysis reporter pBHE573 that can neither access the host cytosol nor avoid bacteriolysis in the host cytosol induce low levels of luciferase production in the host. However, L. monocytogenes mutants with survival defects transfer pBHE573 to the host cytosol and induce high levels of luciferase production. (B) Approximately 6,500 random transposon mutants were tested for increased bacteriolysis within macrophages. Data are normalized to wild-type levels of bacteriolysis. Download FIG S1, EPS file, 0.5 MB (545.5KB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes identified for intracellular survival of L. monocytogenes. Download TABLE S1, DOCX file, 0.05 MB (51KB, docx) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacteriolysis mutants identified in the genetic screen undergo complete bacteriolysis, inside macrophages. (A) Genetic complements of clean deletions and transposon mutants (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages using the bacteriolysis assay. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from six independent experiments. NT, not tested. (B) Bacteriolysis mutants carrying chromosomal bacteriolysis reporter pYL56 (MOI of 10) were tested for bacteriolysis in iIFNAR−/− macrophages 6 h postinfection. Ampicillin (Ap; 1 mg/ml) was added to the wild-type infection as a positive control. All data are presented as mean fluorescence ± SEM from six independent experiments. Download FIG S2, EPS file, 1.2 MB (1.3MB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacteriolysis mutants differentially lyse in Caco-2 and BHKs. (A and B) Bacteriolysis mutants were tested for bacteriolysis in Caco-2 cells (A) or BHKs (B) (MOI of 64 and 100, respectively) using the bacteriolysis assay. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from six independent experiments. Download FIG S3, EPS file, 0.8 MB (869.2KB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Intracellular replication of bacteriolysis in different cell types. (A) Bacteriolysis mutants (MOI of 0.2) were examined for intracellular replication in BMDMs. This panel depicts the data from the experiment in Fig. 2A but displayed as CFU at 0.5, 2, 5, and 8 h postinfection. (B) ΔmenD and ΔmenF strains were grown in minimal media plus 25 ng/ml MK overnight. Infected BMDMs at a starting MOI of 0.2 were then enumerated for CFU at 2 and 5 h postinfection. MK (50 μg/ml) was added exogenously to infected cells to complement growth of MK-deficient strains. Data are calculated as fold change between 2 and 5 h and representative of results from three independent experiments. (C to E) Bacteriolysis mutants were grown in primary IFNAR−/− macrophages (MOI of 0.2) (C), Caco-2 cells (MOI of 5) (D), and BHKs (MOI of 5) (E) and then enumerated for CFU at 2 and 5 h postinfection. Data are calculated as fold change between 2 and 5 h and representative of results of two independent experiments. (B to E) The panels on the right depict the same data but graphed as CFU at 0.5, 2, 5, and 8 h postinfection. Download FIG S4, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacteriolysis mutants do not induce apoptosis. (A) Lactate dehydrogenase assays were performed infecting wild-type (WT) and AIM2−/− BMDMs with bacteriolysis strains (MOI of 1). Data are presented ± SEM of results from two independent experiments. (B and C) yvcJ::Tn (B) and pdhC::Tn (C) strains (MOI of 0.2) were grown in BMDMs and then enumerated for CFU at 2 and 5 h postinfection. Data are calculated as fold change between 2 and 5 h and representative of results of two independent experiments. The panels on the right depict the same data but graphed as CFU at 0.5, 2, 5, and 8 h postinfection (D) iIFNAR−/− macrophages infected with bacteriolysis mutants at a MOI of 10 and measured for caspase-3/7 activation 6 h postinfection. Data are presented as means ± SEM of results from three independent experiments. NS, not significant. Download FIG S5, EPS file, 1.8 MB (1.8MB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
MK-deficient strains are sensitive to ROS in vitro but not ex vivo. (A to C) The menD::Tn and menF::Tn strains were tested for sensitivity to various concentrations of ceftriaxone (CRO), ampicillin (AMP), bacitracin (BAC), and daptomycin (DAP) (A); lysozyme and LL-37 (B); and hydrogen peroxide (C) in vitro to calculate MICs. Data represent median MICs of results from three or more independent experiments. (D) iIFNAR−/− macrophages were treated with 100 µM menadione for 1 h or infected with the wild-type strain or the ΔmenD strain at an MOI of 10 for 6 h. Cells were treated with or without 1mM N-acetylcysteine (NAC) throughout the assay and were then examined for ROS production. (E) The ΔmenD strain (MOI of 10) was examined for bacteriolysis in iIFNAR−/− macrophages 6 h postinfection. NAC (1 mM) was added to infections to scavenge ROS. Data are normalized to wild-type levels of bacteriolysis and presented as means ± SEM of results from four independent experiments. NS, not significant. Download FIG S6, EPS file, 0.7 MB (713.9KB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Electron transport chain mutations are not impaired for intracellular replication. (A) The atpH::Tn strain was grown on BHI plates under oxic and anoxic conditions. (B) cydA::Tn, qoxA::Tn, and atpH::Tn strains (MOI of 0.2) were grown in BMDMs and enumerated for CFU at 2 and 5 h postinfection. Data are calculated as fold change between 2 and 5 h and representative of results from three independent experiments. The panel on the right depicts the same data but graphed as CFU at 0.5, 2, 5, and 8 h postinfection. Download FIG S7, EPS file, 1 MB (1MB, eps) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains and plasmids used in this study. Download TABLE S2, DOCX file, 0.04 MB (43.8KB, docx) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download TABLE S3, DOCX file, 0.1 MB (139.2KB, docx) .
Copyright © 2017 Chen et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.