

Abstract
Multiple proteases in a system hydrolyze target substrates, but recent evidence indicates that some proteases will degrade other proteases as well. Cathepsin S hydrolysis of cathepsin K is one such example. These interactions may be uni‐ or bi‐directional and change the expected kinetics. To explore potential protease‐on‐protease interactions in silico, a program was developed for users to input two proteases: (1) the protease‐ase that hydrolyzes (2) the substrate, protease. This program identifies putative sites on the substrate protease highly susceptible to cleavage by the protease‐ase, using a sliding‐window approach that scores amino acid sequences by their preference in the protease‐ase active site, culled from MEROPS database. We call this PACMANS, Protease‐Ase Cleavage from MEROPS ANalyzed Specificities, and test and validate this algorithm with cathepsins S and K. PACMANS cumulative likelihood scoring identified L253 and V171 as sites on cathepsin K subject to cathepsin S hydrolysis. Mutations made at these locations were tested to block hydrolysis and validate PACMANS predictions. L253A and L253V cathepsin K mutants significantly reduced cathepsin S hydrolysis, validating PACMANS unbiased identification of these sites. Interfamilial protease interactions between cathepsin S and MMP‐2 or MMP‐9 were tested after predictions by PACMANS, confirming its utility for these systems as well. PACMANS is unique compared to other putative site cleavage programs by allowing users to define the proteases of interest and target, and can also be employed for non‐protease substrate proteins, as well as short peptide sequences.
Keywords: bioinformatics, proteases, proteolysis, hydrolysis, MMPs, cathepsins, prediction, mutagenesis
Abbreviations
- CatS
cathepsin S
- CatK
cathepsin K
- MMP‐2
matrix metalloproteinase‐2
- MMP‐9
matrix metalloproteinase‐9
- PACMANS
Protease‐Ase Cleavages from MEROPS ANalyzed Specificities
Introduction
Cysteine cathepsins are a group of lysosomal proteases in the papain superfamily that are involved in intracellular protein degradation as well as degradation of extracellular matrix proteins when secreted by cells.1, 2 Of particular interest, cathepsins K, L, S, and V share 60% sequence homology and among them, include the most potent mammalian collagenase, cathepsin K, and most potent mammalian elastase, cathepsin V.3, 4, 5, 6 These four cathepsins have been implicated in many tissue destructive pathologies such as cancer metastasis,7, 8, 9, 10, 11, 12 osteoporosis,13, 14, 15 atherosclerosis,3, 15, 16, 17, 18, 19 rheumatoid arthritis,20 endometriosis,21 and tendinopathy.22 Consequently, these proteases have redundancy in substrate cleavage preferences.
We recently showed that one cathepsin family member could preferentially degrade another, even in the presence of the substrate, which we termed cathepsin cannibalism.23 It is also important to note that this mechanism occurred between two mature enzymes, with one mature enzyme hydrolyzing the other into inactive fragments, not the well reported propeptide activation that occurs between cathepsins.24, 25, 26 Determining that cathepsin S proteolysis of cathepsin K involved substrate degradation assays, immunoblotting, zymography, and computational modeling to test hypotheses about the cause of the unexpectedly low amount of substrate being degraded when two cathepsins were co‐incubated.
Mass spectrometry can be used to sequence fragments of substrate protein after these cannibalistic hydrolysis, however this can be prohibitive due to cost and requirements of large amounts of purified protein, to test a hypothesis that may or may not be supported. We present here a computer algorithm and program to identify sites where proteases may cleave other proteases or substrate proteins, in an unbiased way, using data automatically gleaned from the online database of peptidases and their inhibitors, MEROPS (http://merops.sanger.ac.uk/).27, 28 We call this new tool PACMANS: Protease‐Ase Cleavages from MEROPS ANalyzed Specificities. PACMANS was validated by studying cannibalism between mature cathepsin S, as the “protease‐ase,” and cathepsin K, as the substrate protease, using this bioinformatically informed site‐directed mutagenesis of cathepsin K to prevent its cannibalistic cleavage by cathepsin S, and then applied to inter‐familial proteolysis of cathepsins by matrix metalloproteinases (MMPs).
Results
Development and instructions for PACMANS
The design goals for PACMANS included (1) ease of use, (2) algorithmically efficient, (3) versatile for protease and substrate sequence input, and (4) automation. With these design goals in mind, a program using a sliding‐window approach was developed, where each 8 residue segment of the amino acid sequence of the substrate protease was assessed for likelihood of cleavage in the active site pocket of the protease‐ase. Individual residue scores were retrieved from MEROPS online database that populates a specificity matrix for each protease that quantifies the likelihood of cleavage of each residue in each subsite of a protease's active site.27, 28 The positions on the protease at the active site are designated with S, and for the substrate as P, and are numbered outward (typically to P4/P4′) from the scissile bond between P1 and P1′ positions,29 where the non‐primed numbers refer to residues on the N‐terminal side and the primed numbers refer to the C‐terminal side of the cleaved bond (Fig. 1). A cumulative likelihood score was calculated by summing individual residue scores for each set of 8 amino acids being analyzed in that sliding window. A higher cumulative likelihood scored sequence would indicate higher specificity of that 8‐amino acid sequence on the substrate protease for the active site in the protease‐ase. Lower cumulative likelihood scores represent sequences with lower specificity for hydrolysis by the protease‐ase for the substrate protease at that 8‐amino acid window. This algorithm was coded and implemented in MATLAB as described in the Methods section, and is available online at platt.gatech.edu/PACMANS.
Figure 1.

