

Abstract
Plant viral nanoparticles (VNPs) are a novel class of nanocarriers with implications for drug delivery in cancer therapy. VNPs are characterized by their highly symmetrical nanoscale structures. Furthermore, plant VNPs are biocompatible, biodegradable, and non-infectious in mammals. VNPs provide a proteinaceous platform technology that can be readily engineered to carry contrast agents and therapies using chemical and genetic modifications. Of particular interest are high aspect ratio, elongated filaments such as the ones formed by potato virus X (PVX, measuring 515 × 13 nm). PVX has demonstrated enhanced tumor homing and penetration properties compared to spherical counterparts. Here, we sought to investigate the potential of PVX as a drug carrier delivering doxorubicin (DOX), a commonly used cancer chemotherapy. We synthesized therapeutic PVX nanoparticles using a simple in-solution mixing protocol; after 5 days of mixing of DOX and PVX and ultra-centrifugal purification, ~1000 DOX per PVX were stably associated with the carrier, most likely based on hydrophobic interaction. Efficacy and drug activity of PVX-DOX were confirmed using a panel of cancer cell lines including ovarian cancer, breast cancer, and cervical cancer. Lastly, we demonstrated treatment of athymic mice bearing human MDA-MB-231 breast cancer xenografts: PVX-DOX treatment resulted in reduced tumor growth in this model. Our results open the door for further development of PVX and other high aspect ratio plant VNPs for applications in cancer therapy.
Graphical Abstract

1. Introduction
A myriad of nanoparticle platform technologies is currently being developed as drug delivery vehicles with potential applications in cancer treatment.1–3 These nanovehicles, generally less than 100 nm, have many advantages that overcome the hurdles in using traditional therapeutics; many small compound therapeutics exhibit short plasma circulation times, poor solubility in aqueous media, and non-specific biodistribution. Nanoparticles hold potential to overcome these challenges and enhance the bioavailability and biodistribution. Having large surface-to-volume ratio, nanoparticles allow for high payload delivery. Furthermore, nanoparticles can be decorated with appropriate surface chemistries to enhance their biocompatibility, tissue-specificity, and therefore overall efficacy.
There are multiple choices ranging from lipid-based micelles, polymeric capsules, inorganic carriers such as iron oxide or gold nanoparticles, and protein-based nanoparticles.4–8 Traditionally, most of the explored systems are spherical. However, mounting data suggest that elongated, filamentous nanomaterials have advantageous properties compared to their spherical counterparts.9–12 Rods with high aspect ratio (AR>5) have demonstrated decreased nonspecific uptake by phagocytic cells and improved extravasation, leading to higher tumor penetration and accumulation.13,14
However, to date it has been proven challenging to manufacture high aspect ratio nanoparticles with good homogeneity – a critical factor in quality control and assurance of translational nanomedicine. Currently available high aspect ratio nanomaterials include filomicelles, carbon nanotubes, and PRINT nanoparticles. Filomicelles are assembled from block copolymers such as polystyrene-b-poly(ethylene oxide) resulting in a worm-like micelles, but these are typically on the micron size scale.15,16 Carbon nanotubes have great control in size and shape but are not biocompatible as they accumulate in the lungs and persist in the body over prolonged time periods.17, 18 While highly sophisticated, challenges remain to manufacture PRINT nanoparticles with dimensions below 50 nm.19 Viruses, which can be regarded as naturally occurring, pre-formed nanoparticles, overcome the manufacturing challenges; mammalian viruses, plant viruses, and bacteriophages come in many shapes and sizes and several platforms form filamentous particles; examples include Ebola virus, potato virus X (PVX), or M13 phage. Here we turned toward the development of PVX – a plant-produced viral nanoparticle (VNP) platform technology.
VNPs, in contrast to other synthetic materials, are formed from the assembly of naturally occurring coat proteins around RNA templates, resulting in highly symmetrical structures and excellent mono-dispersity.20,21 Additionally, plant VNPs are biocompatible, biodegradable, and non-pathogenic in mammals. The proteinaceous scaffold is amenable to chemical and genetic modification, thus allowing the programmable engineering as a drug carrier. Finally, the production is scalable through molecular farming in plants, e.g. Nicotiana benthamiana.
PVX forms flexible filamentous nanocarriers measuring 515 nm in length and 13 nm in width.22 PVX has been shown to target to tumors by passive accumulation via the enhanced permeability and retention (EPR) effect23 or active targeting by means of presentation of receptor-specific ligands.24 Analogous to other elongated materials, PVX exhibits enhanced tumor homing and tissue penetration compared to the spherical cowpea mosaic virus (CPMV) in a variety of cancer models including human tumor xenografts of fibrosarcoma, colon cancer, and breast cancer.23,25 We also demonstrated that the in vivo fate of PVX could be further enhanced by conjugation of stealth polymers such as polyethylene glycol (PEG) via the solvent-exposed lysine side chains.26 Together, these characteristics motivated us to explore the use of PVX platform for drug delivery in chemotherapy.
Here, we demonstrate the use of PVX for delivery of doxorubicin (DOX), an FDA-approved anti-tumor drug used in treatments of various malignancies including breast cancer, ovarian cancer, acute lymphoblastic leukemia, Hodgkin’s lymphoma, and small cell lung cancer.27,28 DOX is an anthracycline that kills cancer cells via three mechanisms of action: (1) intercalating with DNA, (2) inhibiting topoisomerase II, and (3) producing reactive oxygen species.27 However, like all chemotherapies, when administered systemically, it is associated with off-target effects due to non-specific interaction with healthy cells, ranging in severity from hair loss to heart failure.29,30 Herein, we loaded DOX onto the surface of PVX by hydrophobic interactions and characterized the therapeutic nanoparticles (DOX-loaded PVX) using transmission electron microscopy, gel electrophoresis, and UV/visible spectroscopy. The efficacy of the approach was then examined in a panel of cancer cells in vitro. Lastly, we assessed efficacy using a mouse model of triple negative breast cancer using MDA-MB-231 breast cancer xenografts in athymic mice.
