
Figure 1.
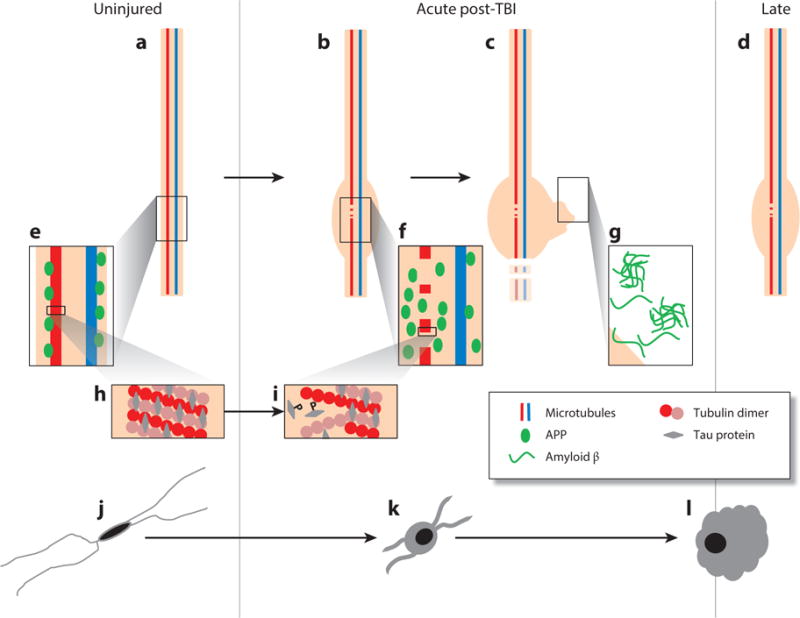
Proposed evolution of axonal and microglial pathologies contributing to late neurodegeneration following traumatic brain injury (TBI). In the intact, uninjured axon (a) microtubules composed of tubulin dimers bound by tau protein (h) transport cargo along the axon, including amyloid precursor protein (APP) and the enzymes responsible for its cleavage to amyloid β (Aβ) (e). As a consequence of dynamic stretch during injury, there is a change in the physical properties of tau protein, resulting in the mechanical breaking of microtubules, tau liberation, and its subsequent phosphorylation (i). At these sites of microtubule breakage, transport interruption follows, with the accumulation of transported cargo (f) resulting in axonal swelling (b), degeneration (c), and the liberation of large pools of Aβ, leading to plaque formation (g), a process that continues beyond the acute phase in a proportion of survivors (d). In parallel with this axonal pathology, there is a notable neuroinflammatory response marked by quiescent, ramified microglia (j) becoming activated (k); these activated and amoeboid microglia (l) persist beyond the acute phase in a proportion of survivors.