

Abstract
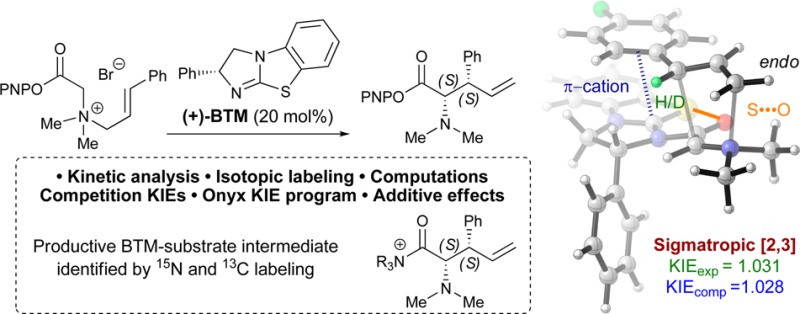
A mechanistic study of the isothiourea-catalyzed enantioselective [2,3]-rearrangement of allylic ammonium ylides is described. Reaction kinetic analyses using 19F NMR and density functional theory computations have elucidated a reaction profile and allowed identification of the catalyst resting state and turnover-rate limiting step. A catalytically relevant catalyst–substrate adduct has been observed, and its constitution elucidated unambiguously by 13C and 15N isotopic labeling. Isotopic entrainment has shown the observed catalyst–substrate adduct to be a genuine intermediate on the productive cycle toward catalysis. The influence of HOBt as an additive upon the reaction, catalyst resting state, and turnover-rate limiting step has been examined. Crossover experiments have probed the reversibility of each of the proposed steps of the catalytic cycle. Computations were also used to elucidate the origins of stereocontrol, with a 1,5-S···O interaction and the catalyst stereodirecting group providing transition structure rigidification and enantioselectivity, while preference for cation−π interactions over C–H···π is responsible for diastereoselectivity.
1. Introduction
The [2,3]-rearrangement of allylic ammonium ylides is a direct and elegant method toward the synthesis of α-amino acid derivatives containing multiple stereocenters.1 The mechanism of this process, and that of the competitive [1,2]-Stevens rearrangement, has been much discussed and disputed within the literature. A concerted thermally allowed sigmatropic process is thought to be operative in the [2,3]-rearrangement, while a radical mechanism involving bond cleavage and recombination is usually favored for [1,2]-rearrangement (Scheme 1A).2
Scheme 1. Rearrangements of Allylic Ammonium Ylides.
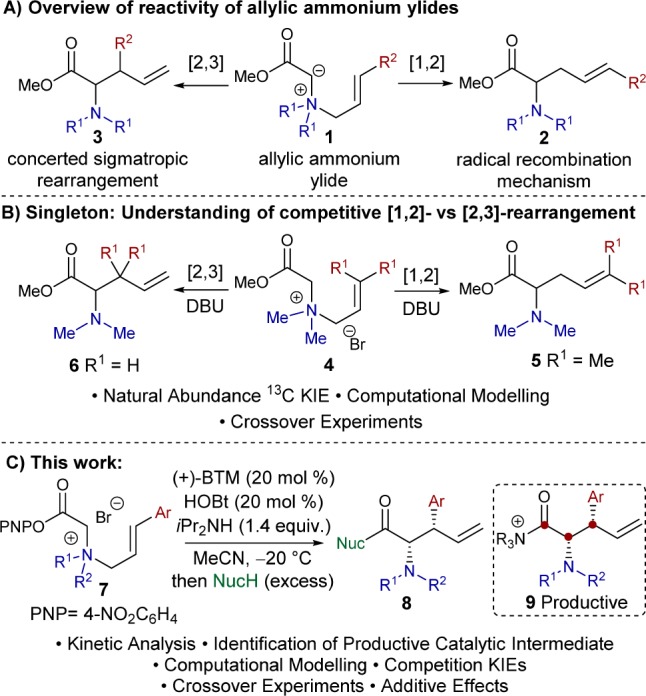
To date, few mechanistic analyses of [2,3]-rearrangements of allylic ammonium ylides have been conducted, although Jacobsen and co-workers have recently reported a detailed mechanistic investigation into the related thiourea-catalyzed [2,3]-Wittig rearrangement.3 The elegant experimental and computational work of Singleton and co-workers concerning the competitive [2,3]- and [1,2]-rearrangements of allylic ammonium ylides promoted by DBU represents the current state-of-the-art (Scheme 1B). Through 13C kinetic isotope effects, crossover experiments, and computation, these studies demonstrate that the origin of competitive [1,2]- and [2,3]-rearrangement is the common loose transition state leading to dynamic bond cleavage.4 The development of both catalytic and stereoselective