
Figure 5. Combination of TGFBRII-disruption and ERK inhibition attenuates aggressive potential of TGFBRII-edited prostate cancer cells.
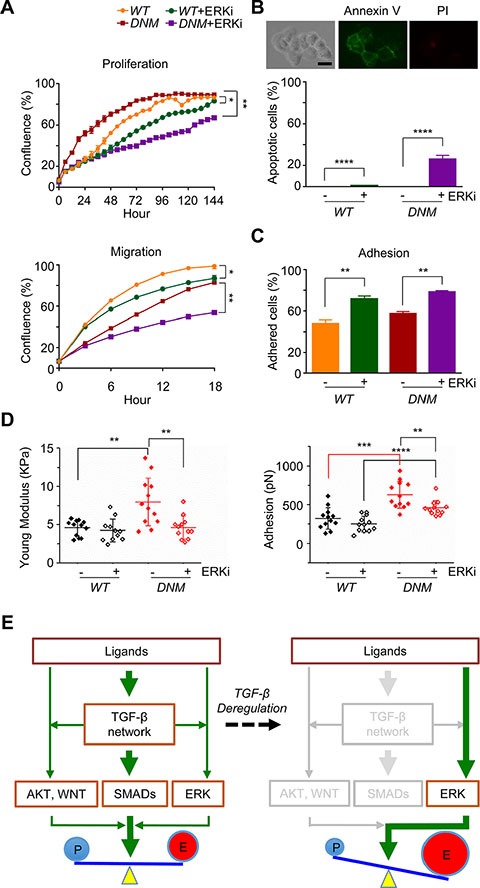
(A) Cell proliferation and migration (wound healing), respectively, using IncuCyte® ZOOM live-cell kinetic imaging system. DU145 TGFBRII WT and DNM cells were treated with 10 μM of ERK inhibitor SCH772984 (ERKi) or vehicle alone. *p < 0.05; **p < 0.01. (B) Cell apoptosis/death analysis. DU145 TGFBRII WT and DNM cells treated with or without ERKi (10 μM) for 24 hours and stained with FITC annexin V and propidium iodide. Annexin V positive cells were counted against total cells. Scale bar: 50 μm. ****p < 0.0001. (C) Cell adhesion assay using IncuCyte® ZOOM live-cell kinetic imaging system. DU145 TGFBRII WT and DNM cells were treated with 10 μM of ERK inhibitor SCH772984 (ERKi) or vehicle alone. **p < 0.01. (D) AFM analysis of cultured DU145 TGFBRII WT and DNM cells treated with or without 3 μM of ERKi for 24 hours. **p < 0.01; ***p < 0.001; ****p < 0.0001. (E) A proposed model of feedback rewiring of deregulated TGF-β signaling sustains EMT of CTCs. In TGFBRII-disrupted prostate cancer cells, both SMADs-dependent and -independent pathways were suppressed with the exception of positive feedback of ERK signaling, resulting uncoupled cell growth and metastasis capability.