

ABSTRACT
Chagas disease is a life-threatening infection caused by a variety of genetically diverse strains of the protozoan parasite Trypanosoma cruzi. The current treatment (benznidazole and nifurtimox) is unsatisfactory, and potential alternatives include inhibitors of sterol 14α-demethylase (CYP51), the cytochrome P450 enzyme essential for the biosynthesis of sterols in eukaryotes and the major target of clinical and agricultural antifungals. Here we performed a comparative investigation of two protozoon-specific CYP51 inhibitors, VNI and its CYP51 structure-based derivative VFV, in the murine models of infection caused by the Y strain of T. cruzi. The effects of different treatment regimens and drug delivery vehicles were evaluated in animals of both genders, with benznidazole serving as the reference drug. Regardless of the treatment scheme or delivery vehicle, VFV was more potent in both genders, causing a >99.7% peak parasitemia reduction, while the VNI values varied from 91 to 100%. Treatments with VNI and VFV resulted in 100% animal survival and 0% natural relapse after the end of therapy, though, except for the 120-day treatment schemes with VFV, relapses after three cycles of immunosuppression were observed in each animal group, and quantitative PCR analysis revealed a very light parasite load in the blood samples (sometimes below or near the detection limit, which was 1.5 parasite equivalents/ml). Our studies support further investigations of this class of compounds, including their testing against other T. cruzi strains and in combination with other drugs.
KEYWORDS: Chagas disease, chemotherapy, sterol 14α-demethylase, inhibitors, VNI, VFV, Trypanosoma cruzi
INTRODUCTION
Chagas disease (CD), or American trypanosomiasis, is a zoonosis caused by multiple strains of the protozoan parasite Trypanosoma cruzi, which are transmitted to more than 150 mammalian species by the triatomine insect vector (kissing bugs). T. cruzi has been infecting humans in South America for at least 9,000 years (1) and was discovered 107 years ago by Carlos Chagas (2). Although, according to the WHO, the number of infected patients has dropped significantly within the past decades, most likely because of successful vector control programs (3), CD still represents an important public health issue, remaining endemic in 21 Latin American countries (more than 6 million patients, with the largest estimated numbers in Argentina, Brazil, and Mexico [4]) and spreading outside the area where CD is endemic as a result of human migration (5–7). The broadening of the area of the insect vector habitat is particularly alarming in the United States (8), where kissing bug bites were reported in 43 states; studies in Louisiana (2007) revealed that 30% of armadillos and 38% of opossums were infected, and although no epidemiological study of humans has been performed, some estimates suggest that the number of chagasic patients in the United States is more than 1 million, most of them remaining undetected (6, 9–12).
The current therapeutic options for CD are limited to two nitroderivatives, benznidazole (Bz) and nifurtimox. These compounds, however, have several limitations, including serious adverse side effects, which lead to treatment discontinuation in 15 to 30% of patients (10, 13), lack of efficacy in the later chronic phase of the disease, and the occurrence of several naturally resistant strains of T. cruzi (14–16). In both nitroderivatives (as well as the nitroimidazole fexinidazole, which was under recent clinical trial for CD), the nitro group is expected to undergo reductive metabolism catalyzed by parasite nitroreductases, leading to the formation of biologically active species exerting their trypanocidal activity (15). Because of their toxicity, neither Bz nor nifurtimox is approved by the FDA and therefore they are not sold or prescribed in the United States. Although the possibility of obtaining these drugs from the Centers for Disease Control (parasites@cdc.gov; www.cdc.gov/parasites/chagas) has been reported (10), most physicians appear to be unaware of this option.
Within the past decades, after the parasitological nature of the chronic stage of CD has been confirmed (17), different alternative therapeutic strategies for the disease have been considered (18), though only a few new candidates entered clinical trials and the results so far have been quite disappointing. For example, a clinical trial of another nitroderivative, fexinidazole, that was very active in preclinical assays (15) has been stopped and the compound was discarded as a drug candidate because of its severe toxic profile in chagasic patients (19). The results of clinical trials of two antifungal agents, posaconazole and ravuconazole, have not met expectations. Thus, although treatment with posaconazole revealed a clear dose-dependent effect in the randomized Chagazol trials, only ∼20% of the patients (versus ∼60% in the Bz group) were found to be negative by follow-up reverse transcription-PCR assays, though T. cruzi DNA was undetectable in the blood of all of the posaconazole-treated patients during the treatment period (16). Currently, largely because of its much better safety profile and clear antiparasitic effect in the Chagasol clinical trials, posaconazole is considered for potential use in combination or in sequential therapies (20). Posaconazole inhibits the fungal sterol 14α-demethylase (CYP51), the cytochrome P450 enzyme that is required for the production of sterols, which in turn serve as essential components of eukaryotic membranes and regulators of the cell cycle and development (21, 22). Posaconazole is used as a systemic clinical drug to treat invasive fungal infections, and its repurposing for CD could be highly advantageous (23). However, the procedure of its synthesis is long and low in yield (24), making posaconazole very costly.