2. Materials and Methods
2.1. Preparation of PVX
PVX was propagated in Nicotiana benthamiana plants and purified as previously reported.31
2.2. Preparation and characterization of DOX-loaded PVX
Doxorubicin HCl (denoted as DOX) was purchased from INDOFINE Chemical Company. To prepare PVX-DOX, PVX (at 2 mg mL−1 in 0.1 M potassium phosphate buffer (KP), pH 7.0) was incubated with a 5000 molar excess of DOX in a 10% (v/v) final concentration of DMSO for 5 days at room temperature, with agitation. PVX-DOX was purified twice over a 30% (w/v) sucrose cushion using ultracentrifugation (212,000 × g for 3 h at 4°C) and resuspended overnight in 0.1 M KP, pH 7.0. The particles were stored in 0.1 M KP, pH 7.0 at 4°C until used.
To prepare therapeutic particles for the in vivo study, we also conjugated polyethylene glycol (PEG) using PEG-succinimidyl ester with a molecular weight of 5000 Da (PEG-NHS, purchased from Nanocs) to formulate PEG-PVX-DOX. PEG-NHS was added to the mixture of PVX and DOX on day 4 at 2000 molar excess of PVX and incubated overnight at room temperature. Following purification as described above, PVX, PVX-DOX, and PEG-PVX-DOX were characterized by transmission electron microscopy (TEM), agarose gel electrophoresis, denaturing gel electrophoresis (SDS-PAGE), and UV/visible spectroscopy.
Transmission electron microscopy (TEM) was performed after DOX loading to confirm integrity of PVX-DOX filaments. PVX-DOX samples (0.1 mg mL−1, in dH2O) were placed on carbon-coated copper grids and negatively stained with 0.2% (w/v) uranyl acetate. Grids were imaged using a Zeiss Libra 200FE transmission electron microscope, operated at 200 kV.
To confirm PVX-DOX association, PVX-DOX filaments were separated using a 0.8% (w/v) agarose gel (in 1x TBE buffer). PVX-DOX or PEG-PVX-DOX and corresponding amounts of free DOX or PVX alone were loaded with 6x agarose loading dye (Fisher Scientific). Samples were run at 100 V for 30 min in TBE. Gels were visualized under UV light and white light before and after staining with 0.25% (w/v) Coomassie Blue (CB) staining, respectively.
SDS-PAGE was carried out to determine the number of PEG chains per PVX filament. 10 μg of denatured protein samples were analyzed on 4–12% NuPage gels (Life Technologies) in 1 x MOPS SDS running buffer (Life Technologies). Protein bands were visualized under UV light and white light before and after staining with CB. Band density analysis was conducted using ImageJ 1.47d (http://imagej.nih.gov/ij).
The number of DOX per PVX filament was determined by UV/visible spectroscopy using a NanoDrop 2000 spectrophotometer. PVX concentration and DOX loading were determined using the Beer-Lambert law and the DOX- (11,500 M−1 cm−1 at 495 nm) and PVX-specific (2.97 mL mg−1 cm−1 at 260 nm) extinction coefficients.
2.3. Cell culture
A2780, a gift from Dr. Analisa DiFeo (Case Western Reserve University) were cultured in Dulbecco’s modified Eagle’s media (DMEM, Life Technologies), supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta Biologicals) and 1% (v/v) penicillin-streptomycin (penstrep, Life Technologies). HeLa (ATCC) were cultured in minimum essential media (MEM, Life Technologies), supplemented with 10% (v/v) FBS, 1% (v/v) penstrep, and 1% (v/v) L-glutamine (Life Technologies). MDA-MB-231, a gift from Dr. Ruth Keri (Case Western Reserve University), were cultured in Roswell Park Memorial Institute (RPMI, Life Technologies) medium supplemented with 10% (v/v) FBS, 1% (v/v) penstrep, and 1% (v/v) L-glutamine. All cells were maintained at 37°C and 5% CO2.
2.4. Cytotoxicity evaluation
Confluent cells were removed with 0.05% (w/v) trypsin-EDTA (Life Technologies), seeded at either 2 × 103 cells/100 μL/well (MDA-MB-231 and HeLa) or 5 × 103 cells/100 μL/well (A2780) in 96-well plates and grown overnight at 37°C, 5% CO2. The next day, cells were washed 2 times with PBS and incubated with free DOX, PVX-DOX, or PEG-PVX-DOX corresponding to 0, 0.01, 0.05, 0.1, 0.5, 1, 5, or 10 μM (A2780, MDA-MB-231) or 0, 0.1, 0.5, 1, 5, 10, 25, or 50 μM (HeLa) DOX for 24 h, in triplicate. A PVX only control corresponded to the amount of PVX in the highest PVX-DOX sample. Following incubation, cells were washed 2 times to remove unbound DOX or particles. Fresh medium (100 μL) was added and cells were returned to the incubator for 48 h. Cell viability was assessed using an MTT proliferation assay (ATCC); the procedure was as the manufacturer suggested.
2.5. Tracking of free DOX and PVX-DOX in cells
A2780 cells were grown to confluency and removed with 0.05% (w/v) trypsin-EDTA. Cells were seeded at 1 × 104 cells/500 μL/well on coverslips in untreated 24-well plates and grown overnight at 37°C, 5% CO2. The next day, cells were washed 3 times and incubated with free DOX or PVX-DOX (1.5 μM DOX) for 2, 4, 12, 24, or 48 h. Following incubation, cells were washed 3 times and fixed in DPBS (Corning) containing 5% (v/v) paraformaldehyde (Electron Microscopy Sciences) and 0.3% (v/v) glutaraldehyde (Polysciences) for 10 min at room temperature. Coverslips were mounted using Fluoroshield (Sigma). Slides were imaged on a Zeiss Axio Observer Z1 motorized FL inverted microscope. Fluorescence intensity was analysed as follows using ImageJ 1.47d (http://imagej.nih.gov/ij). All images were identically processed prior to fluorescence intensity measurement. Nuclei were selected and analyzed for fluorescence intensity using the measure feature of ImageJ. For each sample, a minimum of 3 images was analyzed and at least 4 cells were analyzed per image.