Using PACMANS to score locations of hydrolysis of the substrate protease by the protease‐ase. Input the name of hypothesized protease‐ase and desired substrate protease, specificity matrix for the protease‐ase, and amino acid sequence of desired substrate protease. The specificity matrix quantifies the likelihood that a given amino acid in positions P4 through P4′ will result in a peptide bond cleavage between P1 and P1′ in the enzyme's active site. Using the protease‐ase's specificity matrix and another protease's amino acid sequence as the substrate protease, the potential locations of cleavage were scored by the sum of the individual scores that a given residue has been reported to occur in each subsite of the active site pocket. These putative site of cleavage by protease‐ase were then sorted by score and given to users in text file along with a histogram of all the scores.
Users are required to input five main pieces of information into PACMANS to generate the ranked list of putative cleavage site locations.
User inputs names of the protease‐ase and substrate protease, which will be used to generate the output file name: “PROTEASEASEonSUBSTRATE.txt” [Fig. 1(A)].
User defines the sliding window size of up to 8 subsites for analysis, where a lower bound of 1 would correspond to the residue in the S4 subsite and an upper bound of 8 would correspond to the residue in the S4′ subsite. If these fields are left blank, PACMANS will default to the full 8 (S4‐S4′), but the option is available for a smaller window.
User goes to MEROPS (https://merops.sanger.ac.uk/), click on the “Name” link under “Peptidase” on the left column on the page. Find the putative protease of interest from the Index of Peptidase Names page and click on the “MEROPS ID” given in the right column of the table. Copy the web‐address of the protease‐ase of interest and paste into the link field of PACMANS interface [Fig. 1(B)].
User inputs the FASTA‐formatted amino acid sequence of the substrate protease. Amino acid sequences can be obtained from UniProt (www.uniprot.org) by searching for substrate protease of interest in the search box at the top of the page [Fig. 1(C)]. Select the “Entry” number of substrate protease of interest and scroll down to find the “Sequence” section. Click link to “FASTA” sequence and copy only the amino acid sequence. Be sure to remove spaces before inputting into PACMANS.
User clicks “Load Data” and then “Calculate Scores” to generate a text file of scores.
After execution, PACMANS will return a text file with the substrate protease amino acid sequence, molecular weight, and cleavage site analysis [Fig. 1(D)]. The cleavage site analysis will rank segments by cumulative sum score, and for each segment, list specific amino acid sequence being hydrolyzed, individual scores of residues (i.e. S1‐P1 score), the range and median of those individual scores, as well as expected size of the two fragments of the hydrolyzed substrate protein, assuming one cleavage site. The cumulative likelihood scores will also be plotted in a histogram for quick viewing of high scoring outliers that should be considered first as most probable [Fig. 1(E)].
Application of PACMANS utility: Cathepsin S cannibalism of Cathepsin K
PACMANS was used to determine putative locations of protease‐on‐protease cleavage for the cathepsin S‐cathepsin K relationship. Cathepsin S was input as the protease‐ase and cathepsin K as the substrate protease. Top putative hydrolysis sites were ranked and output into a text file (Supporting Information File S1). PACMANS ranks all possible sequence segments, however only the top 5% of highest scoring sequences (16 sites) were further reviewed for structural cues that would likely affect protease‐ase hydrolysis. In Figure 2, the amino acid sequence segments are presented such that ‘/’ indicates the scissile bond in cathepsin K. The range of the lowest and highest individual scores as well as the median score of each 8 residue sequence is also presented. Schematics of cathepsin K with the location of hydrolysis and the fragment sizes generated were also added to Figure 2. Five of the 16 sites were eliminated because they were in the propeptide region (the first 114 residues of cathepsin K), which would have already been cleaved off during activation to the mature enzyme (numbers 3, 8, 9, 14, and 15 in Fig. 2 with grayed text). The other eleven sites were inspected for their accessibility using Swiss‐Pdb Viewer 4.1.0 of Cathepsin K (PDB ID code: 1ATK) (Fig. 3): residues on cathepsin K's surface exposed to the aqueous environment and residues in unstructured regions were given higher priority over more internal residues because of their accessibility to cleavage by cathepsin S.
Figure 2.
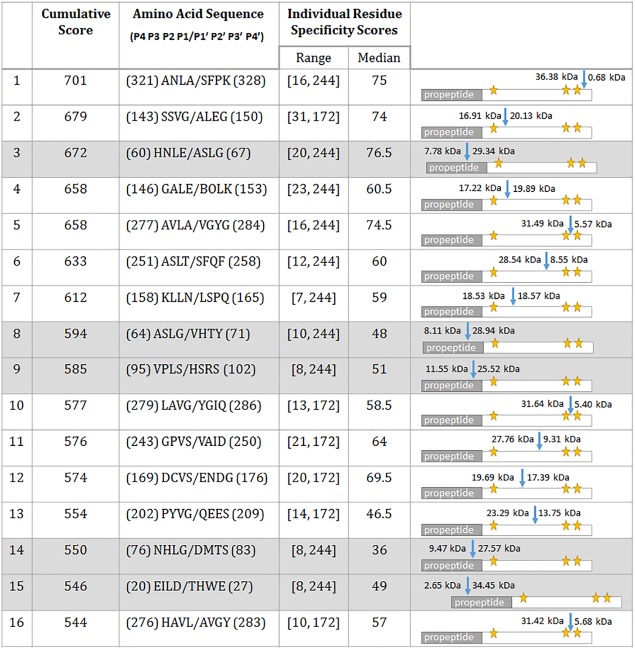
Top 5% of cumulative cleavage scores of cathepsin S cannibalizing cathepsin K. The table includes the highest‐scored regions of where cleavage of cathepsin K by cathepsin S is most likely to occur as well as descriptive information about the range and median residue scores and a schematic of where cleavage was expected to occur within the cathepsin K protein. The pro‐region of cathepsin K includes residues 1 to 114, which were excluded when determining cathepsin K mutants to interrupt cathepsin cannibalism. The ‘/’ in the amino acid sequences indicates the scissile bond that would be hydrolyzed by cathepsin S. Schematics in the right column have a blue arrow indicating the hydrolysis would occur along with subsequence fragment sizes. Yellow stars indicate the active site residues for cathepsin K.