Recently, we have shown that a potent inhibitor of Tulahuen T. cruzi CYP51 (25), VNI {lsqb]N-[(1R)-1-(2,4-dichlorophenyl)-2-imidazol-1-yl-ethyl]-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide}, given orally at 25 mg/kg for 30 days, cures Tulahuen T. cruzi infection with 100% efficacy in (BALB/c) mouse models of both acute and chronic CD (26). VNI is easy to synthesize and nontoxic (25, 27), has favorable pharmacokinetics (26), and does not upregulate CYP51 gene expression (28). However, VNI was found to be less potent against infection with the Y strain of T. cruzi (short-term treatment) (29). Sequencing of the genomic DNA of the Y strain of T. cruzi revealed the presence of two CYP51 genes, gene A (NCBI accession no. JQ434483) and gene B (accession no. JQ434484). While CYP51B was identical to CYP51B in the CL-Brener strain of T. cruzi (NCBI accession number XP_821219.1), CYP51A was found to carry one amino acid sequence variation (P355S) that involves the surface of interaction between the enzyme and its inhibitors. Gene A was expressed in Escherichia coli, and the CYP51A protein was purified and characterized, confirming lower susceptibility to inhibition by VNI, posaconazole, and all of the other compounds tested, except for VFV (30). VFV [(R)-N-(1-(3,4′-difluorobiphenyl-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide] is the CYP51 structure-based VNI derivative designed to fill the deepest portion of the CYP51 substrate binding cavity (Fig. 1) to broaden its spectrum of activity. Similar to VNI, VFV was proven to be 100% efficient in curing Tulahuen T. cruzi infection and displayed higher potency than VNI in a mouse model of visceral leishmaniasis (31). In this study, we compared the effects of VNI and VFV in mouse models of infection with the Y strain of T. cruzi by using both genders (32), different drug delivery vehicles, and different treatment schemes.
FIG 1.
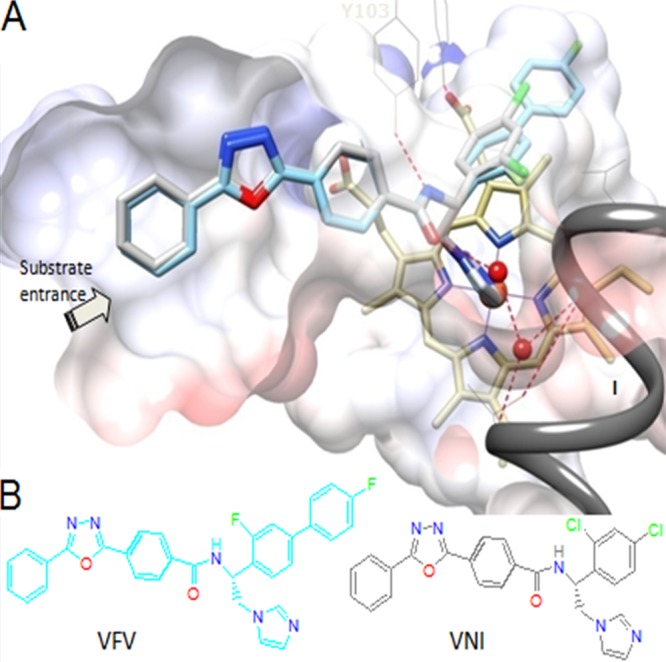
(A) VNI (gray) and VFV (cyan) bound in the CYP51 active site (Protein Data Bank codes 3GW9 and 4G7G, respectively). Shown is a slice through the semitransparent protein surface, distal cytochrome P450 view. The heme is depicted in yellow; the catalytic iron atom is presented as an orange sphere. The H-bond network connecting the carboxamide fragment of the inhibitors with the CYP51 B′ helix (Y103) and helix I (proton delivery route) is displayed as dashed red lines, and water molecules are shown as red spheres. (B) Structural formulas. The color code for the atoms is the same as in panel A.
RESULTS
The rapid acute toxicity assay in vivo (48 h after administration per os) demonstrated a low toxicity profile of VFV (no-observed-adverse-effect level [NOAEL] of 200 mg/kg), with no detectable side effects assayed by clinical observation (animal behavior and body weight analysis) and biochemical plasma measurements (Table 1). The comparative testing of VNI and VFV in the mouse models of infection with the Y strain of T. cruzi was performed by using both genders, different drug delivery vehicles, and different treatment schemes (Table 2). Both genders were included in the experiments because male mice infected with the Y strain of T. cruzi display about 2-fold greater parasite loads at the peak of parasitemia (which corresponds to day 8 after infection with the Y strain of T. cruzi in this experimental mouse model) and appeared to be more suitable for screening of antiparasitic compounds (32). Indeed, VNI, particularly if the treatment was started 5 days postinfection (dpi) (3 days before peak parasitemia), caused a >91% peak parasitemia reduction in all male mouse groups versus a >99% peak parasitemia reduction in the female mouse groups (Table 2). Interestingly, although it is well known that CYP51 inhibitors have a relatively slower mode of action (since several cycles of pathogen multiplication are necessary to exhaust the preexisting resource of cellular sterols), the suppressive effect of VFV was >99.8% in both genders even when the treatment was started 5 dpi and reached 100% if the treatment was started 24 h after infection (Table 2).