variants of the [2,3]-rearrangement of allylic ammonium ylides has been a significant synthetic challenge. Tambar and co-workers have reported a tandem ammonium salt formation and diastereoselective [2,3]-rearrangement process, exploiting Pd-catalyzed allylic substitution to form the reactive ammonium salt in situ, giving (±)-anti-α-amino acid derivatives with excellent diastereocontrol via proposed transition state 13 (Scheme 2A).5 The observed diastereoselectivity of this [2,3]-rearrangement process, and indeed most [2,3]-rearrangements, can be rationalized through the exo- or endo- transition states 15 and 17 initially described by Houk and Marshall for the related [2,3]-Wittig rearrangement (Scheme 2B).6
Scheme 2. Stereochemical Models for [2,3]-Rearrangements.

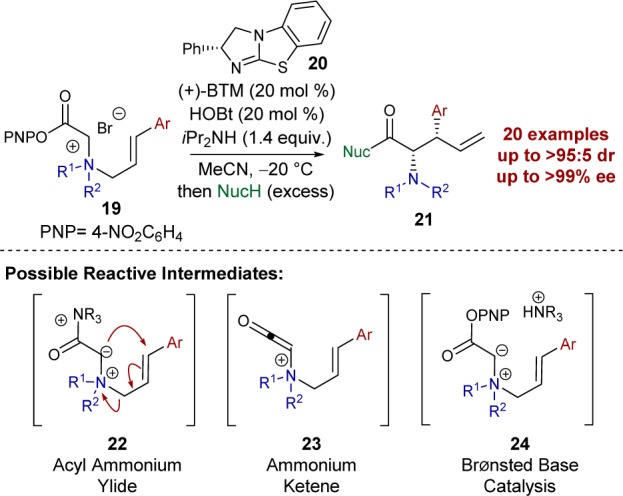
Prior to our studies within this area, only limited methods capable of imparting enantiocontrol in the [2,3]-rearrangement of allylic ammonium ylides had been developed. Sweeney first demonstrated a chiral auxiliary approach to allow access to enantiomerically enriched α-amino acid derivatives,7 while the use of a superstoichiometric chiral Lewis acid promoter was subsequently reported by Somfai.8 Catalytic enantioselective variants were unknown until 2014, when our laboratory reported an isothiourea-catalyzed9 [2,3]-rearrangement of allylic quaternary ammonium salts to give syn-α-amino acid derivatives with excellent levels of diastereo- and enantiocontrol (Scheme 3).10 Treatment of quaternary ammonium salts 19 bearing an activated p-nitrophenol ester, either isolated or generated in situ, with catalytic (+)-benzotetramisole ((+)-BTM) 20, cocatalytic hydroxybenzotriazole (HOBt), and iPr2NH, gave stereoselective [2,3]-rearrangement into syn-α-amino acid derivatives with excellent levels of stereocontrol. This process can be performed in the absence of HOBt; however, its addition provides a subtle enhancement in both diastereo- and enantioselectivity. We tentatively proposed a Lewis base catalytic cycle, initiated by nucleophilic addition of (+)-BTM 20 into the activated ester substrate to form an acyl ammonium intermediate prior to the formation of ammonium ylide 22. However, alternative mechanistic pathways using either Lewis or Brønsted base catalysis proceeding via different intermediates can be envisaged (Scheme 3). For example, assuming Lewis base catalysis is operative, the reaction could proceed through initial formation of a ketene intermediate 23 en route to acyl ammonium ylide 22. Furthermore, the origin of the observed diastereo- and enantiocontrol in the rearrangement process is currently unknown.