2.6. In vivo efficacy of PEG-PVX-DOX
All experiments were conducted in accordance with Case Western Reserve University’s Institutional Animal Care and Use Committee. Female NCR nu/nu mice (6 weeks old) were injected subcutaneously into the right flank with 2 × 106 MDA-MB-231 cells suspended in 100 μL of media and Matrigel (Corning) at a 1:1 ratio. When tumors reached 100–200 mm3, a dosage of 1.0 mg/kg body weight DOX was injected intravenously (this dosage corresponds to 1.0 – 2.0 mg PVX depending on batches). Groups of 4 animals each were treated with PEG-PVX-DOX, free DOX, or PBS. The dosage was normalized to the DOX content. Treatments were performed every 5 days, and a total of 5 treatments was given. Tumors were measured daily and total volume was calculated using the formula: ν = (l × w2)/2. Mice were euthanized following 25 days of treatment.
3. Results
3.1. Synthesis and characterization of DOX-loaded PVX
PVX was produced through farming in N. benthamiana plants and purified using established protocols.31 From 100 g of infected leaf material, about 20 mg of pure PVX was extracted. DOX was loaded onto PVX by incubating excess DOX with the filaments in 0.1 M KP buffer (pH 7.0, final 10% (v/v) DMSO) for 5 days at room temperature (Fig. 1A). We used a 5000 molar excess of DOX per PVX; this was determined to be the optimized ratio that allowed homogenous mixing without aggregation. Further prolonged incubation time did not increase loading capacity (data not shown). The reaction was purified twice via ultracentrifugation. Purified DOX-loaded PVX (denoted as PVX-DOX) was characterized by TEM, agarose gel electrophoresis, and UV/visible spectroscopy (Fig. 1B–D).
Fig. 1.

Synthesis and characterization of DOX-loaded PVX (PVX-DOX). (A) Scheme of doxorubicin (DOX) loading onto potato virus X (PVX). (B) TEM image of negatively stained PVX-DOX. (C) Agarose gel electrophoresis of PVX (lane 1), PVX-DOX (lane 2), and DOX (lane 3) under UV light (top) and after Coomassie Blue (CB) staining (bottom). (D) UV/visible spectra of PVX-DOX at 1x and 5x dilution vs. DOX (at the equivalent concentration).
First, TEM was used to confirm the particle integrity after loading DOX. The filamentous structure remained unchanged as seen from the TEM image (Fig. 1B). Agarose gel electrophoresis was used to confirm that DOX was associated to PVX and not free in solution. DOX is fluorescent when visualized under UV light and PVX was visualized under white light after Coomassie Blue (CB) staining. PVX is too large to migrate through the gel in 30 min, and remains confined to the well. Under UV light, no signal was seen in the PVX only lane, while PVX-DOX signal was seen directly within the well (Fig. 1C). The free DOX control, due to its positive charge, migrated towards the cathode. Imaging of the gels after staining with CB confirmed the presence of PVX and PVX-DOX in their respective wells; gel electrophoresis also indicates that DOX is stably associated with PVX. Finally, UV/visible spectroscopy was used to determine the number of DOX associated per PVX. The Beer-Lambert law, in conjunction with the PVX- and DOX-specific extinction coefficients, was used to determine the concentrations of both PVX and associated DOX in solution. The ratio of DOX to PVX concentration was then used to determine DOX loading. It was determined that PVX was loaded with 850–1000 DOX (Fig. 1D); the range indicates batch-to-batch variability. Noteworthy, the peak derived from DOX in PVX-DOX was red-shifted, indicating the association of DOX to PVX (Fig. 1D). PVX-DOX stability was confirmed after 1 month of storage at 4°C; DOX release was not apparent (data not shown).
3.2. PVX-DOX maintains cell killing ability in vitro
We next determined whether doxorubicin maintained its cell killing ability after being loaded onto PVX. Increasing amounts of PVX-DOX and corresponding free DOX were evaluated in a panel of cell lines: A2780 (ovarian cancer), MDA-MB-231 (breast cancer), and HeLa (cervical cancer) (Fig. 2A–D). The calculated IC50 values of PVX-DOX were 0.84, 0.94, and 5.8 μM for A2780, MDA-MB-231, and HeLa, respectively (Fig. 2D). Meanwhile, free DOX showed slightly lower values (indicating higher efficacy) at each corresponding cell line: 0.22, 0.13, and 1.6 μM (Fig. 2D). The calculated IC50 values for free DOX are similar to previously reported values.32–34 PVX itself at the highest corresponding concentration exhibited no toxicity, thus attributing the cell killing function to the doxorubicin activity. A similar trend was observed in all tested cell lines: PVX-DOX was therapeutically active, but with decreased efficacy compared to free DOX (p<0.05).
Fig. 2.

Efficacy of DOX (black line) vs. PVX-DOX (blue line) in a panel of cell lines including (A) A2780 human ovarian cancer, (B) MDA-MB-231 human breast cancer, and (C) HeLa human cervical cancer as determined by MTT assay. Cells were treated by DOX or PVX-DOX corresponding to 0, 0.01, 0.05, 0.1, 0.5, 1, 5, or 10 μM (A2780 and MDA-MB-231) or 0, 0.1, 0.5, 1, 5, 10, 25, or 50 μM (HeLa) in 24 h. (D) IC50 values of DOX vs. PVX-DOX were determined using GraphPad Prism software (DOX vs. PVX-DOX p<0.05).