Figure 3.
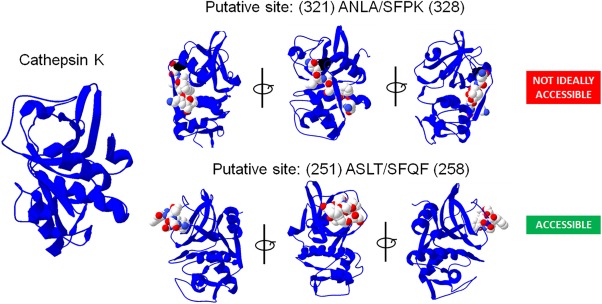
Cathepsin K putative sites of cleavage locations reviewed for accessibility to cleavage. A cathepsin K protein data bank (PDB) model was downloaded (PDB ID: 1ATK) and sequences identified in Figure 2 were reviewed for external accessibility by a protease‐ase using Swiss‐Pdb Viewer 4.1.0.
Sequence #6 ASLT/SFQF and #12 DCVS/ENDG met these criteria. L253 and V171were identified as key locations that fit all criteria and the most probable sites of cathepsin K cannibalism by cathepsin S.
Validation of PACMANS predictions with site‐directed mutagenesis
To validate that these amino acid residues were important for hydrolysis of cathepsin K by cathepsin S, simulated mutations in PACMANS were analyzed. Mutating those two positions greatly reduced the overall cumulative score (L253A: 633 to 411; L253V: 633 to 561; V171A: 574 to 424) of those sequences. Site‐directed mutagenesis at L253 and V171 was completed. They were mutated to alanine and valine30 for small residues, whereas mutating to a bulky, charged residue, such as glutamic acid, would reduce the PACMANS overall score even more (L253E: 633 to 391), but could be more disruptive to protein folding which we wanted to retain.
Next, mutations were introduced to test the hypothesis that mutating PACMANS identified sites (L253, V171) would yield cannibalism‐resistant cathepsin K mutants. Mutations were introduced into the cathepsin K sequence using overlap extension PCR on a plasmid with six tandem histidine residues (6xHis‐tagged) at the C‐terminal end of cathepsin K, confirmed by sequencing, then transfected into HEK293T cells. Active cathepsin K in eukaryotic cell lysates in zymography yields a signal at 37 kDa under non‐reducing conditions to preserve disulfide bonds in cathepsin structure, but that same sample under reducing conditions would yield a 29 kDa protein detected in the Western blot. This was validated in our previous publications establishing the use of zymography for cathepsin activity.31 Translation of full length mutant cathepsin K proteins were verified by immunoblotting (reducing SDS‐PAGE), and cathepsin zymography (a non‐reducing SDS‐PAGE method) was used to test if they were proteolytically active [Fig. 4(A)]. L253A, V171A, and L253V were expressed as full length proteins but, L253V did not yield an enzymatically active protein in these studies, but L253A and V171A did, as confirmed by the zymogram [Fig 4(A)]. There is a ∼15kDa immunoreactive band detected, which may be a degradation product, but it appears both with and without co‐incubation with cathepsin S, so is not due to cannibalism. Mature cathepsin K does not appear in the Western blots, but the active/mature form is verified by zymogram, and we have previously shown that zymography has a lower limit of detection than Western blotting.32
Figure 4.
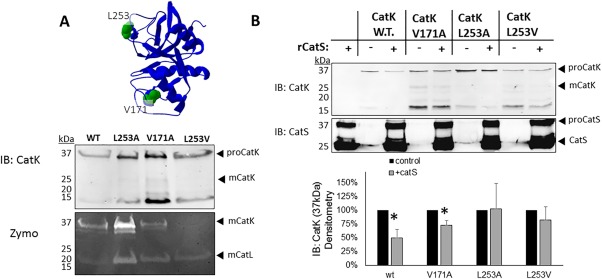
Expression of cleavage‐site cathepsin K mutants yield full length protein and are protected from degradation by cathepsin S. (A) HEK293T cells were stably transfected with a plasmid for each of the His‐tagged cathepsin K mutants. Cell pellets were collected and lysed and prepared for immunoblotting to determine if protein was produced, and cathepsin zymography to test for active cathepsin K with the mutations. Active cathepsin K in zymography yields a signal at 37 kDa under non‐reducing conditions, but that same sample run under reducing conditions would yield a 29 kDa protein detected in the Western blot. (B) One picomole of recombinant cathepsin S was co‐incubated with purified cathepsin K wildtype or cleavage site cathepsin K mutants for 2 hours at 37°C. Cathepsin S degraded 50% of wildtype (w.t.) cathepsin K (n = 3, P < 0.05). Mutations at V171 showed partial protection of 25% (n = 3, P < 0.05), but mutations at L253 protected cathepsin K with no statistically significant difference, in the presence or absence of cathepsin S (n = 3). Densitometry is quantified in the graph below.
Recombinant cathepsin K mutants were also purified from cell lysates and incubated in the presence or absence of recombinant cathepsin S to determine resistance to degradation. Quantification of immunoblots after two hours of co‐incubation with cathepsin S indicated a 50% reduction in wildtype (WT) cathepsin K (n = 3, P < 0.02), compared to when incubated alone, only a 27% reduction in V171A cathepsin K protein (n = 3, P < 0.02), and no significant reduction in protein for the L253A or the L253V cathepsin K mutants (n = 3) [Fig. 4(B)]. This indicates that PACMANS was correctly able to identify a putative site of cleavage of cathepsin K by cathepsin S, which could be mitigated by bioinformatically informed mutagenesis.