Scheme 3. Catalytic Enantioselective [2,3]-Rearrangement.
Herein we report experimental and computational investigations into the mechanism and origins of stereocontrol in the isothiourea-catalyzed [2,3]-rearrangement of allylic ammonium ylides (Scheme 1C). In situ NMR analysis has allowed a reaction profile to be elucidated, while isotopic-labeling studies have unambiguously identified a genuine productive catalytic intermediate. Kinetic analysis has given insight into the overall process, and crossover studies have provided information about the reversibility of each step. Kinetic isotope effects have also been used to probe the stereodetermining [2,3]-rearrangement step of the process. Computational reaction coordinate modeling provides deeper insight into the catalytic cycle, and transition state modeling reveals the origins of stereochemical control.
2. Results and Discussion
2.1. Mechanistic Studies
2.1.1. Temporal Concentration Profiles
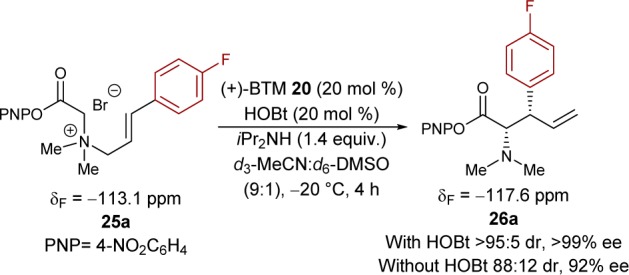
Initial studies aimed to establish the kinetics of the [2,3]-rearrangement and identify any reaction intermediate(s) or catalyst resting states. Ammonium salt 25a (34 mM) rearranges to 26a in d3-MeCN/d6-DMSO (9:1) at −20 °C, catalyzed by (+)-BTM 20 (20 mol %) (Scheme 4). The presence of various salts in the reaction medium causes extensive line broadening in the 1H NMR spectrum, making it unsuitable for in situ analysis of the [2,3]-rearrangement. However, the 19F{1H} and 13C{1H} NMR spectra were tractable, and the 4-fluoro substituent in 25a allows quantitative monitoring of the process by in situ19F{1H} NMR (δF = −113.1 ppm), with PhCF3 as an internal standard.11 After an initial burst phase (<1000 s), ammonium salt 25a is converted into 26a (>80% 26a, δF = −117.6 ppm) with pseudo-first-order kinetics, vide infra, over a period of 4 h. During the reaction evolution, a transient species (δF = −117.0 ppm) was detected, accumulating to a maximum concentration of ∼5.2 mM in the early stages of catalysis and then depleting as substrate 25a was consumed. Rearrangement in the absence of HOBt resulted in a similar reaction profile, but afforded higher concentrations of the same species (δF = −117.0 ppm), over a longer period, assisting in its analysis.
Scheme 4. System Chosen for in Situ19F NMR Study.
2.1.2. Identification of an On-Cycle Catalytic Intermediate
2.1.2.1. Catalyst Speciation
To determine if the transient species (δF = −117.0 ppm) involves the catalyst, a fluorinated variant,12 (+)-F-BTM 27, was prepared.11In situ monitoring of the [2,3]-rearrangement of 25a, catalyzed by 27 (Figure 1), confirmed conversion of free (+)-F-BTM 27 (δF = −123.3 ppm) into a catalyst-derived species (δF = −113.4 ppm), which was also transient, reaching a maximum concentration of 4.6 mM at ∼2500 s, and decaying as the reaction proceeds to completion (see inset graph to Figure 1). The comparable temporal intensities of the two signals (δF = −117.0 ppm and −113.4 ppm) strongly suggest they arise from a single transient intermediate containing both 25a and 27 in a formal 1:1 combination. Based on reference to isothiouronium salt 28 (δF = −113.3 ppm), the 19F chemical shift of the catalyst-derived component in transient species 29 suggested it to be an N-acylated isothiourea.
Figure 1.