3.3. Tracking of DOX and PVX-DOX in A2780 cell line
The decreased efficacy of PVX-DOX vs. free DOX could be due to decreased uptake or inefficient release and trafficking of the PVX-DOX complex. To address these questions, we performed fluorescence microscopy studies to monitor DOX accumulation in the nucleus of treated cells. A2780 cells were incubated with 1.5 μM of DOX (either as PVX-DOX or free DOX) for 2, 4, 12, 24, or 48 h and imaged using a fluorescence microscope. DOX fluorescence per cell was analysed using ImageJ software (Fig. 3). As reported in previous studies, DOX accumulated within the nucleus, where it was visible via fluorescence.35,36 DOX within the cytoplasm is not fluorescent due to scattering and absorption by cellular components.37 Additionally, DOX is more concentrated within the nucleus than when it is dispersed throughout the cytoplasm; increased concentration of DOX is correlated to increases in fluorescence intensity.38 Up until 24 h, the accumulation of DOX in the nucleus increased over time, as shown by increased fluorescence observed in cells treated with either PVX-DOX or free DOX (Fig. 3A). At 48 h, treated cells were undergoing apoptosis leading to reduced DOX fluorescence – drug-induced apoptosis was apparent for cells treated with either PVX-DOX or free DOX attesting to the efficacy of the PVX-based delivery system. Quantitative analysis of the images indicated that PVX-DOX exhibited lower nuclear accumulation compared to free DOX at all time points tested (Fig. 3B). However, the decrease was only statistically significant (p<0.05) at 24 and 48 h.
Fig. 3.

Nuclear accumulation of DOX in A2780 cells after incubation with DOX or PVX-DOX over a 48 h time course. DOX accumulation was imaged (A) and quantified (B) using fluorescence microscopy. Scale bar = 20 μm. For quantification, at least 3 images were analyzed per sample, with at least 4 cells per image considered. Fluorescence intensity (FI) was quantified by measuring the nuclei using ImageJ software.
3.4. Preparation of PEGylated PEG-PVX-DOX nanoparticles
To assess the efficacy of PVX-DOX in vivo, we prepared a PEGylated formulation. Like other nanoparticles, PVX is cleared by the mononuclear phagocyte system (MPS) when administered systemically. Conjugation of stealth polymers such as PEG, however, decreases serum protein adsorption, resulting in the “stealth properties” that enhance circulation time and decrease accumulation in liver and spleen. We previously reported that PEGylated PVX coated with linear PEG chains with a molecular weight of 5000 Da demonstrates enhanced in vivo fates and favourable tumor accumulation.25,26
To prepare PEG-PVX-DOX, PEG-NHS (using a 2000 molar excess of PEG to PVX) was added into the mixture of PVX and DOX on day 4 (Fig. 4A). Excess DOX and PEG were removed by two rounds of ultracentrifugation. We used agarose gel and denaturing gel (SDS-PAGE) electrophoresis to confirm the loading of DOX and attachment of PEG. Agarose gel electrophoresis indicated that free DOX was completely removed from the solution and the fluorescence was only derived from DOX associated with PVX (Fig. 4B). Additionally, SDS-PAGE (Fig. 4C) confirmed successful conjugation of PEG to the PVX coat protein. Under UV light, DOX, released from its PVX carrier, is detectable at the bottom of gel. Nevertheless, dim fluorescence was observed from the PVX coat proteins (~25 kDa) indicating that some amounts of DOX remained associated with the protein even after exposure to denaturing conditions and electrophoretic separation. Analysis by ImageJ indicates that 95–97% of DOX was released from the denatured PVX. After staining by CB, the PVX coat protein and an additional band at a higher molecular weight corresponding to the PEGylated coat protein was observed in PEG-PVX-DOX but not PVX-DOX. To estimate the degree of PEGylation (number of PEG chains per PVX particle), band density analysis was performed using ImageJ. About 10% of PVX coat proteins were modified with PEG. Finally, the UV/visible spectrum indicated that about 1000–1500 DOX were loaded per particle (Fig. 4D); the range indicates the batch-to-batch variability. A red shift of the DOX-derived absorbance peak was also observed for PEG-PVX-DOX (as in PVX-DOX), indicating the association of DOX with the PEGylated PVX (Fig. 4D). Data show that higher loading of DOX to the PEGylated vs. native PVX is achieved. The increase in DOX loading might be because attached PEG chains further entrapped free DOX.
Fig. 4.

(A) Schematic of the PEGylated, DOX-loaded PEG-PVX-DOX. (B–D) Characterization of PEG-PVX-DOX. (B) Agarose gel electrophoresis of PVX (lane 1), PEG-PVX-DOX (lane 2), and DOX (lane 3); the gel was imaged under UV light (top) and after Coomassie Blue (CB) stain under white light (bottom). (C) SDS-PAGE of PVX (lane 1), PVX-DOX (lane 2), and PEG-PVX-DOX (lane 3) under UV light (left) and after staining with CB (right) photographed under white light. M: SeeBlue Plus2 Protein standard. Released DOX, PVX coat proteins (CP) and PEGylated PEG-CP are detected. (D) UV/visible spectra of PEG-PVX-DOX at 1x and 5x dilution vs. DOX (at the equivalent concentration).
3.5. In vitro and in vivo efficacies of PEG-PVX-DOX
We chose a model of triple negative breast cancer to assess the efficacy of PEG-PVX-DOX. First, in vitro efficacy was tested using MDA-MB-231 cells to confirm cell killing of the PVX-based drug carrier after PEGylation. Increasing amounts of PEG-PVX-DOX and free DOX were added to the culture media for 24 h then cells were washed and efficacy determined using an MTT assay. Analogous to PVX-DOX, PEG-PVX-DOX had cell killing ability but decreased efficacy compared to free DOX (Fig. 5A). The efficacy of PEG-PVX-DOX was 0.78 μM, which is about 6-fold higher than that of free DOX (0.13 μM). There is no significant difference in efficacies with and without PEG (Fig. 2D and 5A).