Inter‐familial protease‐on‐protease interactions: cathepsin S, MMP‐2, and MMP‐9
In our design of PACMANS, we wanted to provide versatility for protease selection of any protease with information in MEROPS, not just cysteine cathepsins. PACMANS was applied to study interfamilial proteolysis. Matrix metalloproteinases (MMPs) are another family of proteases implicated in similar tissue destructive diseases as cysteine cathepsins, MMP‐2 and MMP‐9 in particular,4, 33, 34, 35, 36 and interfamilial proteolysis can impact substrate degradation similarly to intrafamilial proteolysis. We used PACMANS to test the hypothesis that cathepsin S could degrade MMP‐2 and MMP‐9. To do so, cathepsin S was input into PACMANS as the protease‐ase and either MMP‐2 or MMP‐9 as the substrate protease, the maximum cumulative scores of potential protease‐on‐protease interactions between cathepsin S and MMP‐2 and 9 were plotted [Fig. 5(A)]. This procedure was repeated for MMP‐2 [Fig. 5(B)] and MMP‐9 [Fig. 5(C)] as the protease‐ase as well, with cathepsin S being the substrate. Cumulative likelihood scores for each protease‐ase's hydrolysis of type 1 collagen and elastin were also analyzed by PACMANS to compare with likelihood scores of substrate proteases as a comparative metric for likelihood of cleavage.
Figure 5.
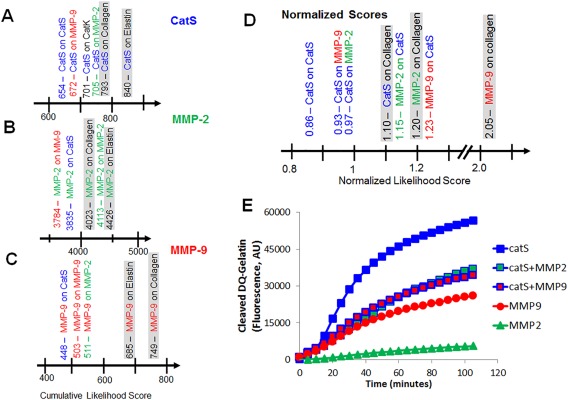
PACMANS predictions and validation of protease‐on‐protease interactions causing a reduction in gelatinase activity of cathepsin S when co‐incubated with MMP‐2 or MMP‐9. Using cathepsin S, MMP‐2, and MMP‐9's specificity matrices and the amino acid sequence of other enzymes as the substrate protease, the maximum cumulative cleavage likelihood were plotted, specifically focusing on (A) cathepsin S, (B) MMP‐2, and (C) MMP‐9 as the protease‐ases of interest. PACMANS was further refined to calculate normalized scores to allow users to make comparisons among cathepsin S, MMP‐2, and MMP‐9 with their collagen scores as markers (D). Fluorogenic gelatin was incubated individually and in pairs of cathepsin S (catS), MMP‐2, and MMP‐9 (E). Cathepsin S alone degraded the most gelatin, but when co‐incubated with equal amounts of either MMP‐2 or MMP‐9, the total gelatin degradation was approximately half that of cathepsin S alone. Collagen alpha‐1(I) chain (COL1A1) was used for comparative analysis. The FASTA sequences was retrieved from UniProt with the following link: http://www.uniprot.org/uniprot/P02452.
The initial intention was to use the PACMANS scores from traditional extracellular matrix substrates collagen I and elastin as a normalization strategy to compare between protease‐ases, but the magnitude of the scores would have different ranges based on the number of papers published on the protease‐ase. Maximum score was 840 for cathepsin S, 4426 for MMP‐2, and 749 for MMP‐9. The reason for this is that the MEROPS specificity matrix contains the frequency that a given residue has been reported in publications for each subsite. This was not constant when comparing across different proteases in this study; MMP‐2 has more published substrate specificities analyzed and therefore generally have higher frequency values in their specificity matrix than other proteases such as cathepsin V with less known substrate preferences published. As a result, the magnitude of the cumulative score was greater for proteases with a greater number of known substrates and preferences. To enable comparisons across protease‐ases, an effective normalization strategy was developed. By dividing the individual likelihood score for a position by the total possible score of that subsite for all amino acids; , the normalized score for a residue in subsite , can be described mathematically as:
where is amino acid (row), is subsite (column), and is specificity matrix. Norm means normalized. The sum of the normalized scores, , can be used to more accurately compare cleavage potential across multiple protease‐ases, as shown below.
Using normalized scores, cathepsin S hydrolysis of MMP‐2 (score of 0.97) scored higher than cathepsin K (score of 0.95), which was higher than MMP‐9 (score of 0.93). MMP‐2, as the protease‐ase, was more likely to hydrolyze cathepsin S (score of 1.25), than MMP‐9 (score of 0.88) [Fig. 5(D)]. This suggested that MMP‐2 would preferentially hydrolyze cathepsin S more than vice versa and that co‐incubation would reduce cathepsin S's degradation ability. PACMANS‐based hypotheses were then tested experimentally by co‐incubating cathepsin S with MMP‐2 or MMP‐9 and monitoring cleavage of a fluorescently quenched gelatin substrate; increased fluorescence indicates substrate degradation. By itself, cathepsin S degraded more gelatin than the other proteases with MMP‐2 degrading 10.3% as much and MMP‐9 degrading 46.0% as much gelatin as cathepsin S alone [Fig 5(E)]. When co‐incubating with cathepsin S, only 65.2% of total gelatin was degraded with MMP‐2, and only 60.5% with MMP‐9 compared to cathepsin S alone (100%), even though there was initially twice the molar amount of proteases in the system [Fig. 5(E)]. If no interactive proteolysis was occurring, then the results from co‐incubation would be expected to be the sum of each individual protease's activity. This suggests that MMP‐2 and MMP‐9 were reducing the amount of active cathepsin S in the system by cleaving it, or were distracting cathepsin S from degrading the gelatin serving as an alternative substrate for cathepsin S to bind, or both, which supports the hypothesis from PACMANS that MMP‐2 and MMP‐9 could cleave cathepsin S when co‐incubated, and to some degree, vice versa.