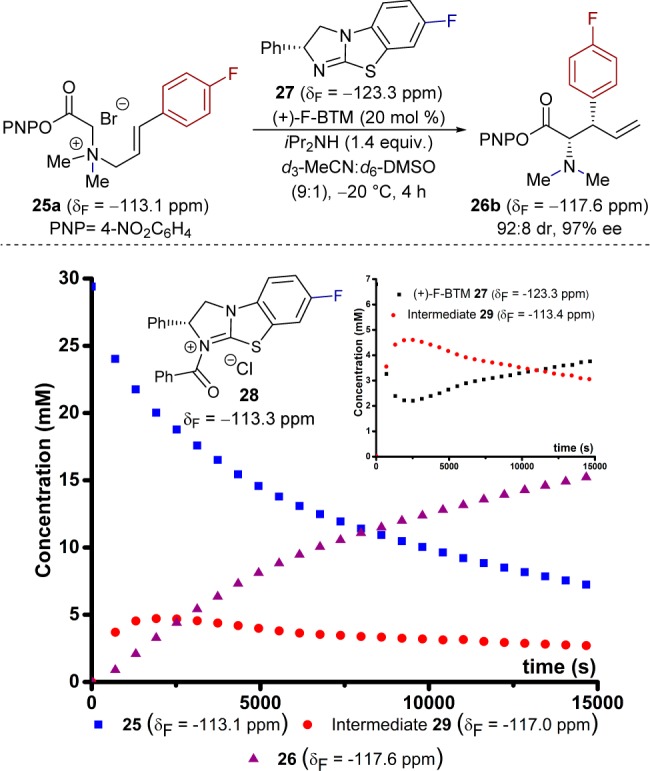
Temporal concentration data for [2,3]-rearrangement of 25a using (+)-F-BTM 27. Conditions: 25a (29.5 mM), (+)-F-BTM 27 (6.8 mM), iPr2NH (47 mM), d3-MeCN/d6-DMSO (9:1), −20 °C. Inset: monitoring of catalyst-derived species.
2.1.2.2. Atom Connectivity: 13C/15N Labeling
Isotopically labeled substrates and catalyst (1-[13C1]-25a, 1,2-[13C2]-25a, 1,2,3′-[13C3]-25b (Figure 2A) and (±)-[15N1]-27) were prepared11 to deduce connectivity between various atoms in the intermediate and probe for reversibility in its generation. Rearrangement of 1,2-[13C2]-25a (34 mM) catalyzed by (±)-[15N1]-27 (6.8 mM, 20 mol %) was monitored by 13C{1H} NMR (Figure 2B). Reducing the iPr2NH concentration to 23.8 mM prolonged the lifetime of the intermediate, [15N1,13C2]-29a, allowing detailed analysis of the 13C=O region, Figure 2C. The characteristic doublets (1JCC = 52 Hz) arising from the adjacent 13C labels, C(1)–C(2), in the substrate (1,2-[13C2]-25a) and the product (1,2-[13C2]-26a) are replaced by a double–doublet in [15N1, 13C2]-29a (δC = 173.1 ppm).13 The magnitude of the C–C coupling (1JCC = 52 Hz) confirms that C(2) remains sp3-hybridized. The magnitude of the additional coupling constant (1JCN = 5.2 Hz)13 indicates that C(1) is directly bound to the 15N-labeled atom in the catalyst, confirming 29a to be an N-acylated isothiourea (Figure 2).
Figure 2.