Fig. 5.
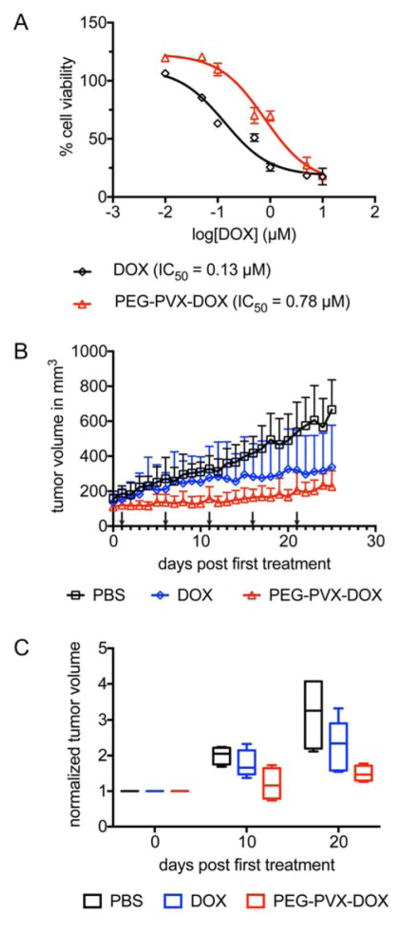
(A) In vitro efficacy of PEG-PVX-DOX vs. DOX determined by MTT assay. DOX or PEG-PVX-DOX corresponding to 0, 0.01, 0.05, 0.1, 0.5, 1, 5, or 10 μM were incubated with MDA-MB-231 cells. IC50 values were determined using GraphPad Prism software (DOX vs. PVX-DOX p<0.05). (B,C) Treatment of MDA-MB-231 tumors in an athymic mouse model using PEG-PVX-DOX vs. DOX and PBS. Treatment started when tumors reached 100–200 mm3; arrows indicate the treatment schedule. Groups of 4 animals were treated with a dosage of 1.0 mg/kg body weight DOX or PEG-PVX-DOX normalized to the DOX dose. (B) Daily monitored tumor volume growth in mice of different groups and (C) tumor volumes normalized to initial tumor volumes at day 0, 10, and 20 post first treatment (middle bars represent median values and the whiskers denote the minimum and maximum values).
Next, we investigated the capacity of PEG-PVX-DOX to inhibit tumor growth using MDA-MB-231 tumor-bearing athymic mouse model. Treatments were started when the tumor volume reached 100–200 mm3. The dosage of DOX on the carrier as well as free DOX was 1 mg/kg body weight. Intravenous injections into the tail vein were performed every 5 days for a total of 5 injections. Tumor volumes were measured daily. Statistically significant differences in mice treated with PBS or free DOX were only observed after day 22 post first treatment (Fig. 5B). In contrast, statistical significant differences between PBS vs PEG-PVX-DOX were apparent from day 7 post first treatment (p<0.05) (Fig. 5B). At the end of the treatment, tumor volumes in PEG-PVX-DOX treated group were 2.6 times smaller, while ones in DOX treated group were 2.1 times smaller than the PBS control. While data did not show statistical significance between DOX vs. PEG-PVX-DOX at the end of the treatment, a trend was apparent indicating that tumor volumes were smaller when mice were treated with PEG-PVX-DOX compared to the controls; this trend was observed throughout the study and indicates effective inhibition of tumor growth mediated by the plant virus-based drug delivery formulation (Fig. 5B,C).
4. Discussion
From a material point of view, potato virus X (PVX) provides a promising candidate for drug delivery. PVX is assembled from identical coat proteins to generate a highly homogeneous soft matter nanostructure with a flexible filamentous shape. Filamentous nanotechnologies have been shown to exhibit enhanced tissue penetration and tumor homing compared to their spherical counterparts.23 The precise, genetically-controlled size and shape of the PVX-based platform technology provides an advantage over some synthetic nanotechnologies, because large-scale manufacture of filamentous nanotechnologies with dimensions at the nanoscale remains technologically challenging, in particular when demanding high aspect ratio materials with narrow size distribution. In the case of PVX, the production of this nanomaterial can be easily scaled up through molecular farming with some commercial facilities already established, such as Medicago39 or Bioprodex.40 PVX is amenable to chemical and genetic modification for functionalization thus providing an array of synthetic and chemical biology approaches to produce a library of formulations with distinct functionality. Finally, the PVX platform technology is biocompatible, biodegradable, and non-infectious in mammals – thus adding a layer of safety compared to mammalian viral vectors. In this study, we demonstrated the use of this filamentous nanomaterial for delivering doxorubicin (DOX), a key therapeutic used in cancer treatments.
