

Abstract
Rationale:
Pycnodysostosis is a rare autosomal recessive skeletal dysplasia characterized by short stature, craniofacial dysmorphism, acro-osteolysis, osteosclerosis, and brittle bone with poor healing. Pycnodysostosis results from the deficient activity of cathepsin K, a lysosomal cysteine protease that is encoded by CTSK.
Patient concerns:
We report a Korean adult patient with pycnodysostosis and atypical femur fracture whose diagnosis was confirmed by next-generation sequencing (NGS) of candidate genes. A 41-year-old female patient was presented with a left femur fracture after falling down. Underlying sclerotic bone disease was suspected as a radiographic skeletal survey showed thickened cortical bones, and the total body bone density was increased (T score was 5.3, and Z score was 4.9).
Diagnoses:
We performed candidate gene sequencing of various sclerotic bone diseases for the differential molecular diagnosis of underlying sclerosing bone disease. Two heterozygous variants of CTSK were detected. One was a frameshift variant in exon 5, c.426delT (p.Phe142Leufs∗19), which was previously reported, and the other was a novel missense variant in exon 6, c.755G>A (p.Ser252Asn). Sanger sequencing of CTSK confirmed the 2 heterozygous variants and thus the patient was diagnosed with pycnodysostosis.
Interventions:
The patient had emergency surgery for subtrochantic femoral fracture.
Outcomes:
After 4 months of surgery, the patient had almost a full range of hip and knee movements and radiographs show the substantial bridging callus across the fracture.
Lessons:
Candidate gene sequencing could be a useful diagnostic tool for the genetically heterogeneous skeletal dysplasia group, especially in cases with a mild or atypical clinical phenotype.
Keywords: atypical femur fracture, candidate gene sequencing, cathepsin K, CTSK, pycnodysostosis
1. Introduction
Pycnodysostosis (OMIM 265800) is a rare autosomal recessive skeletal dysplasia with an estimated prevalence of 1 to 1.7 per million,[1] first described in 1962 by Maroteaux and Lamy.[2] Pycnodysostosis is presented with distinct clinical features including short stature, craniofacial dysmorphism, acroosteolysis of the terminal phalanges, osteosclerosis, and unusually brittle bone with poor healing. Craniofacial dysmorphism includes frontal bossing, opened fontanels, delayed closure of the cranial sutures, and an obtuse mandible angle. The abnormal skeletal morphology and bone composition in pycnodysostosis result from loss of cathepsin K activity, a lysosomal cysteine protease that is highly expressed in osteoclasts.[3] Cathepsin K is encoded by the CTSK gene located at chromosome 1q21.[4–6]
The phenotypes of pycnodysostosis are highly variable and overlap among various groups of skeletal dysplasia.[7,8] A specific diagnosis based on clinical manifestation is challenging in cases with atypical or mild clinical presentations. Molecular diagnostic approaches can be a useful diagnostic tool in these cases.
Here, we described the case of a Korean adult female patient with pycnodysostosis who sustained an atypical subtrochanteric femoral fracture with an impending subtrochanteric fracture in the contralateral femur. Because of her atypical presentation, a genetic diagnosis was carried out by next-generation sequencing (NGS) of candidate genes and later confirmed performing by Sanger sequencing. To the best of our knowledge, this is the first genetically documented case of pycnodysostosis in Korea.
2. Case report
A 41-year-old female patient fell from a chair and had sustained pain in her left thigh. Plain radiographs revealed a displaced left subtrochanteric transverse fracture with lateral beaking and thickened cortices as well as higher bone density in general. The contralateral femur also showed signs of cortical thickening and lateral beaking, raising concern for an impending fracture (Fig. 1A and B). She had previous traumatic fractures of the left tibia after a car accident 9 years ago and of the right clavicle 2 years ago that were completely healed. She was born from nonconsanguineous Korean parents. There was no family history of bone diseases. Her height was 160 cm and weight was 59 kg. A prominent forehead with frontal bossing and bitemporal narrowing were observed. However, her anterior fontanel was closed, mandibular angles appeared normal, and her fingers were not stubby. Serum laboratory tests revealed low osteocalcin and relatively low alkaline phosphatase considering concurrent bone fracture: calcium 8.7 mg/dL (normal range 8.6–10.2), phosphorus 3.1 mg/dL (2.5–4.5), PTH 52 pg/mL (11–62), 25(OH)D 13.2 ng/mL (9.0–37.6), alkaline phosphatase 43 U/L (35–104), and osteocalcin 6.1 ng/mL (8.0–36.0). The 24-hour urine calcium level was 40 mg (100–300), with inorganic phosphorus at 970 mg (400–1300) and creatinine at 1200 mg (740–1570).