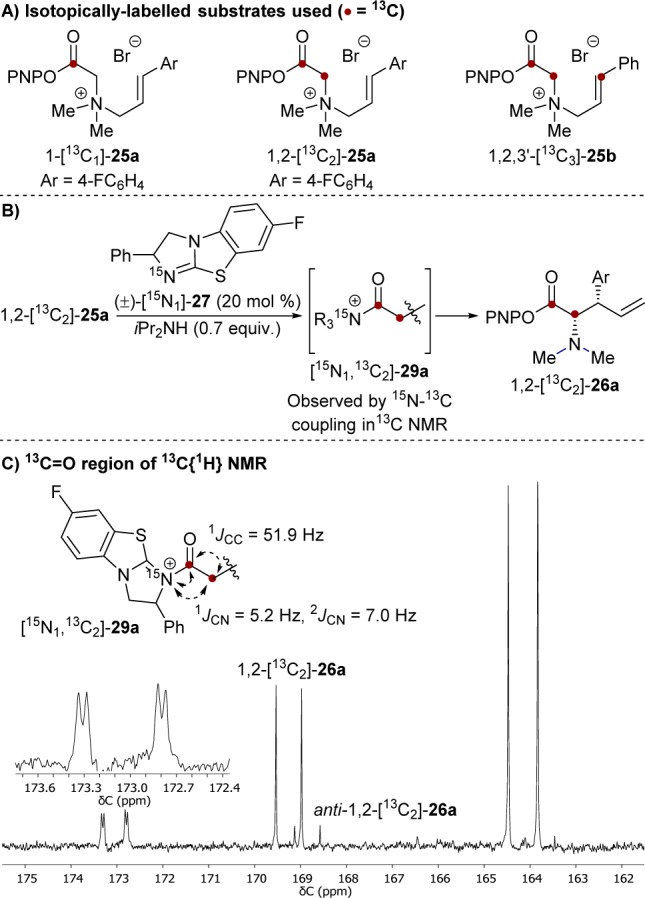
(A) Isotopically labeled substrates. (B) Generation of [15N1, 13C2]-labeled intermediate 29a. (C) 13C{1H} NMR subspectrum (101 MHz, d3-MeCN/d6-DMSO (9:1), 273 K) of the 13C=O region. Conditions: 1,2-[13C2]-25a (34 mM), (±)-15N-F-BTM [15N1-27] (6.8 mM), iPr2NH (23.8 mM).
Attempts to identitify the atom adjacent to C(2) in [15N1, 13C2]-29a from its 1H-coupled 13C NMR signal were thwarted by line broadening.13 Instead, the 13C{1H} NMR spectrum of intermediate 1,2,3-[13C3]-29b, generated from 1,2,3′-[13C3]-25b, was analyzed (Figure 3). The C(2) signal of the resulting [2,3]-rearrangement product 1,2,3-[13C3]-26b (δC = 70.5 ppm) displayed the expected double–doublet coupling arising from C(2)–C(3) bond formation. However, this coupling pattern was also evident in intermediate 1,2,3-[13C3]-29b (δC = 68.0 ppm, 1JCC 51.4 Hz, 1JCC 35.8 Hz) with the magnitude of the C(2)–C(3) coupling indicative of sp3-hybdrization at both centers.13 Overall the data confirms that the intermediate (29) is a catalyst-bound, post-[2,3]-rearrangement, acyl ammonium salt (Figure 3).
Figure 3.
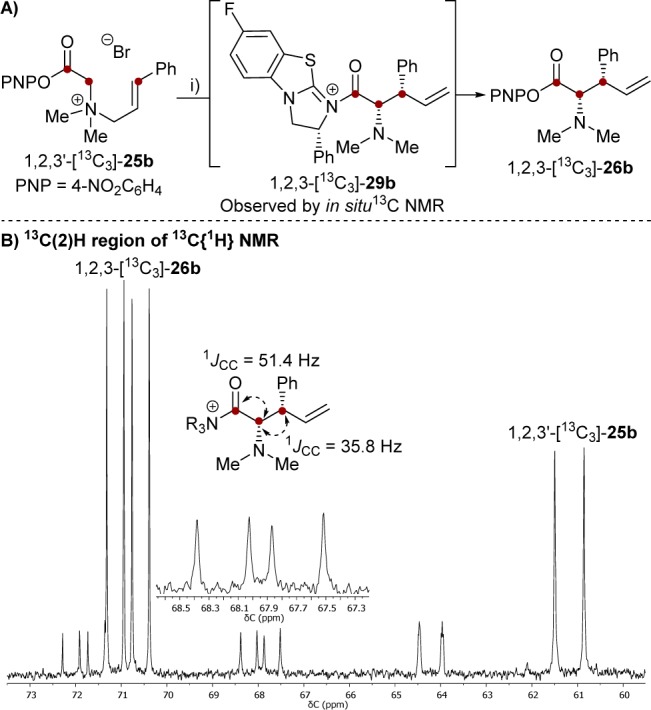
Catalyst-bound [2,3]-rearrangement product, 13C{1H} NMR subspectrum (101 MHz, d3-MeCN/d6-DMSO (9:1), 293 K) of the 13C(2)-H region. Conditions: 1,2,3′-[13C3]-25b (34 mM), (+)-F-BTM 27 (6.8 mM), iPr2NH (23.8 mM).