Discussion
PACMANS uses a bioinformatics approach to analyze the primary structure of a protease (substrate protease) and determines possible locations that could be vulnerable to hydrolysis by another protease (here termed, a protease‐ase), PACMANS is unique compared to other putative site cleavage programs, by allowing users to define the protease of interest, and utilizing the specificity matrix from MEROPS to rank the most likely locations of cleavage. Although this program was initially conceived to analyze proteases cleaving other proteases, which could be bidirectional, it can also be used for traditional, non‐protease substrate proteins, as well as short peptide sequences. Putative sites on cathepsin K susceptible to cleavage by cathepsin S were identified by PACMANS, confirming its ability to score sites based on protease‐ase preferences. Cathepsin S prefers moderately‐sized, branched hydrophobic residue (such as leucine in L253) in the P2,37, 38, 39 and these were detected in the top 5% of putative sites on cathepsin K. Introducing mutations into cathepsin K at sites L253 and V171, protected cathepsin K from cleavage by cathepsin S (Fig. 4). This validated PACMANS as a useful tool for predicting locations of protease‐on‐protease interactions and cleavage. Taken together, this confirms cathepsin cannibalism between cathepsin S and cathepsin K and that its outcomes can be modified by mutational analysis and application.
Other protease families may undergo similar proteolytic interactions between family members, but we also used PACMANS to demonstrate the possibility of inter‐familial regulation of proteases using cathepsin S/MMP‐2,‐9 studies. PACMANS predicted interactions and preferential hydrolysis directions between cathepsin S vs. MMP‐2 and MMP‐9 (which are both gelatinases), were then tested experimentally by monitoring gelatinase activity of these proteases in isolation, in concert, or against each other. PACMANS analysis indicates that both MMP‐2 and −9 have greater preference for cleaving cathepsin S, then cathepsin S has for cleaving them, and we observed experimentally that cathepsin S gelatinase activity was reduced when co‐incubated with MMP‐2 or MMP‐9 compared to its incubation by itself indicating interactions between MMP‐2 and cathepsin S and of MMP‐9 with cathepsin S were reducing cathepsin S activity. Cathepsin S could be acting as a decoy or distraction for MMPs, causing them to bind to and hydrolyze cathepsin S preferentially over the gelatin substrate, reducing the amount of gelatin being degraded over the same unit time. If MMP‐2/‐9 are cleaving cathepsin S, there is the additional consequence of inactivating a fraction of cathepsin S, preventing that amount of active enzyme from degrading gelatin resulting in a net reduction in total gelatinase activity. This reduced gelatinase activity observed occurred even when there was twice the molar amount of active protease present since equimolar amounts were added for each of the experimental conditions [Fig. 5(E)].
Expanding PACMANS to test inter‐familial protease relationships between cathepsin S and MMP‐2/MMP‐9, it became clear that the initial scores calculated from MEROPS were not comparable across protease‐ases. The protease‐ase scores clustered depending on the magnitude of scores in the MEROPS specificity matrices, rather than just biological preference. For example, MMP‐9 is known to be a strong collagenase,4 however, based solely on the scores, MMP‐2 preference for collagen, elastin, as well as cathepsin S, MMP‐2, and MMP‐9 was greater than MMP‐9's score for degrading collagen. To compare across cathepsin S, MMP‐2, and MMP‐9, a normalized score was developed, which normalized the specificity matrix by the total number of substrates used to generate it. After normalization, protease‐ase preferences could be more directly compared and conclusions made based on experimental data [Fig. 5(D), 5(E)]. MMP‐9 was then deemed the strongest collagenase (score 2.05) followed by MMP‐2 (score 1.20) and cathepsin S (score 1.10), which better approximates published comparisons.4
PACMANS customizability allows users to input amino acid sequences and proteases of interest. Other programs developed to determine proteolytic cleavage sites, more commonly have a predefined, set list of proteases available for analysis. PROSPER40 and PeptideCutter41 are two programs that differ from PACMANS in that they analyze a set list of enzymes to cleave the amino acid sequence, while PACMANS's only limit is that the protease must have a specificity matrix characterized in MEROPS (which as of the time of this submission is 263). PeptideCutter did not have any of the cysteine cathepsins, and PROSPER only included cathepsin K.
Prediction of Protease Specificity (PoPS) is a robust program similar to PACMANS, but users must manually type each subsite's specificities for each amino acid by hand (which can be error‐prone)42 instead of directly linking to MEROPS like PACMANS. Furthermore, PoPS has the user choose between scaling methods. PACMANS normalizes the specificity matrix and computes a normalized score for comparison. PoPS gave similar results to PACMANS with the sequence (143) SSVG/ALEG (150) having a maximum score of 17.83, which was the second entry in PACMANS results (Fig. 2). However, the sequences containing L253 (rank 9, score: 9.28) and V171 (rank 19, score: 5.85) in the P2 locations were lower ranked in the top scores from PoPS and were shown from the PACMANS identified sites as key locations for interrupting hydrolysis.