Figure 1.
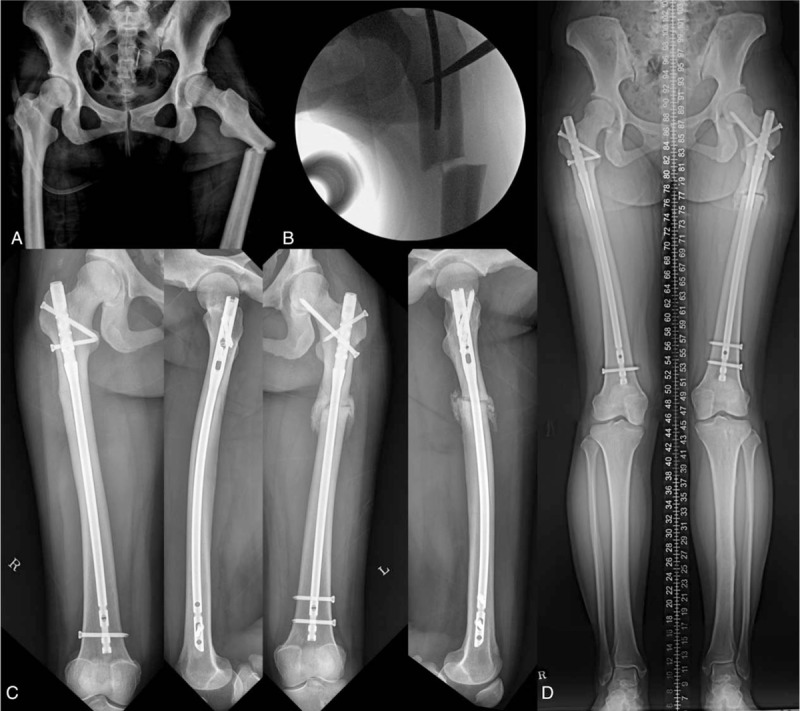
Atypical femur fracture of the patient. (A) Preoperative radiographs of the patient with pycnodysostosis. (B) Intraoperative C-arm image shows a long osteotome used to open and widen the intramedullary canal. (C) Four months postoperatively, radiographs show that the left femur exhibited substantial bridging callus across the fracture, and the right femur was healed. (D) Scanogram shows good alignment with an incomplete union of the fracture.
On the day of her arrival at the emergency room, she underwent closed reduction and internal fixation with an antegrade femoral nail on the affected side and prophylactic internal fixation with the same implant on the contralateral side of the femur. The patient was placed supine on a fracture table under general anesthesia. The trochanter was inserted with a guide wire and opening reamer. It was impossible to pass the ball-tip guide wire into the intramedullary canal because the patient had extremely sclerotic bone and a narrow canal. Both cannulated powered and hand reamers did not work. The canal was opened with a long curved osteotome that was inserted into the intramedullary canal through a previously opened entry portal, and the ball-tip guide wire was positioned. The canal was then enlarged with flexible power reamers to a final diameter of 10.5 mm. A 9.3 mm ×380 mm lateral-entry nail (Zimmer Natural Nail, Zimmer, Warsaw, IN) was placed over the guide wire and locked using 2 proximal screws and 2 distal screws. Prophylactic fixation was performed on the contralateral side of the femur (Fig. 1C). Rehabilitation started on the first postoperative day with sitting and continuous passive motion of the knee and hip joints. Standing and ambulation using walking aids with tolerable weight bearing were allowed after 3 days following surgery.
After her general condition had recovered, a radiological skeletal survey was performed and generalized increased density of the bones in the entire skeleton was observed (Fig. 2A–D). A thickened base of the skull and mild scoliosis and spondylolysis of the L5 vertebrae were observed in whole spine radiography. X-ray assessment of the hand was relatively normal. Abnormal increased uptakes in both distal femurs, both tibiae, and both humeri were observed from a Tc-99m whole body bone scan (Fig. 2E). Total body bone densitometry using dual-energy X-ray absorptiometry (DEXA) showed an abnormal elevation of her bone density (T score was 5.3, and Z score was 4.9). Based on the clinical phenotype, adult-type osteopetrosis was initially suspected.