2.1.2.3. Productivity of the Intermediate
The data presented so far does not discriminate between the N-acylated isothiourea species (29) being peripheral to the productive catalytic cycle (Figure 4, case A) or an integral part of it (cases B–D). The following isotopic entrainment test distinguishes these four possibilities. A catalytic reaction employing 1-[13C1]-25a (17 mM) was allowed to evolve until 1-[13C1]-29a had reached its maximum concentration (∼5 mM). A further 1.0 equiv (17 mM) of a differently labeled substrate, 1,2-[13C2]-25a, was then rapidly added, resulting in an isotopic perturbation of the system. At the point that 1,2-[13C2]-25a is added, there has been 32% net conversion of 25a ([13C1]- and [13C2]-). However, neither of the [2,3]-rearrangement products (29a and 26a) yet contain any of the [13C2]-label: all of this resides in unreacted [13C1,2]-25a, which comprises 0.28 [13C1]/0.72 [13C2]. The key features are the changes in 13C2-populations in the substrate 25a, intermediate 29a and product 26a, as the reaction evolves. Irrespective of the pathway (A–D) the population in the final product (26a) must ultimately rise from 0% to 50%, as dictated by the equal proportions of 1-[13C1]-25a and 1,2-[13C2]-25a added overall. For case A, where 29a is not productive, the isotope population in 29a will depend only on that of the final product (26a; max 50% 13C2) and at all stages will be lower or equal to it. For case B, where the intermediate is productive, but is in equilibrium with 25a, the 13C2-population in 25a will be reduced, in the limit from 72% to 55%. For cases C and D, where the [2,3]-rearrangement to 29a is irreversible, the isotope population in 25a is constant (72% 13C2-) and the 13C2-content in 29a rises from 0% to a maximum of 72% as it is repopulated from 25a.14 However, for case C, equilbration of 29a with product 26a will attenuate the rise in 13C2-population in 29a, in the limit to 50%. Only for case D will the 13C2 isotope population in 29a rise, in advance of 26a, to reach a maximum 72% 13C2.14 Comparison of the predicted and experimentally determined 13C2-populations as a function of net conversion (Figure 4) confirms that 26 arises from two irreversible sequential first-order interconversions (25 → 29 → 26) where 29 is the productive catalytic intermediate (case D).14 Kinetic modeling confirms that the impact of heavy-atom (12C/13C) KIEs on the isotope-entrainment are negligible.11
Figure 4.