In summary, PACMANS is now added to other tools that can be used to test basic scientific questions for proteolysis, as well as design interventions in tissue‐destructive diseases. PACMANS also can be used to design mutant proteins that can be therapeutically resistant or enhanced for protease‐on‐protease interactions. For example, if design goals were for cathepsin K to be degraded quickly for some biotechnology application or for some biologic manufacturing tasks, then instead of expressing L253A mutants resistant to cannibalism, one could express T254G or Q257P; both of these mutants would increase likelihood scores to enhance its degradation by cathepsin S. Understanding protease‐on‐protease interactions can also allow for accurate dosing of protease inhibitors, considering the activities of protease‐on‐protease interactions that can generate off‐target side effects caused by miscalculations of protease concentrations in pharmacological analyses. PACMANS offers an inexpensive in silico method to perform first pass analyses prior to the mutagenesis or more costly methods to confirm, which will save time and money for proteolysis researchers.
Materials and Methods
Algorithm and program workflow
Protease‐Ase Cleavages from MEROPS ANalyzed Specificities (PACMANS) is a program that determines putative cleavage site locations of a dominant protease (protease‐ase) hydrolyzing a substrate protease (Fig. 1). Putative locations were determined by point‐by‐point comparison of a substrate protease's amino acid sequences with a protease‐ase's active site specificity matrix (available from the MEROPS online peptidase database) to score the frequency each amino acid has been found in subsite P4 through P4′, and is susceptible to cleavage in the protease‐ase's active site.27
PACMANS was developed using a sliding‐window approach, where individual sub‐sequences that fill the P4 to P4′ active site pocket were scored. This pocket slides along the length of the substrate amino acid sequence and all possible sub‐sequences were scored and then sorted by score. This program was implemented using MATLAB. Pseudocode for the program flow of PACMANS is given below:
Main inputs: sequence (text string of protein under analysis) and specmat (protease‐ase matrix of specificity values)
Determine pocket size using starting and end points (testlen)
Determine number of 1‐8 amino acid sequences to be analyzed (numsequences)
for i = 1:numsequences
Determine current sequence (curseq)
for j = 1:length(curseq)
Determine the individual amino acid preference score for the j‐th amino acid in the current sequence and store these scores in a matrix
Add individual scores together to determine cumulative sequence score
end
Calculate the range and median of individual residue scores as well as subsequent fragment size for each sequence
end
Scores matrix was then sorted by maximum cumulative score and printed out to a text file
Specialized MATLAB functions utilized in these calculations included molweight()[from the Bioinformatics Toolbox], which calculates the molecular weight of a given amino acid sequence and quantile() [from the Statistics and Machine Learning Toolbox], which returns the quantiles of a dataset, in this case the scores, to determine the top five‐percent of scores for further analysis. The computational complexity of this algorithm is O(n), where n is the number of amino acids in the input sequence.
The functionality of PACMANS was further extended when used to compare substrate preferences between different protease‐ases. The code was modified to calculate a normalized score, where the individual residue scores were normalized to the number of substrates used to fill the specificity matrix and then summed, like the original cumulative scoring method.
The code has been packaged into a graphical user interface (GUI) using MATLAB's GUIDE Toolbox to allow use by a broader audience, which will be able to be downloaded and run as a MATLAB Application and the code was also re‐implemented in PHP to host online (platt.gatech.edu/PACMANS). The functionality of the code was also adapted to allow the user to vary the size of the active site pocket, to a subset of the P4 to P4′ used in this work.
Cathepsin K mutagenesis
pCMVA6 plasmid containing the gene for human cathepsin K was originally purchased from OriGene (CTSK (NM_000396) Human cDNA clone). The mutant cathepsins were made by site‐directed mutagenesis using overlap extension polymerase chain reaction (PCR) with mutagenic primers to introduce point‐mutations into cathepsin K gene (Supporting Information Table S1),43 which was then ligated into pcDNA 4/TO/A plasmid (Invitrogen) that contains a C‐terminal polyhistidine tag for nickel‐column purification. Recombined plasmid was amplified in competent bacteria cells (Strain Zymo 5α, Zymo Research) and successful mutagenesis was confirmed with DNA sequencing by Genewiz.
Stable expression and purification of mutant cathepsin K
HEK293T cells were stably transfected using Lipofectamine 2000 (Thermo‐Fisher Scientific) and zeocin (Invivogen) as the selection marker for steady production of mutant cathepsins. C‐terminal 6X polyhistidine tagged cathepsin K (wildtype and mutants) were purified from cell lysates with the ProBond Purification System (Thermo‐Fisher Scientific).
Multiplex cathepsin zymography
The multiplex cathepsin zymography assay was used to determine the amount of active cathepsin by the protocol previously established by our lab.31, 32
Western blotting
Western blots were performed on proteins either from cell lysate (extracted using modified RIPA buffer) or purified protein in co‐incubation experiments to determine levels of detectable protein. To test the cannibalism‐resistance of cleavage site mutants, purified cathepsin mutants were co‐incubated with or without 1 pmol of recombinant cathepsin S (Enzo) in a phosphate buffer (pH 6.0), 2mM dithiothreitol (DTT), and 1mM ethylenediamine tetraaceticacid (EDTA, Fischer Scientific) for two hours at 37°C. The reaction was quenched by the additional of a reducing loading dye and then samples were boiled for 5 minutes before loading into Western blot gels. SDS‐PAGE was performed using a 12.5% SDS polyacrylamide (National Diagnostics) gel and resolved at 150V at room temperature to separate proteins by molecular weight and then transferred onto a nitrocellulose membrane (Bio‐Rad) using a Trans‐Blot SD Semi‐Dry Transfer Cell (Bio‐Rad). Membranes were blocked for one hour with blocking buffer (Odyssey, diluted 1:1 with PBS) and incubated over night with either a monoclonal mouse primary cathepsin K antibody (Millipore) or a polyclonal goat primary cathepsin S antibody (R&D Biosystems). Proteins were imaged on a LI‐COR Odyssey scanner using donkey anti‐mouse or anti‐goat secondary antibodies (Invitrogen) tagged with an infrared fluorophore. Densitometry to quantify band intensity (indicating protein amount) was performed using ImageJ (NIH). Results are presented with a mean ± standard error of the mean (SEM). Statistical analysis performed was a two‐tailed, Student's t‐test.