Figure 2.
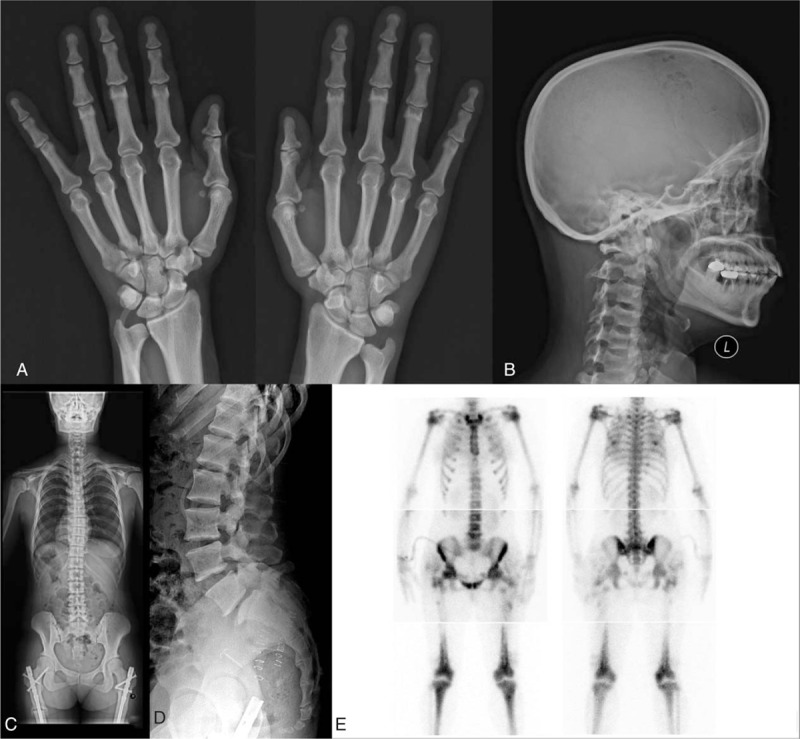
Skeletal survey of the patient. (A) Acroosteolysis of the distal phalanges was not observed in the hand radiograph. (B) Lateral cranial film shows thickening at the base of the skull and frontal bossing. (C) Mild scoliosis was observed on the whole spine posteroanterior (PA) radiograph. (D) Spondylolysis of the L5 vertebrae was observed on the lateral lumbar spine radiograph. (E) Multiple hot uptake was observed on the whole body bone scan.
For the differential molecular diagnosis of underlying sclerosing bone disease, we performed NGS of candidate genes for sclerotic bone diseases including various types of osteopetrosis (CLCN7, LRP5, CA2, TCIRG1, TNFSF11, PLEKHM1, and SNX10), pycnodysostosis (CTSK), craniometaphyseal dysplasia (ANKH), and craniodiaphyseal dysplasia (SOST). Briefly, genomic DNA was extracted from the peripheral blood. Library was prepared using the TruSight One Sequencing Panel (Illumina, Inc., San Diego, CA), which enriched a 12-Mb region spanning candidate genes with clinical relevance. Massively parallel sequencing was performed on the Illumina NextSeq platform. Average coverage of depth of the entire panel was 77×, and 97% of targeted bases were covered by 10× sequence reads. Coverage of the genes of interest is shown in Table 1. Sequence reads were aligned to hg19 with Burrow-Wheeler Aligner (version 0.7.12, MEM algorithm). Duplicate reads were removed by using Picard-tools1.96. Local realignment and base quality recalibration was done by The Genome Analysis Toolkit (GATK version 3.5). Variant calling was performed by GATK HaplotypeCaller. Variants were annotated by Variant Effect Predictor and dbNSFP. Candidate gene sequencing revealed 2 heterozygous variants in the CTSK gene that were confirmed by Sanger sequencing; 1 was a frameshift variant in exon 5, c.426delT (p.Phe142Leufs∗19), which was previously reported in patients with pycnodysostosis,[9] and the other was a novel missense variant in exon 6, c.755G>A (p.Ser252Asn) (reference sequence: NM_000396.3) (Fig. 3). The novel variant was not found in population databases such as 1000 Genomes, ESP6500, and ExAC. In silico analysis of p.Ser252Asn revealed a contradictory prediction result: damaging effect by SIFT and MutationTaster and tolerable effect by PolyPhen-2 HumVar. Codon 252 is highly conserved among species. Through targeted Sanger sequencing analysis of the patient's family members, it is likely that 2 different mutations are located on 2 different alleles because the daughter of the patient was a heterozygous carrier of the c.755G>A variant, and the son of the patient was a heterozygous carrier of the c.426delT mutation (Fig. 4). Unaffected family members had none of the 2 variants or only 1 heterozygous variant (Fig. 4). According to the American College of Medicine and Genetics guidelines for the interpretation of sequence variation,[10] the p.Ser252Asn variant could be classified as a likely pathogenic variant based on the following evidences absence from control population (PM2), trans-configuration with a pathogenic variant in autosomal recessive disorders (PM3), in silico prediction, and patient's phenotype. There was no pathogenic variant in the remaining 9 genes. Therefore, the patient was diagnosed with pycnodysostosis, and genetic counseling was performed.