TABLE 1.
Plasma biochemical analysis of female mice after 48 h of VFV administration
| Enzyme | Mean level ± SDa after treatment with VFV at (mg/kg): |
|||||
|---|---|---|---|---|---|---|
| 0b | 12.5 | 25 | 50 | 100 | 200 | |
| ALT | 106 ± 39 | 72 ± 21 | 73 ± 13 | 62 ± 0 | 83 ± 38 | 161 ± 118 |
| CK | 396 ± 32 | 1,000 ± 30 | 446 ± 102 | 540 ± 255 | 369 ± 228 | 407 ± 336 |
| AST | 123 ± 6.4 | 168 ± 14 | 115 ± 16 | 92 ± 0 | 104 ± 40 | 70 ± 0 |
Values of two independent assays are shown. Reference values (CECAL/Fiocruz): ALT, up to 132 U/liter; AST, up to 247 U/liter; CK, up to 1,070 U/liter.
Results of one representative assay are shown.
TABLE 2.
Effects of Bz, VNI, and VFV on male and female Swiss mice infected with the Y strain of T. cruzi and subjected to different treatment schemes with different drug delivery vehicles
| Gender and treatment scheme | Drug | % suppression at peak parasitemia | % animal survival | No. of mice with natural relapse at end of therapy/surviving animals | No. of mice with relapse after Cy immunosuppression/surviving animals |
|---|---|---|---|---|---|
| Males | |||||
| 6 dpi, 30 days | Bza | 54 | 100 | 0/5 | 0/5 |
| 5 dpi, 30 days | VNIb | ||||
| DGAT | 93.4 | 100 | 0/4 | 1/4 | |
| HPCD | 94 | 100 | 0/3 | 2/3 | |
| DGA | 91 | 100 | 0/4 | 0/4 | |
| 1 dpi, 30 days | DGAT | 99.8 | 100 | 0/4 | 2/4 |
| 5 dpi, 120 days | HPCD | 98 | 100 | 0/5 | 4/5 |
| 5 dpi, 30 days | VFVb | ||||
| DGAT | 99.9 | 100 | 0/5 | 4/5 | |
| HPCD | 99.9 | 100 | 0/5 | 4/5 | |
| DGA | 99.9 | 100 | 0/4 | 3/4 | |
| 1 dpi, 30 days | DGAT | 100 | 100 | 0/4 | 2/4 |
| 5 dpi, 120 days | HPCD | 99.8 | 100 | 0/5 | (0/4)c |
| Bza | 99.6 | 100 | 0/5 | 0/5 | |
| Females | |||||
| 6 dpi, 30 days | Bza | >90 | 100 | 0/5 | 0/5 |
| 5 dpi, 30 days | VNIb | ||||
| DGAT | 99.7 | 100 | 0/4 | 1/4 | |
| HPCD | 99.8 | 100 | 0/5 | 0/5 | |
| DGA | NTd | ||||
| 1 dpi, 30 days | DGAT | 100 | 80 | 0/4 | 4/4 |
| 5 dpi, 120 days | HPCD | 99.8 | 100 | 0/5 | 1/5 |
| 5 dpi, 30 days | VFVb | ||||
| DGAT | 100 | 100 | 0/5 | 4/5 | |
| HPCD | 99.9 | 100 | 0/5 | 3/5 | |
| DGA | NT | ||||
| 1 dpi, 30 days | DGAT | 100 | 80 | 0/4 | 0/4 |
| 5 dpi, 120 days | HPCD | 100 | 100 | 0/5 | 0/5 |
| Bza | 100 | 100 | 0/5 | 0/5 |
Administered at 100 mg/kg.
Administered at 25 mg/kg/bid.
One mouse was euthanized after it lost an eye because of mouse aggression.
NT, not tested.