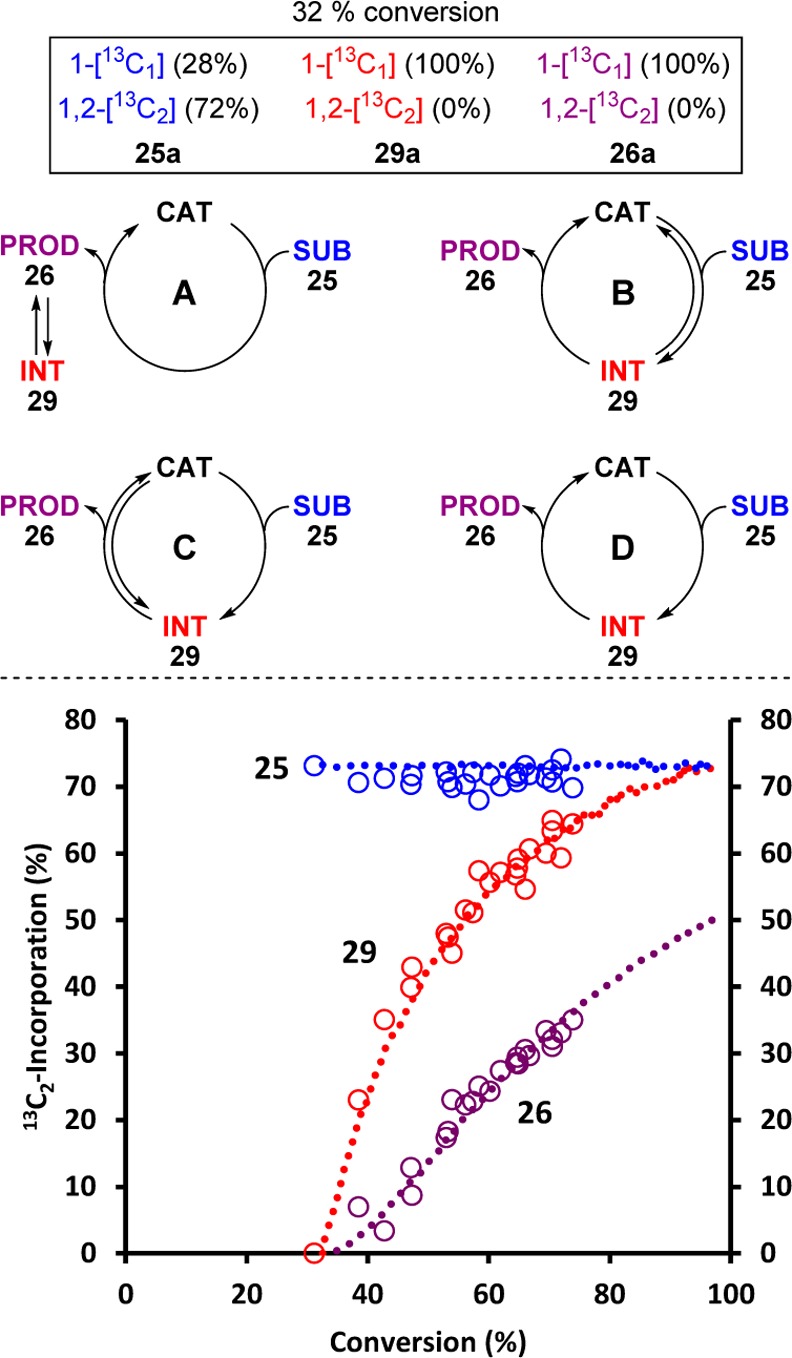
13C2 Entrainment into the catalytic cycle. Conditions: 1-[13C1]-25a (17 mM), (+)-BTM (6.8 mM), iPr2NH (47.6 mM), d3-MeCN/d6-DMSO (9:1), with 1,2-[13C2]-25a (17 mM) added at 32% net conversion of 25a. Open circles: experimental (13C{1H} NMR) data for 13C2-incorporation (%) versus net conversion (%). Dashed lines: kinetic simulation where 29 is a productive intermediate in two irreversible sequential pseudo-first-order interconversions (25 → 29 → 26, Case D; with rate ratio 0.407).14
2.1.3. HOBt Cleavage of Acyl Ammonium Intermediate
HOBt provides optimal diastereo- and enantiocontrol (Scheme 4); however, its role within the catalytic cycle is unclear. To probe if the HOBt enhances stereocontrol via suppression of the base-mediated background reaction, the iPr2NH-mediated rearrangement of 25a was examined. Reaction of 25a with iPr2NH (47 mM) in the absence of the BTM catalyst resulted in slow formation of racemic 26a with low diastereocontrol (79:21 dr) and a kobs of 2.74 × 10–5 s–1. The addition of a catalytic amount of HOBt (6.8 mM) resulted in no change in rate (kobs 2.70 × 10–5 s–1). This rules out the role of HOBt as improving stereocontrol through direct suppression of the rate of the background reaction.