Table 1.
Depth of coverage of genes with interest.
Figure 3.
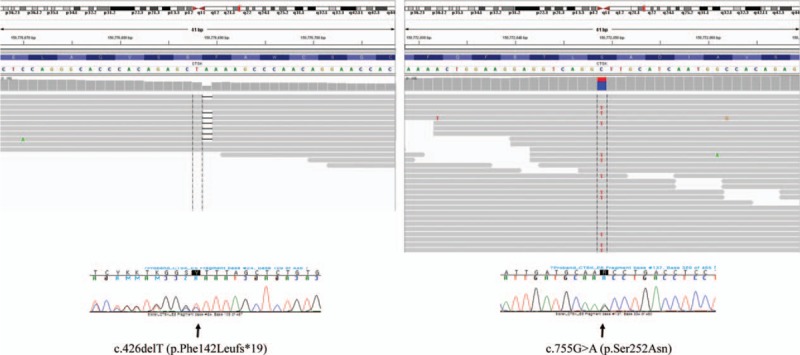
CTSK variants detected by massively parallel sequencing and Sanger sequencing. Two heterozygous variants of CTSK were detected by next-generation sequencing of candidate genes and confirmed by Sanger sequencing. One was a frameshift variant in exon 5, c.426delT (p.Phe142Leufs∗19), and the other was a novel missense variant in exon 6, c.755G>A (p.Ser252Asn).
Figure 4.
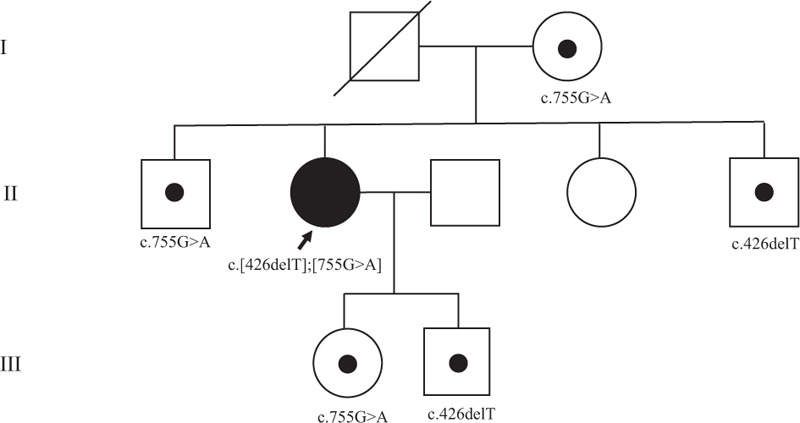
Pedigree of the patient and mutation analysis of the patient's family. Mutation analysis of the patient's family revealed that the daughter of the patient was a heterozygous carrier of the c.755G>A variant, and the son of the patient was a heterozygous carrier of the c.426delT mutation. Her mother was a heterozygous carrier of the c.755G>A variant. Her 2 unaffected brothers were heterozygous carriers, and 1 sister had none of the 2 variants.