Preparation of DOX-loaded PVX is simple. Without the need for chemical reaction, DOX was loaded onto PVX by mixing the drug and the nanocarrier at room temperature for 5 days. Purification was performed by ultracentrifugation to remove excess DOX. The filamentous nature of PVX-DOX was not altered after loading and purification (as demonstrated by TEM imaging, Fig. 1B). We hypothesize that DOX is associated to PVX via hydrophobic interactions. In fact, the anthracycline has been shown to interact with many naturally occurring proteins such as human serum albumin or human α-1 acid glycoprotein via hydrophobic interactions and stabilized by a network of hydrogen bonding.41,42 Furthermore, the binding of DOX to another plant virus, specifically red clover necrotic mosaic virus (RCNMV), has also been observed.43 Although the crystal structure of PVX is not known, from the protein sequence, it is apparent that there are numerous nonpolar aromatic amino acids such as phenylalanine, tyrosine, and tryptophan, which are crucial for protein stability and often form hydrophobic interactions via stacking between them.44 The ring structure of DOX has a high tendency to interact with the benzene rings of these amino acids via π-π stacking. Red shift of DOX spectra after loading onto PVX provides indication of the association between DOX and the PVX scaffold (Fig. 1D, 4D). Besides, DOX can also interact via π-π stacking with other DOX molecules.45 Additionally, from the cryo-electron microscopy structure of PVX, it is evident that the filament has a grooved surface,46 which may foster DOX binding. From the UV/visible spectra, we have observed a red shift of DOX calculated 850–1000 DOX molecules per PVX particle, thus indicating that ~70–80% of the coat proteins of PVX were loaded. Interestingly, we observed that PEGylated formulations had increased DOX loading ranging from 1000 to 1500 DOX per PVX (Fig. 4D). For either formulation, DOX association was found stable: DOX was not released in solution after a month of storage. Together, these results suggest that PVX could be used for loading other hydrophobic drugs in the future.
We tested the efficacy of PVX-DOX in vitro in a panel of DOX-sensitive cell lines: A2780 (ovarian cancer), MDA-MB-231 (breast cancer), and HeLa (cervical cancer) using the MTT assay. There was a similar trend in these models: PVX-DOX exhibited cell killing, but with decreased efficacy compared to free DOX. However, these results are not unexpected; similar trends have been reported with synthetic47 and virus-based nanoparticles for DOX delivery.48,49 The reason for the lower efficacy may be explained by the lower uptake of DOX in the nucleus when delivered as PVX-DOX vs. free DOX (as was shown through longitudinal cell imaging using A2780 cells, see Fig. 3A). Differences in DOX uptake could be explained by uptake mechanisms of free DOX versus PVX-delivered DOX. Free DOX is taken up via diffusion across the cell membrane, while VNPs such as PVX are likely taken up by endocytosis or macropinocytosis.50,51 Thus, it is possible that PVX-delivered DOX is not as readily taken up as free DOX, resulting in decreased accumulation over time. Indeed, nuclear fluorescence intensity from PVX-DOX is approximately 2/3 of the nuclear fluorescence intensity measured for free DOX after 24 h of incubation. In comparison, DOX killed A2780 cells 4 times more effectively than PVX-DOX. The non-linear relationship between the imaging results and the MTT assay may be explained by the differences in experimental set up: nuclear fluorescence intensity was determined 24 h post-exposure to DOX or PVX-DOX; however, for cell viability, cells were incubated with DOX or PVX-DOX for 24 h followed by an additional 48 h incubation time. Thus, the differences between DOX and PVX-DOX may be more profound in the MTT assay vs. the imaging study. Nevertheless, the in vitro assays do not reflect the complexity of the in vivo situation. In vivo additional biological barriers must be considered – and biodistribution and clearance for free and PVX-formulated DOX will not follow the same trend. The conclusion to be drawn from the in vitro studies are that even though the IC50 indicates reduced efficacy of the nanoparticle formulation vs. free DOX, the PVX-DOX formulations maintains cell killing efficacy.
Finally, we prepared the stealth therapeutic nanoparticle PEG-PVX-DOX for evaluation of the in vivo efficacy in the MDA-MB-231-bearing athymic mouse model. PEGylation did not alter the cell killing ability (Fig. 5A). In a mouse model of triple negative breast cancer, we observed that PEG-PVX-DOX outperformed free DOX (Fig. 5B,C), highlighting the potential of PVX as a drug carrier.
Overall, the drug delivery properties of PVX shown in this study are comparable to other VNP systems in literature. DOX loading via non-covalent or covalent attachments on various icosahedral (sphere-like) VNPs has been reported (reviewed in 52). For example, DOX has been covalently attached to 30 nm-sized CPMV (up to 270 DOX per VNP) or Qβ bacteriophage (up to 500 DOX per VNP).53,54 DOX has also been loaded into the interior cavity of cucumber mosaic virus (CMV) via infusion yielding a 60% encapsulating efficiency (or 1500 DOX per CMV).55 Another interesting approach is the use of red clover necrotic mosaic virus (RCNMV); using this platform loading therapeutics such as DOX can be controlled using a pH- and salt-responsive gating mechanism.43 Up to 1000–1200 DOX can be infused into RCNMV. Recently, Francis et al. described a new approach using disk-shaped cargos assembled from double arginine mutant of recombinantly expressed tobacco mosaic virus (TMV) coat protein.56 Additionally, TMV, as a rigid soft-matter nanorod, has also been reported as a carrier for DOX delivery.57,58 Chemically conjugated via the available glutamic acid residues in the interior channel of TMV allowed more than 5000 DOX loading per particle.57 While many of these platforms have been studied extensively in vitro, only few in vivo studies have been reported. The field has grown out of its infancy and thus there is a need to be a push toward in vivo evaluation to gain insights into efficacy, pharmacodynamics and toxicity. Our study is the first step in this direction.
5. Conclusions
This work presents the use of potato virus X (PVX), a flexible filamentous plant virus, for delivering doxorubicin (DOX), an anti-tumor drug. We demonstrate that DOX can be loaded onto the surface of PVX through simple in-solution mixing followed by purification using ultracentrifugation. In vitro, DOX-loaded PVX demonstrates efficacy in a panel of cancer cells including ovarian cancer, breast cancer, and cervical cancer, although at lower efficacy than free DOX. Stealth polymers, such as PEG, can also be used to coat therapeutic PVX nanoparticles for enhanced in vivo properties enabling the treatment of MDA-MB-231 breast cancer xenografts in athymic mice. Tumor growth rates of mice treated with the PEGylated, DOX-loaded PVX formulation showed significant decreased tumor growth compared to PBS controls. Tumor volumes of PEG-PVX-DOX-treated mice were smaller compared to tumors from mice who received free DOX. These results lay the groundwork and open the door for the development of plant virus-based filamentous vehicles for use in cancer treatment.