No drastic alterations in the course of parasitemia or mortality rates were observed during the use of different drug delivery vehicles in these experiments, but dimethyl sulfoxide and gum arabic (DGA) resulted in greater parasite loads measured by real-time quantitative PCR (qPCR) assay of the blood of VNI-treated male animals (Fig. 2 and 3A). In this male mouse model, all three formulations of VFV produced a 99.9% peak parasitemia decline, though for VNI, DGA appeared to be slightly weaker (91% peak parasitemia suppression versus 94% and 93.4% for hydroxypropyl-β-cyclodextrin (HPCD) and 5% DGA plus 0.5% Tween 80 (DGAT), respectively (Table 2 and Fig. 2A), all three being more efficient than when VNI was added from 10% DMSO alone (see Fig. 4 in reference 29). Because of the weaker effect of VNI in DGA, only DGAT and HPCD formulations were tested in the female mouse model.
FIG 2.

Parasitemia levels in experimental male (A) and female (B) mouse models of Y strain T. cruzi infection treated with VNI and VFV (25 mg/kg/day) with different vehicles for 30 days starting at 5 dpi.
FIG 3.

Cure assessment by qPCR analysis of the load parasite burdens (parasite equivalents per milliliter) of male and female mice infected with the Y strain of T. cruzi and submitted or not to VNI and VFV therapies started at the onset of parasitemia (5 dpi) or 24 h after parasite infection (1 dpi) and continued for 30 days (A, log) or 120 days (B). *, statistically significant at P = 0.02. Dashed lines: limit of qPCR detection (1.5 parasite equivalents/ml). Treatment of female and male mice with Bz did not result in qPCR positivity or parasite load detection.
All of the treatment schemes used protected against animal death, and there was no natural relapse of parasitemia after completion of the 30-day therapies. However, after a 1-month waiting period, when the animals were immunosuppressed with 12 injections of cyclophosphamide (Cy), most of them relapsed and the presence of parasite DNA was confirmed by qPCR analysis of blood samples (Fig. 3A). Although it is impossible to draw any solid conclusions because of the high variability of the qPCR data (quite expected, as outbred animal models were used), but the qPCR results suggest that in the case of the treatments with VNI (opposite to its effects on peak parasitemia suppression), there was no major decrease in the parasite burden when the treatment was started 1 versus 5 dpi, while in the case of VFV, the early treatment scheme appears to be more beneficial (at least in the female model) (Fig. 3A). In the hope that longer drug exposure would produce better outcomes, the treatment period was extended to 120 days (HPCD vehicle, starting at 5 dpi, both genders). All of the animals survived the treatment and showed no natural relapse after the completion of therapy, and in both VFV-treated groups (males and females), no relapses after immunosuppression were detected by light microscopy, while in the VNI-treated groups, four out of five males and one out of five females still relapsed (Table 2 and Fig. 3B). qPCR analysis, however, still displayed residual traces of parasite DNA in blood samples, 2.2 ± 3.6 parasite equivalents/ml for VFV-treated males and 0.94 ± 0.49 parasite equivalents/ml for VFV-treated females (Fig. 3B). Quite peculiarly, in the blood samples of VNI-treated animals (where the presence of the parasite was detected by light microscopy [see above]), qPCR assays revealed lighter loads of T. cruzi DNA (0.001 ± 0 parasite equivalents/ml in males and 0.095 ± 0.14 parasite equivalents/ml in females). Overall, all of the values determined by qPCR after the 120-day therapies were below or near the limit of parasite detection (1.5 parasite equivalents/ml). The statistical analysis only demonstrated a significant difference in the amount of parasite DNA when females were treated for 120 days with VFV compared to VNI (P = 0.02). Treatment of mice (both genders) with Bz for 120 days resulted in no qPCR amplification in any of the mice tested.
DISCUSSION
The fact that Bz will now have a much broader use in the treatment of CD is, overall, highly positive. However, we tend to agree with the notion (13, 20, 33) that searches for new drugs should not be discouraged and less toxic alternatives for chagasic patients must still be considered, especially regarding the combination approach of two drugs targeting different mechanisms. CYP51 inhibitors remain one of them, because unlike Bz (or any nitroderivatives), whose very mode of action is based mainly on the generation of toxic nitroreduction intermediates, CYP51 inhibitors are potentially much less harmful, as they act selectively. Thus, although negative follow-up PCR results after the Chagazol clinical trials were achieved in only 20% of the posaconazole-treated patients, none of the patients had to discontinue therapy because of adverse side effects, which was not the case for the Bz-treated group (16). Longer treatment periods and greater doses of posaconazole were recommended (16), and variability of the T. cruzi population was suggested as a possible reason for the poor treatment outcomes (13, 20).
It is well known that there are more than 70 different strains of T. cruzi (see http://www.dbbm.fiocruz.br/TcruziDB/strain.html). Depending on the strain, the infection varies in its progression (the times of onset and peak parasitemia), the severity of the acute stage, tissue tropism, and chronic symptoms (cardiac versus gastrointestinal), etc. Some of the T. cruzi strains, including Y, CL, and Colombiana, similar to filamentous fungi (34), have been found to carry two CYP51 genes (29, 30), and the high CYP51 sequence variability across the strains suggests that they should really be regarded as different species (30).