One month postoperatively, the patient was able to bear full weight, and implants in both femurs were properly positioned with some sign of callus in the left femur. At the follow-up visit after 4 months, the patient was pain-free and had almost a full range of hip and knee movements, and radiographs show that the left femur exhibited substantial bridging callus across the fracture (Fig. 1D).
3. Discussion
Atypical subtrochanteric femoral fractures, as observed in our case, have been previously described in patients with pycnodysostosis.[11–14] Atypical femoral fractures are associated with high complication rates with operative fixation using conventional open reduction and internal fixation technique.[15] Several authors[16] have recommended the use of intramedullary nail systems for the management of atypical femoral fractures. However, intramedullary nailing in pycnodysostosis sometimes requires a high degree of skill because of the difficulties in reaming sclerotic bone.[11] In our case, a great deal of labor was required to accomplish intramedullary nailing. We used a long curved osteotome to widen the medullary canal, and it was effective. Poor healing of the bone structure is also an important clinical feature of pycnodysostosis.[17] This patient also suffered from an incomplete union of the femur fracture after 4 months of surgery. Bone fracture healing requires not only active bone formation to produce osteotylus but also degradation of excess bone matrix to restore the structure of the bone.[17] Cathepsin K plays a key role in osteoclast-mediated bone resorption during the healing process of bone fracture.[18,19]
A large number and overlapping phenotypes of various groups of skeletal dysplasia often result in challenges in performing a specific molecular diagnosis.[7,8] The role of NGS in the diagnosis of genetically heterogeneous disorders is well described.[20,21] Furthermore, NGS can be a useful diagnostic tool when clinical manifestations are atypical or mild. Therefore, we performed NGS of candidate genes for the differential diagnosis of this patient's underlying skeletal dysplasia. A precise molecular diagnosis of skeletal dysplasia is crucial for providing a prognosis of the patient and genetic counseling for the family.
Xue et al[1] reported that the most common phenotype of pycnodysostosis was short stature, which was presented in 95.9% of the 97 previously reported cases, followed by increased bone density, presented in 88.7% cases. However, pycnodysostosis exhibits similar clinical manifestations as adult-type osteopetrosis, including increased bone density, low bone turnover markers, and fragility fractures. Indeed, Pangrazio et al[20] reported that exome sequencing revealed 6 patients with pycnodysostosis among 27 patients who were clinically diagnosed with mild osteopetrosis. The typical phenotypes of pycnodysostosis include short stature, a peculiar facial appearance with frontal bossing and bitemporal narrowing, hypoplasia of the maxilla, absence of the mandibular angle, and stubby hands and feet with acroosteolysis.[1,22] Nevertheless, Pangrazio et al[20] reported that the obtuse mandibular angle and obvious acroosteolysis were not present in some genetically confirmed pycnodysostosis patients. We also initially suspected osteopetrosis based on the increased bone density of this patient. The phenotypic evidence for a diagnosis of pycnodysostosis seemed insufficient for this patient as her height was in a normal range and she only experienced 2 fractures until her forties. Radiological evaluation did not indicate obvious acroosteolysis on the distal phalanges or obtuse mandibular angle. However, candidate gene sequencing and Sanger sequencing revealed 2 heterozygous variants of CTSK. It is likely the 2 different mutations are located on the 2 different alleles because targeted Sanger sequencing analysis of the patient's family members showed a trans-configuration of the 2 variants (Fig. 4), which led to a diagnosis of pycnodysostosis. The mild phenotype of this patient could be possibly resulted from presence of other genes compensating the mutate CTSK gene, or uncertain compensatory regulation of other genes, which may inspire genomic investigation and developing treatment in the future.
4. Conclusions
In conclusion, we report a Korean adult patient with pycnodysostosis and atypical femur fracture whose diagnosis was confirmed by molecular genetic analysis. We suggest that NGS of candidate genes could be a useful diagnostic tool for the genetically heterogeneous skeletal dysplasia group, especially in cases with a mild or atypical clinical phenotype.
Footnotes
Abbreviations: DEXA = dual-energy X-ray absorptiometry, NGS = next-generation sequencing.
Funding: This study was supported by the new faculty research fund of Ajou University School of Medicine.
Ethical approval and patient's consent: Since this is a case report, institutional review board approval is not sought. However, written informed consent was obtained from patient for the publication.
The authors have no conflicts of interest to disclose.