Acknowledgments
This work was funded by a Research Scholar Award from the American Cancer Society (128319-RSG-15-144-01-CDD to NFS) and National Institutes of Health (NCI R25 CA148052 to KLL, NIBIB T32 EB007509 to KLL). Dr. A. M. Wen (CWRU) is thanked for assistance with TEM imaging.
Notes and references
- 1.Lammers T, Kiessling F, Hennink WE, Storm G. J Control Release. 2012;161:175–187. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 2.Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. Nanomedicine. 2013;9:1–14. doi: 10.1016/j.nano.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Adv Drug Deliv Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishra B, Patel BB, Tiwari S. Nanomedicine. 2010;6:9–24. doi: 10.1016/j.nano.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Bansal V, Zelikin AN, Caruso F. Nano Lett. 2008;8:1741–1745. doi: 10.1021/nl080877c. [DOI] [PubMed] [Google Scholar]
- 6.Maeng JH, Lee DH, Jung KH, Bae YH, Park IS, Jeong S, Jeon YS, Shim CK, Kim W, Kim J, Lee J, Lee YM, Kim JH, Kim WH, Hong SS. Biomaterials. 2010;31:4995–5006. doi: 10.1016/j.biomaterials.2010.02.068. [DOI] [PubMed] [Google Scholar]
- 7.Cheng Y, CSA, Meyers JD, Panagopoulos I, Fei B, Burda C. J Am Chem Soc. 2008;130:10643–10647. doi: 10.1021/ja801631c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elzoghby AO, Samy WM, Elgindy NA. Journal of Controlled Release. 2012;161:38–49. doi: 10.1016/j.jconrel.2012.04.036. [DOI] [PubMed] [Google Scholar]
- 9.Cai S, Vijayan K, Cheng D, Lima EM, Discher DE. Pharm Res. 2007;24:2099–2109. doi: 10.1007/s11095-007-9335-z. [DOI] [PubMed] [Google Scholar]
- 10.Chauhan VP, Popovic Z, Chen O, Cui J, Fukumura D, Bawendi MG, Jain RK. Angew Chem Int Ed Engl. 2011;50:11417–11420. doi: 10.1002/anie.201104449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SY, Ferrari M, Decuzzi P. Nanotechnology. 2009;20:495101. doi: 10.1088/0957-4484/20/49/495101. [DOI] [PubMed] [Google Scholar]
- 12.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shukla S, Eber FJ, Nagarajan AS, DiFranco NA, Schmidt N, Wen AM, Eiben S, Twyman RM, Wege C, Steinmetz NF. Adv Healthc Mater. 2015;4:874–882. doi: 10.1002/adhm.201400641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toy R, Peiris PM, Ghaghada KB, Karathanasis E. Nanomedicine (Lond) 2014;9:121–134. doi: 10.2217/nnm.13.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oltra NS, Swift J, Mahmud A, Rajagopal K, Loverde SM, Discher DE. Journal of Materials Chemistry B. 2013;1:5177–5185. doi: 10.1039/c3tb20431f. [DOI] [PubMed] [Google Scholar]
- 16.Simone EA, Dziubla TD, Discher DE, Muzykantov VR. Biomacromolecules. 2009;10:1324–1330. doi: 10.1021/bm900189x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Zhang Z, Zhang Y. Nanoscale Res Lett. 2011;6:555. doi: 10.1186/1556-276X-6-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamilton RF, Jr, Wu Z, Mitra S, Shaw PK, Holian A. Part Fibre Toxicol. 2013;10:57. doi: 10.1186/1743-8977-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu J, Wong DH, Byrne JD, Chen K, Bowerman C, DeSimone JM. Angew Chem Int Ed Engl. 2013;52:6580–6589. doi: 10.1002/anie.201209145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinmetz NF. Nanomedicine. 2010;6:634–641. doi: 10.1016/j.nano.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wen AM, Steinmetz NF. Chem Soc Rev. 2016;45:4074–4126. doi: 10.1039/c5cs00287g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinmetz NF, Mertens ME, Taurog RE, Johnson JE, Commandeur U, Fischer R, Manchester M. Nano Lett. 2010;10:305–312. doi: 10.1021/nl9035753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shukla S, Ablack AL, Wen AM, Lee KL, Lewis JD, Steinmetz NF. Mol Pharm. 2013;10:33–42. doi: 10.1021/mp300240m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chariou PL, Lee KL, Wen AM, Gulati NM, Stewart PL, Steinmetz NF. Bioconjugate Chemistry. 2015;26:262–269. doi: 10.1021/bc500545z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shukla S, Wen AM, Ayat NR, Commandeur U, Gopalkrishnan R, Broome AM, Lozada KW, Keri RA, Steinmetz NF. Nanomedicine (Lond) 2014;9:221–235. doi: 10.2217/nnm.13.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee KL, Shukla S, Wu M, Ayat NR, El Sanadi CE, Wen AM, Edelbrock JF, Pokorski JK, Commandeur U, Dubyak GR, Steinmetz NF. Acta Biomaterialia. 2015;19:166–179. doi: 10.1016/j.actbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 28.Sabeti B, Noordin MI, Mohd S, Hashim R, Dahlan A, Javar HA. Biomed Res Int. 2014;2014:765426. doi: 10.1155/2014/765426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carelle N, Piotto E, Bellanger A, Germanaud J, Thuillier A, Khayat D. Cancer. 2002;95:155–163. doi: 10.1002/cncr.10630. [DOI] [PubMed] [Google Scholar]