The results of the present study confirm that infection with the Y strain of T. cruzi is less susceptible to treatment with CYP51 inhibitors than is infection with the Tulahuen strain (26) and show that although VFV is more potent than VNI in killing the parasite at the peak of infection (>99% versus >91% peak parasitemia reduction), a much longer treatment period is required to prevent a relapse of parasitemia after immunosuppression. One more possible reason for the various susceptibilities of T. cruzi strains to CYP51 inhibitors may be connected to the differences in the propensity of parasites to form metabolically quiescent amastigotes (20). Hidden within different organs and tissues to evade the host immune system, these dormant forms cannot be targeted efficiently by CYP51 inhibitors because, as is well known in mycology, CYP51 inhibitors act largely on metabolically active cells that require a permanent supply of newly synthesized sterols to build their membranes and regulate intracellular processes. In a similar sense, if these supposedly latent intracellular forms could more likely be trypomastigotes and as CYP51 inhibitors have less of an effect on these nonproliferative forms, this could also result in different outcomes with different parasite strains.
Studies including infections with a broader variety of T. cruzi strains, testing greater dosages of VNI and VFV (e.g., 50 mg/kg), and perhaps the use of mild immunosuppression during the treatment period (as was done in the case of successful treatment of immunosuppressed chronic human CD with posaconazole [35]) should provide useful information on the prospective treatment of CD. In the meantime, combination therapy appears to be the most obvious step to proceed because, since it is by now quite clear that a decrease in the parasite burden leads to a significant reduction in the development of the symptomatic chronic form of CD (13). CYP51 inhibitors would definitely fulfill this function and besides, given in combination with Bz, they should allow lowering of the Bz dosage and shortening of the therapy period, which, in turn, may decrease the severity of side effects.
MATERIALS AND METHODS
Compounds.
VNI and VFV were synthesized at Vanderbilt University as described previously (27). For in vivo analysis, the compounds were diluted with three different vehicles, (i) 20% HPCD (CTD, Inc., USA) (36), (ii) 10% DMSO and 5% DGA (as reported in reference 29 for Colombiana T. cruzi infection), and (iii) 5% DMSO and DGAT (26). Cy (Genuxal) was purchased from Baxter Oncology (Frankfurt, Germany) and prepared in sterile distilled water (32). Bz was purchased from Laboratório Farmacêutico do Estado de Pernambuco, LAFEPE, Brazil, and dissolved in sterile distilled water supplemented with 3% Tween 80 as described previously (32).
Parasites.
Bloodstream trypomastigotes (BTs) of the Y strain (discrete typing unit II) of T. cruzi were obtained from the blood of infected male Swiss mice at the peak of parasitemia (37). Immediately after the purification step, the parasites were resuspended in RPMI 1640 medium (pH 7.2 to 7.4) without phenol red (Gibco BRL) and supplemented with 10% fetal bovine serum and 2 mM glutamine as reported previously (38).
In vivo acute toxicity.
To determine the NOAEL, increasing doses of the test compounds (up to 200 mg/kg of body weight) were injected by the intraperitoneal (i.p.) route individually into female Swiss Webster mice (20 to 23 g, two per assay, two assays). Treated animals were inspected for toxic and subtoxic symptoms according to the Organization for Economic Cooperation and Development guidelines. Forty-eight hours after compound injection, the NOAELs were determined as reported previously (27).
Biochemical analysis.
Forty-eight hours after compound administration, mouse blood was collected and immediately submitted to biochemical analysis for determination of plasma tissue markers, including, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and creatine kinase (CK), which was performed at the animal facilities of the Oswaldo Cruz Foundation (Rio de Janeiro, Brazil, CECAL/Fiocruz platform) with Vitros 250 (Ortho Clinical-Johnson & Johnson) as reported previously (27).
In vivo infection.
Female and male Swiss Webster mice (18 to 20 g) were obtained from the Fundação Oswaldo Cruz (FIOCRUZ) animal facilities (CECAL/FIOCRUZ, Rio de Janeiro, Brazil). Mice were housed at a maximum of five per cage and kept in a conventional room at 20 to 24°C under a 12-h light–12-h dark cycle. The animals were provided with sterilized water and chow ad libitum. Infection was performed by i.p. injection of 104 BTs of the Y strain. Age-matched uninfected mice were maintained under identical conditions (29).
Treatment schemes.
The animals (female and male) were divided into the following groups (five animals per group): uninfected (uninfected and nontreated), untreated (infected but treated only with each respective vehicle), and treated (infected and treated with the test compounds). Therapy, given orally (VNI and VFV at 25 mg/kg/day twice a day [bid] and Bz at 100 mg/kg/day once a day), was performed starting at 1 or 5 dpi. For the 5-dpi treatment schemes, only mice with positive parasitemia were used in the infected groups.
Parasitemia and mortality rates.