References
- [1].Xue Y, Cai T, Shi S, et al. Clinical and animal research findings in pycnodysostosis and gene mutations of cathepsin K from 1996 to 2011. Orphanet J Rare Dis 2011;6:20.doi: 10.1186/1750-1172-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Maroteaux P, Lamy M. The Malady of Toulouse-Lautrec. JAMA 1965;191:715–7. [DOI] [PubMed] [Google Scholar]
- [3].Turan S. Current research on pycnodysostosis. Intractable Rare Dis Res 2014;3:91–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gelb BD, Edelson JG, Desnick RJ. Linkage of pycnodysostosis to chromosome 1q21 by homozygosity mapping. Nat Genet 1995;10:235–7. [DOI] [PubMed] [Google Scholar]
- [5].Polymeropoulos MH, Ortiz De Luna RI, Ide SE, et al. The gene for pycnodysostosis maps to human chromosome 1cen-q21. Nat Genet 1995;10:238–9. [DOI] [PubMed] [Google Scholar]
- [6].Gelb BD, Shi GP, Chapman HA, et al. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996;273:1236–8. [DOI] [PubMed] [Google Scholar]
- [7].Makitie O. Molecular defects causing skeletal dysplasias. Endocr Dev 2011;21:78–84. [DOI] [PubMed] [Google Scholar]
- [8].Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015;167a:2869–92. [DOI] [PubMed] [Google Scholar]
- [9].Fujita Y, Nakata K, Yasui N, et al. Novel mutations of the cathepsin K gene in patients with pycnodysostosis and their characterization. J Clin Endocrinol Metab 2000;85:425–31. [DOI] [PubMed] [Google Scholar]
- [10].Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kundu ZS, Marya KM, Devgan A, et al. Subtrochanteric fracture managed by intramedullary nail in a patient with pycnodysostosis. Joint Bone Spine 2004;71:154–6. [DOI] [PubMed] [Google Scholar]
- [12].Nakase T, Yasui N, Hiroshima K, et al. Surgical outcomes after treatment of fractures in femur and tibia in pycnodysostosis. Arch Orthop Trauma Surg 2007;127:161–5. [DOI] [PubMed] [Google Scholar]
- [13].Hashem J, Krochak R, Culbertson MD, et al. Atypical femur fractures in a patient with pycnodysostosis: a case report. Osteoporos Int 2015;26:2209–12. [DOI] [PubMed] [Google Scholar]
- [14].Yates CJ, Bartlett MJ, Ebeling PR. An atypical subtrochanteric femoral fracture from pycnodysostosis: a lesson from nature. J Bone Miner Res 2011;26:1377–9. [DOI] [PubMed] [Google Scholar]
- [15].Prasarn ML, Ahn J, Helfet DL, et al. Bisphosphonate-associated femur fractures have high complication rates with operative fixation. Clin Orthop Relat Res 2012;470:2295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shane E, Burr D, Ebeling PR, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2010;25:2267–94. [DOI] [PubMed] [Google Scholar]
- [17].Huang X, Qi X, Li M, et al. A mutation in CTSK gene in an autosomal recessive pycnodysostosis family of Chinese origin. Calcif Tissue Int 2015;96:373–8. [DOI] [PubMed] [Google Scholar]
- [18].Fratzl-Zelman N, Valenta A, Roschger P, et al. Decreased bone turnover and deterioration of bone structure in two cases of pycnodysostosis. J Clin Endocrinol Metab 2004;89:1538–47. [DOI] [PubMed] [Google Scholar]
- [19].Novinec M, Lenarcic B, Cathepsin K. a unique collagenolytic cysteine peptidase. Biol Chem 2013;394:1163–79. [DOI] [PubMed] [Google Scholar]
- [20].Pangrazio A, Puddu A, Oppo M, et al. Exome sequencing identifies CTSK mutations in patients originally diagnosed as intermediate osteopetrosis. Bone 2014;59:122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sule G, Campeau PM, Zhang VW, et al. Next-generation sequencing for disorders of low and high bone mineral density. Osteoporos Int 2013;24:2253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Soliman AT, Ramadan MA, Sherif A, et al. Pycnodysostosis: clinical, radiologic, and endocrine evaluation and linear growth after growth hormone therapy. Metabolism 2001;50:905–11. [DOI] [PubMed] [Google Scholar]