Fluorescent gelatinase activity assay
Five pmol of recombinant cathepsin S (Enzo), MMP2 (Enzo), or MMP9 (Enzo) was incubated at 37°C individually or in combinations with 50 μg/ml of DQ gelatin (Thermo‐Fisher) in phosphate buffer, pH 6.0, and 2mM DTT. Changes in fluorescence were measured at an excitation in 485nm and emission of 525nm with a SpectraMax Plus microplate reader (Molecular Devices).
Supporting information
Supporting Information Table 1.
Supporting Information.
Impact Statement: Proteases are enzymes that hydrolyze peptide bonds, and multiple proteases interact with each other in cellular environments. PACMANS is a program to help identify sequences where one protease degrades another to elucidate these protease‐on‐protease interactions that can alter expected kinetics of enzyme‐substrate systems, and to motivate experiments to test the predictions. From PACMANS analysis, scientists can engineer proteins to counteract or exploit the effects of these interactions.
References
- 1. Brix K, Dunkhorst A, Mayer K, Jordans S (2008) Cysteine cathepsins: cellular roadmap to different functions. Biochimie 90:194–207. [DOI] [PubMed] [Google Scholar]
- 2. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824:68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chapman HA, Riese RJ, Shi GP (1997) Emerging roles for cysteine proteases in human biology. Annu Rev Physiol 59:63–88. [DOI] [PubMed] [Google Scholar]
- 4. Garnero P, Borel O, Byrjalsen I, Ferreras M, Drake FH, McQueney MS, Foged NT, Delmas PD, Delaisse JM (1998) The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J Biol Chem 273:32347–32352. [DOI] [PubMed] [Google Scholar]
- 5. Bromme D, Li Z, Barnes M, Mehler E (1999) Human cathepsin V functional expression, tissue distribution, electrostatic surface potential, enzymatic characterization, and chromosomal localization. Biochemistry 38:2377–2385. [DOI] [PubMed] [Google Scholar]
- 6. Aguda AH, Panwar P, Du X, Nguyen NT, Brayer GD, Bromme D (2014) Structural basis of collagen fiber degradation by cathepsin K. Proc Natl Acad Sci U S A 111:17474–17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Littlewood‐Evans AJ, Bilbe G, Bowler WB, Farley D, Wlodarski B, Kokubo T, Inaoka T, Sloane J, Evans DB, Gallagher JA (1997) The osteoclast‐associated protease cathepsin K is expressed in human breast carcinoma. Cancer Res 57:5386–5390. [PubMed] [Google Scholar]
- 8. Brubaker KD, Vessella RL, True LD, Thomas R, Corey E (2003) Cathepsin K mRNA and protein expression in prostate cancer progression. J Bone Miner Res 18:222–230. [DOI] [PubMed] [Google Scholar]
- 9. Mohamed MM, Sloane BF (2006) Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 6:764–775. [DOI] [PubMed] [Google Scholar]
- 10. Chen B, Platt MO (2011) Multiplex zymography captures stage‐specific activity profiles of cathepsins K, L, and S in human breast, lung, and cervical cancer. J Transl Med 9:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park KY, Li G, Platt MO (2015) Monocyte‐derived macrophage assisted breast cancer cell invasion as a personalized, predictive metric to score metastatic risk. Sci Rep 5:13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olson OC, Joyce JA (2015) Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat Rev Cancer 15:712–729. [DOI] [PubMed] [Google Scholar]
- 13. Lutgens E, Lutgens SP, Faber BC, Heeneman S, Gijbels MM, de Winther MP, Frederik P, van der Made I, Daugherty A, Sijbers AM, Fisher A, Long CJ, Saftig P, Black D, Daemen MJ, Cleutjens KB (2006) Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation 113:98–107. [DOI] [PubMed] [Google Scholar]
- 14. Lecaille F, Bromme D, Lalmanach G (2008) Biochemical properties and regulation of cathepsin K activity. Biochimie 90:208–226. [DOI] [PubMed] [Google Scholar]
- 15. Lafarge JC, Naour N, Clement K, Guerre‐Millo M (2010) Cathepsins and cystatin C in atherosclerosis and obesity. Biochimie 92:1580–1586. [DOI] [PubMed] [Google Scholar]
- 16. Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, Kodama T, Tsimikas S, Witztum JL, Lu ML, Sakara Y, Chin MT, Libby P, Shi GP (2003) Deficiency of cathepsin S reduces atherosclerosis in LDL receptor‐deficient mice. J Clin Invest 111:897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Platt MO, Ankeny RF, Shi GP, Weiss D, Vega JD, Taylor WR, Jo H (2007) Expression of cathepsin K is regulated by shear stress in cultured endothelial cells and is increased in endothelium in human atherosclerosis. Am J Physiol Heart Circ Physiol 292:H1479–H1486. [DOI] [PubMed] [Google Scholar]
- 18. Samokhin AO, Lythgo PA, Gauthier JY, Percival MD, Bromme D (2010) Pharmacological inhibition of cathepsin S decreases atherosclerotic lesions in Apoe‐/‐ mice. J Cardiovasc Pharmacol 56:98–105. [DOI] [PubMed] [Google Scholar]
- 19. Platt MO, Shockey WA (2016) Endothelial cells and cathepsins: Biochemical and biomechanical regulation. Biochimie 122:314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hou WS, Li W, Keyszer G, Weber E, Levy R, Klein MJ, Gravallese EM, Goldring SR, Bromme D (2002) Comparison of cathepsins K and S expression within the rheumatoid and osteoarthritic synovium. Arthritis Rheum 46:663–674. [DOI] [PubMed] [Google Scholar]