Parasitemia was individually checked by direct microscopic counting of parasites in 5 μl of blood, and mortality rates were checked daily until 30 days posttreatment and expressed as percent cumulative mortality as described before (32).
Immunosuppression.
Mice that presented consistent negative parasitemia up to 30 days posttreatment were submitted to three cycles of immunosuppression with Cy (50 mg/kg/day), with each cycle including 4 consecutive days of Cy administration (i.p.) and a 3-day interval (32).
Cure assessment.
Cure assessment criteria were based on negative blood parasitemia observed by light microscopy, and blood samples from consistently negative mice were subjected to real-time qPCR. For qPCR, 500 μl of blood was diluted 1:2 in a volume of guanidine solution (6 M guanidine-HCl, 0.2 M EDTA) and heated for 90 s in boiling water. Guanidine-EDTA blood (GEB) samples were processed with the QIAamp DNA minikit (Qiagen) as previously described (39). Multiplex real-time qPCR assays targeting the T. cruzi satellite nuclear DNA and the exogenous internal amplification control (plasmid pZErO-2 containing an insert from the Arabidopsis thaliana aquaporin gene, 40 amplification cycles) were performed as previously described (40). The standard curves for absolute quantification were constructed with 1/10 serial dilutions of total DNA obtained from a negative GEB sample spiked with 105 parasite (Y strain) equivalents/ml of blood.
Ethics.
All procedures were carried out in accordance with the guidelines established by the FIOCRUZ Committee of Ethics for the Use of Animals (CEUA LW16/14).
Statistical analysis.
The data represent means ± standard deviations from two experiments run in duplicate, and statistical analysis was performed by analysis of variance with the level of significance set at P ≤ 0.05 (40).
ACKNOWLEDGMENTS
We thank the Rede de Plataformas Fiocruz (PDTIS-Fiocruz) for the facilities on the real-time PCR RPT09A platform and RPT11G.
The present study was supported by grants from Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Oswaldo Cruz, PDTIS, PAEF/CNPq/Fiocruz, CAPES. M.N.C.S. and C.B. are research fellows of CNPq. M.N.C.S. and C.B. are CNE researchers. O.C.M. is a JCNE researcher. G.I.L. was supported by National Institutes of Health grant GM067871 through the NIGMS.
REFERENCES
- 1.Aufderheide AC, Salo W, Madden M, Streitz J, Buikstra J, Guhl F, Arriaza B, Renier C, Wittmers LE, Fornaciari G, Allison M. 2004. A 9,000-year record of Chagas' disease. Proc Natl Acad Sci U S A 101:2034–2039. doi: 10.1073/pnas.0307312101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chagas C. 1909. Nova tripanozomiase humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem Inst Oswaldo Cruz 1:159–218. doi: 10.1590/S0074-02761909000200008. [DOI] [Google Scholar]
- 3.WHO. 2015. The world health report. World Health Organization, Geneva, Switzerland: http://www.who.int/en/. [Google Scholar]
- 4.Pérez-Molina JA, Perez AM, Norman FF, Monge-Maillo B, López-Vélez R. 2015. Old and new challenges in Chagas disease. Lancet Infect Dis 15:1347–1356. doi: 10.1016/S1473-3099(15)00243-1. [DOI] [PubMed] [Google Scholar]
- 5.Basile L, Jansa JM, Carlier Y, Salamanca DD, Angheben A, Bartoloni A, Seixas J, Van Gool T, Canavate C, Flores-Chavez M, Jackson Y, Chiodini PL, Albajar-Vinas P. 2011. Chagas disease in European countries: the challenge of a surveillance system. Euro Surveill 16:19968 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19968. [PubMed] [Google Scholar]
- 6.Hotez P, Dumonteil E, Betancourt Cravioto M, Bottazzi M, Tapia-Conyer R. 2013. An unfolding tragedy of Chagas disease in North America. PLoS Negl Trop Dis 7:e2300. doi: 10.1371/journal.pntd.0002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein N, Hurwitz I, Durvasula R. 2012. Globalization of Chagas disease: a growing concern in nonendemic countries. Epidemiol Res Int 2012:1–13. https://www.hindawi.com/journals/eri/2012/136793/. [Google Scholar]
- 8.Leslie M. 2011. Drug developers finally take aim at a neglected disease. Science 333:933–935. doi: 10.1126/science.333.6045.933. [DOI] [PubMed] [Google Scholar]
- 9.Barry MA, Bezek S, Serpa JA, Hotez PJ, Woc-Colburn L. 2012. Neglected infections of poverty in Texas and the rest of the United States: management and treatment options. Clin Pharmacol Ther 92:170–181. doi: 10.1038/clpt.2012.85. [DOI] [PubMed] [Google Scholar]