- 30.Volkova M, Russell R., 3rd Curr Cardiol Rev. 2011;7:214–220. doi: 10.2174/157340311799960645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee KL, Uhde-Holzem K, Fischer R, Commandeur U, Steinmetz NF. Methods Mol Biol. 2014;1108:3–21. doi: 10.1007/978-1-62703-751-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao J, DeHaven J, Lamm D, Weissman DN, Malanga CJ, Rojanasakul Y, Ma JK. Drug Deliv. 2001;8:71–76. doi: 10.1080/107175401750177007. [DOI] [PubMed] [Google Scholar]
- 33.Dufes C, Muller JM, Couet W, Olivier JC, Uchegbu IF, Schatzlein AG. Pharm Res. 2004;21:101–107. doi: 10.1023/b:pham.0000012156.65125.01. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q, Xiang G, Zhang Y, Yang K, Fan W, Lin J, Zeng F, Wu J. J Pharm Sci. 2006;95:2266–2275. doi: 10.1002/jps.20714. [DOI] [PubMed] [Google Scholar]
- 35.Dai X, Yue Z, Eccleston ME, Swartling J, Slater NK, Kaminski CF. Nanomedicine. 2008;4:49–56. doi: 10.1016/j.nano.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Shen F, Chu S, Bence AK, Bailey B, Xue X, Erickson PA, Montrose MH, Beck WT, Erickson LC. J Pharmacol Exp Ther. 2008;324:95–102. doi: 10.1124/jpet.107.127704. [DOI] [PubMed] [Google Scholar]
- 37.Chen NT, Wu CY, Chung CY, Hwu Y, Cheng SH, Mou CY, Lo LW. PLoS One. 2012;7:e44947. doi: 10.1371/journal.pone.0044947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohan P, Rapoport N. Mol Pharm. 2010;7:1959–1973. doi: 10.1021/mp100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Aoust MA, Couture MMJ, Charland N, Trepanier S, Landry N, Ors F, Vézina LP. Plant biotechnology journal. 2010;8:607–619. doi: 10.1111/j.1467-7652.2009.00496.x. [DOI] [PubMed] [Google Scholar]
- 40.Charudattan R, Hiebert E. Outlooks on Pest Management. 2007;18:167. [Google Scholar]
- 41.Agudelo D, Bourassa P, Bruneau J, Berube G, Asselin E, Tajmir-Riahi HA. PLoS One. 2012;7:e43814. doi: 10.1371/journal.pone.0043814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Husain N, Agbaria RA, Warner IM. The Journal of Physical Chemistry. 1993;97:10857–10861. [Google Scholar]
- 43.Cao J, Guenther RH, Sit TL, Opperman CH, Lommel SA, Willoughby JA. Small. 2014;10:5126–5136. doi: 10.1002/smll.201400558. [DOI] [PubMed] [Google Scholar]
- 44.Huisman MJ, Linthorst HJ, Bol JF, Cornelissen JC. J Gen Virol. 1988;69(Pt 8):1789–1798. doi: 10.1099/0022-1317-69-8-1789. [DOI] [PubMed] [Google Scholar]
- 45.Missirlis D, Kawamura R, Tirelli N, Hubbell JA. Eur J Pharm Sci. 2006;29:120–129. doi: 10.1016/j.ejps.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Kendall A, McDonald M, Bian W, Bowles T, Baumgarten SC, Shi J, Stewart PL, Bullitt E, Gore D, Irving TC, Havens WM, Ghabrial SA, Wall JS, Stubbs G. J Virol. 2008;82:9546–9554. doi: 10.1128/JVI.00895-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoo HS, Lee KH, Oh JE, Park TG. J Control Release. 2000;68:419–431. doi: 10.1016/s0168-3659(00)00280-7. [DOI] [PubMed] [Google Scholar]
- 48.Ren Y, Wong SM, Lim LY. Bioconjug Chem. 2007;18:836–843. doi: 10.1021/bc060361p. [DOI] [PubMed] [Google Scholar]
- 49.Lockney DM, Guenther RN, Loo L, Overton W, Antonelli R, Clark J, Hu M, Luft C, Lommel SA, Franzen S. Bioconjugate Chemistry. 2011;22:67–73. doi: 10.1021/bc100361z. [DOI] [PubMed] [Google Scholar]
- 50.Wen AM, Infusino M, De Luca A, Kernan DL, Czapar AE, Strangi G, Steinmetz NF. Bioconjug Chem. 2015;26:51–62. doi: 10.1021/bc500524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamba SA, Ismail M, Hussein-Al-Ali SH, Ibrahim TA, Zakaria ZA. Molecules. 2013;18:10580–10598. doi: 10.3390/molecules180910580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee EJ, Lee NK, Kim IS. Adv Drug Deliv Rev. 2016;106:157–171. doi: 10.1016/j.addr.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 53.Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. Mol Pharm. 2013;10:3–10. doi: 10.1021/mp3002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pokorski JK, Breitenkamp K, Liepold LO, Qazi S, Finn MG. J Am Chem Soc. 2011;133:9242–9245. doi: 10.1021/ja203286n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zeng Q, Wen H, Wen Q, Chen X, Wang Y, Xuan W, Liang J, Wan S. Biomaterials. 2013;34:4632–4642. doi: 10.1016/j.biomaterials.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 56.Finbloom JA, Han K, Aanei IL, Hartman EC, Finley DT, Dedeo MT, Fishman M, Downing KH, Francis MB. Bioconjug Chem. 2016 doi: 10.1021/acs.bioconjchem.6b00424. [DOI] [PubMed] [Google Scholar]
- 57.Bruckman MA, Czapar AE, VanMeter A, Randolph LN, Steinmetz NF. J Control Release. 2016;231:103–113. doi: 10.1016/j.jconrel.2016.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tian Y, Gao S, Wu M, Liu X, Qiao J, Zhou Q, Jiang S, Niu Z. ACS Appl Mater Interfaces. 2016;8:10800–10807. doi: 10.1021/acsami.6b02801. [DOI] [PubMed] [Google Scholar]