- 21. Porter KM, Wieser FA, Wilder CL, Sidell N, Platt MO (2016) Cathepsin Protease Inhibition Reduces Endometriosis Lesion Establishment. Reprod Sci 23:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seto SP, Parks AN, Qiu Y, Soslowsky LJ, Karas S, Platt MO, Temenoff JS (2015) Cathepsins in Rotator Cuff Tendinopathy: Identification in Human Chronic Tears and Temporal Induction in a Rat Model. Ann Biomed Eng 43:2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barry ZT, Platt MO (2012) Cathepsin S cannibalism of cathepsin K as a mechanism to reduce type I collagen degradation. J Biol Chem 287:27723–27730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McQueney MS, Amegadzie BY, D'Alessio K, Hanning CR, McLaughlin MM, McNulty D, Carr SA, Ijames C, Kurdyla J, Jones CS (1997) Autocatalytic activation of human cathepsin K. J Biol Chem 272:13955–13960. [DOI] [PubMed] [Google Scholar]
- 25. Menard R, Carmona E, Takebe S, Dufour E, Plouffe C, Mason P, Mort JS (1998) Autocatalytic processing of recombinant human procathepsin L. Contribution of both intermolecular and unimolecular events in the processing of procathepsin L in vitro. J Biol Chem 273:4478–4484. [DOI] [PubMed] [Google Scholar]
- 26. Vasiljeva O, Dolinar M, Pungercar JR, Turk V, Turk B (2005) Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans. FEBS Lett 579:1285–1290. [DOI] [PubMed] [Google Scholar]
- 27. Rawlings ND, Waller M, Barrett AJ, Bateman A (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 42:D503–D509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rawlings ND, Barrett AJ, Finn R (2016) Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 44:D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schechter I, Berger A (1967) On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 425:497–502. [DOI] [PubMed] [Google Scholar]
- 30. Bordo D, Argos P (1991) Suggestions for “safe” residue substitutions in site‐directed mutagenesis. J Mol Biol 217:721–729. [DOI] [PubMed] [Google Scholar]
- 31. Wilder CL, Park KY, Keegan PM, Platt MO (2011) Manipulating substrate and pH in zymography protocols selectively distinguishes cathepsins K, L, S, and V activity in cells and tissues. Arch Biochem Biophys 516:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li WA, Barry ZT, Cohen JD, Wilder CL, Deeds RJ, Keegan PM, Platt MO (2010) Detection of femtomole quantities of mature cathepsin K with zymography. Anal Biochem 401:91–98. [DOI] [PubMed] [Google Scholar]
- 33. Abdul‐Hussien H, Soekhoe RG, Weber E, von der Thusen JH, Kleemann R, Mulder A, van Bockel JH, Hanemaaijer R, Lindeman JH (2007) Collagen degradation in the abdominal aneurysm: a conspiracy of matrix metalloproteinase and cysteine collagenases. Am J Pathol 170:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emmert‐Buck MR, Roth MJ, Zhuang Z, Campo E, Rozhin J, Sloane BF, Liotta LA, Stetler‐Stevenson WG (1994) Increased gelatinase A (MMP‐2) and cathepsin B activity in invasive tumor regions of human colon cancer samples. Am J Pathol 145:1285–1290. [PMC free article] [PubMed] [Google Scholar]
- 35. Mason SD, Joyce JA (2011) Proteolytic networks in cancer. Trends Cell Biol 21:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Platt MO, Xing Y, Jo H, Yoganathan AP (2006) Cyclic pressure and shear stress regulate matrix metalloproteinases and cathepsin activity in porcine aortic valves. J Heart Valve Dis 15:622–629. [PubMed] [Google Scholar]
- 37. Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik CS, Alves FM, Hirata IY, Gouvea IE, Alves MF, Meldal M, Bromme D, Juliano L, Juliano MA, Bromme D, Bonneau PR, Lachance P, Storer AC, Menard R, Carmona E, Plouffe C, Bromme D, Konishi Y, Lefebvre J, Storer AC (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem 281:12824–12832. [DOI] [PubMed] [Google Scholar]
- 38. Menard R, Carmona E, Plouffe C, Bromme D, Konishi Y, Lefebvre J, Storer AC (1993) The specificity of the S1' subsite of cysteine proteases. FEBS Lett 328:107–110. [DOI] [PubMed] [Google Scholar]
- 39. Bromme D, Klaus JL, Okamoto K, Rasnick D, Palmer JT (1996) Peptidyl vinyl sulphones: a new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem J 315 (Pt 1):85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Song J, Tan H, Perry AJ, Akutsu T, Webb GI, Whisstock JC, Pike RN (2012) PROSPER: an integrated feature‐based tool for predicting protease substrate cleavage sites. PLoS One 7:e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gasteiger EH, C.; Gattiker, A. ; Duvaud, S. ; Wilkins, M.R. ; Appel, R.D. , Bairoch, A. Protein identification and analysis tools on the ExPASy server In: Walker JM, Ed. (2005) The proteomic protocols handbook. Humana Press, Totowa, New Jersey, pp. 571–607. [Google Scholar]
- 42. Boyd SE, Pike RN, Rudy GB, Whisstock JC, Garcia de la Banda M (2005) PoPS: a computational tool for modeling and predicting protease specificity. J Bioinform Comput Biol 3:551–585. [DOI] [PubMed] [Google Scholar]
- 43. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site‐directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table 1.
Supporting Information.