- 10.Bern C, Kjos S, Yabsley MJ, Montgomery SP.. 2011. Trypanosoma cruzi and Chagas' disease in the United States. Clin Microbiol Rev 24:655–681. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanford EJ, Zhan FB, Lu Y, Giordano A. 2007. Chagas disease in Texas: recognizing the significance and implications of evidence in the literature. Soc Sci Med 65:60–79. doi: 10.1016/j.socscimed.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 12.Hotez PJ. 2008. Neglected infections of poverty in the United States of America. PLoS Negl Trop Dis 2:e256. doi: 10.1371/journal.pntd.0000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urbina JA. 2015. Recent clinical trials for the etiological treatment of chronic Chagas disease: advances, challenges and perspectives. J Eukaryot Microbiol 62:149–156. doi: 10.1111/jeu.12184. [DOI] [PubMed] [Google Scholar]
- 14.Soeiro MN, de Castro SL. 2009. Trypanosoma cruzi targets for new chemotherapeutic approaches. Expert Opin Ther Targets 13:105–121. doi: 10.1517/14728220802623881. [DOI] [PubMed] [Google Scholar]
- 15.Bahia MT, Nascimento AFS, Mazzeti AL, Marques LF, Gonçalves KR, Mota LWR, Diniz LdF Caldas IS, Talvani A, Shackleford DM, Koltun M, Saunders J, White KL, Scandale I, Charman SA, Chatelain E. 2014. Antitrypanosomal activity of fexinidazole metabolites, potential new drug candidates for Chagas disease. Antimicrob Agents Chemother 58:4362–4370. doi: 10.1128/AAC.02754-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molina I, Gómez i Prat J, Salvador F, Treviño B, Sulleiro E, Serre N, Pou D, Roure S, Cabezos J, Valerio L, Blanco-Grau A, Sánchez-Montalvá A, Vidal X, Pahissa A. 2014. Randomized trial of posaconazole and benznidazole for chronic Chagas' disease. N Engl J Med 370:1899–1908. doi: 10.1056/NEJMoa1313122. [DOI] [PubMed] [Google Scholar]
- 17.Tarleton RL, Zhang L, Downs MO. 1997. “Autoimmune rejection” of neonatal heart transplants in experimental Chagas disease is a parasite-specific response to infected host tissue. Proc Natl Acad Sci U S A 94:3932–3937. doi: 10.1073/pnas.94.8.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lepesheva GI. 2013. Design or screening of drugs for the treatment of Chagas disease: what shows the most promise? Expert Opin Drug Discov 8:1479–1489. doi: 10.1517/17460441.2013.845554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molina I, Salvador F, Sánchez-Montalvá A. 2016. Actualización en enfermedad de Chagas. Enferm Infecc Microbiol Clín 34:132–138. [DOI] [PubMed] [Google Scholar]
- 20.Molina I, Salvador F, Sánchez-Montalvá A. 2015. The use of posaconazole against Chagas disease. Cur Opin Infect Dis 28:397–407. doi: 10.1097/QCO.0000000000000192. [DOI] [PubMed] [Google Scholar]
- 21.Lepesheva GI, Waterman MR. 2007. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta 1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lepesheva GI, Waterman MR. 2011. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr Top Med Chem 11:2060–2071. doi: 10.2174/156802611796575902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clayton J. 2010. Chagas disease: pushing through the pipeline. Nature 465:S12–S15. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- 24.Dobish MC, Villalta F, Waterman MR, Lepesheva GI, Johnston JN. 2012. Organocatalytic, enantioselective synthesis of VNI: a robust therapeutic development platform for Chagas, a neglected tropical disease. Org Lett 14:6322–6325. doi: 10.1021/ol303092v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lepesheva GI, Ott RD, Hargrove TY, Kleshchenko YY, Schuster I, Nes WD, Hill GC, Villalta F, Waterman MR. 2007. Sterol 14 alpha-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem Biol 14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villalta F, Dobish MC, Nde PN, Kleshchenko YY, Hargrove TY, Johnson CA, Waterman MR, Johnston JN, Lepesheva GI. 2013. VNI cures acute and chronic experimental Chagas disease. J Infect Dis 208:504–511. doi: 10.1093/infdis/jit042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hargrove TY, Kim K, de Nazaré Correia Soeiro M, da Silva CF, da Gama Jaen Batista D, Batista MM, Yazlovitskaya EM, Waterman MR, Sulikowski GA, Lepesheva GI. 2012. CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int J Parasitol Drugs Drug Resist 2:178–186. doi: 10.1016/j.ijpddr.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lepesheva GI, Villalta F, Waterman MR. 2011. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Adv Parasitol 75:65–87. doi: 10.1016/B978-0-12-385863-4.00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soeiro MDC, de Souza EM, da Silva CF, Batista DDJ Batista MM, Pavão BP, Araújo JS, Lionel J, Britto C, Kim K, Sulikowski G, Hargrove TY, Waterman MR, Lepesheva GI. 2013. In vitro and in vivo studies of the antiparasitic activity of sterol 14α-demethylase (CYP51) inhibitor VNI against drug-resistant strains of Trypanosoma cruzi. Antimicrob Agents Chemother 57:4151–4163. doi: 10.1128/AAC.00070-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cherkesova TS, Hargrove TY, Vanrell MC, Ges I, Usanov SA, Romano PS, Lepesheva GI. 2014. Sequence variation in CYP51A from the Y strain of Trypanosoma cruzi alters its sensitivity to inhibition. FEBS Lett 588:3878–3885. doi: 10.1016/j.febslet.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lepesheva GI, Hargrove TY, Rachakonda G, Wawrzak Z, Pomel S, Cojean S, Nde PN, Nes WD, Locuson CW, Calcutt MW, Waterman MR, Daniels JS, Loiseau PM, Villalta F. 2015. VFV as a new effective CYP51 structure-derived drug candidate for Chagas disease and visceral leishmaniasis. J Infect Dis 212:1439–1448. doi: 10.1093/infdis/jiv228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guedes-da-Silva FH, Batista DGJ, da Silva CF, Meuser MB, Simões-Silva MR, de Araújo JS, Ferreira CG, Moreira OC, Britto C, Lepesheva GI, Soeiro MdNC. 2015. Different therapeutic outcomes of benznidazole and VNI treatments in different genders in mouse experimental models of Trypanosoma cruzi infection. Antimicrob Agents Chemother 59:7564–7570. doi: 10.1128/AAC.01294-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urbina JA, McKerrow JH. 2015. Drug susceptibility of genetically engineered Trypanosoma cruzi strains and sterile cure in animal models as a criterion for potential clinical efficacy of anti-T. cruzi drugs. Antimicrob Agents Chemother 59:7923–7924. doi: 10.1128/AAC.01714-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hargrove TY, Wawrzak Z, Lamb DC, Guengerich FP, Lepesheva GI. 2015. Structure-functional characterization of cytochrome P450 sterol 14α-demethylase (CYP51B) from Aspergillus fumigatus and molecular basis for the development of antifungal drugs. J Biol Chem 290:23916–23934. doi: 10.1074/jbc.M115.677310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinazo MJ, Espinosa G, Gallego M, Lopez-Chejade PL, Urbina JA, Gascon J. 2010. Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am J Trop Med Hyg 82:583–587. doi: 10.4269/ajtmh.2010.09-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buckner F, Bahia MT, Suryadevara PK, White KL, Shackleford DM, Chennamaneni NK, Hulverson MA, Laydbak JU, Chatelain E, Scandale I, Verlinde CL, Charman SA, Lepesheva GI, Gelb MH. 2012. Pharmacological characterization, structural studies, and in vivo activity of anti-Chagas disease lead compounds derived from tipifarnib. Antimicrob Agents Chemother 56:4914–4921. doi: 10.1128/AAC.06244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batista DG, Batista MM, de Oliveira GM, do Amaral PB, Lannes-Vieira J, Britto CC, Junqueira A, Lima MM, Romanha AJ, Sales Junior PA, Stephens CE, Boykin DW, Soeiro MN. 2010. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas' disease treatment. Antimicrob Agents Chemother 54:2940–2952. doi: 10.1128/AAC.01617-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Timm BL, da Silva PB, Batista MM, da Silva FH, da Silva CF, Tidwell RR, Patrick DA, Jones SK, Bakunov SA, Bakunova SM, Soeiro MN. 2014. In vitro and in vivo biological effects of novel arylimidamide derivatives against Trypanosoma cruzi. Antimicrob Agents Chemother 58:3720–3726. doi: 10.1128/AAC.02353-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreira OC, Ramírez JD, Velázquez E, Melo MF, Lima-Ferreira C, Guhl F, Sosa-Estani S, Marin-Neto JA, Morillo CA, Britto C. 2013. Towards the establishment of a consensus real-time qPCR to monitor Trypanosoma cruzi parasitemia in patients with chronic Chagas disease cardiomyopathy: a substudy from the BENEFIT trial. Acta Trop 125:23–31. doi: 10.1016/j.actatropica.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 40.Duffy T, Cura CI, Ramirez JC, Abate T, Cayo NM, Parrado R, Bello ZD, Velazquez E, Muñoz-Calderon A, Juiz NA, Basile J, Garcia L, Riarte A, Nasser JR, Ocampo SB, Yadon ZE, Torrico F, de Noya BA, Ribeiro I, Schijman AG. 2013. Analytical performance of a multiplex real-time PCR assay using TaqMan probes for quantification of Trypanosoma cruzi satellite DNA in blood samples. PLoS Negl Trop Dis 7:e2000. doi: 10.1371/journal.pntd.0002000. [DOI] [PMC free article] [PubMed] [Google Scholar]