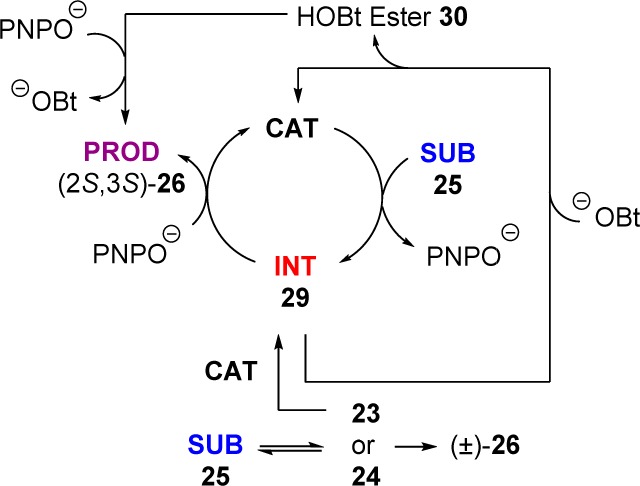
The (+)-BTM-catalyzed rearrangement of 25a in the presence of stoichiometric HOBt (34 mM) was studied by 19F{1H} NMR. The presence of HOBt strongly suppressed accumulation of acyl ammonium intermediate 29c ((+)-BTM replaces (+)-F-BTM) and resulted in the formation of the corresponding HOBt ester 30 (δF = −117.2 ppm, confirmed by comparison with an authentic sample),11 in addition to the PNPO ester product 26a. Addition of HOBt once the acyl ammonium intermediate 29c reached the pseudo steady-state (5 mM, t = 3920 s, Figure 5) resulted in immediate formation of HOBt ester 30 and consumption of acyl ammonium intermediate 29c. HOBt thus shifts the catalyst speciation to be strongly dominated by free (+)-BTM (Figure 5). The background iPr2NH-mediated reaction that converts 25 into racemic (±)-26 presumably involves the generation of ylide intermediate 24. Interception of this species by free (+)-BTM would also generate intermediate 29, thus leading to nonracemic 26. The higher the concentration of free BTM, the more effective this interception.
Figure 5.
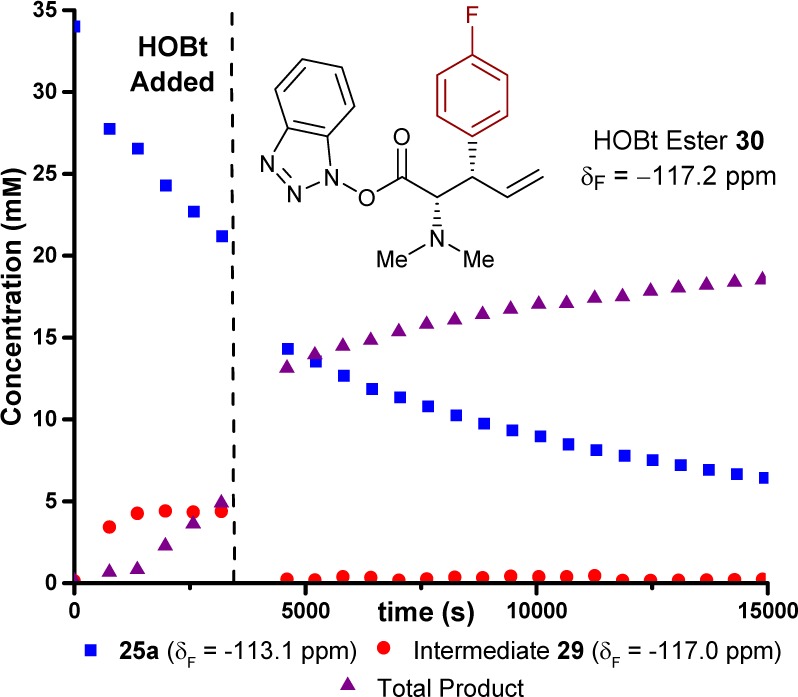
Addition of stoichiometric HOBt at t = 3920 s. Conditions: 25a (34 mM), (+)-BTM (6.8 mM), iPr2NH (47.6 mM), d3-MeCN/d6-DMSO (9:1), HOBt (34 mM). Total Product refers to both HOBt ester 30 and 26a.
Overall, the beneficial effect of HOBt on the selectivity may arise from both a change in catalyst speciation to favor free BTM and by diversion of the background reaction onto the enantioselective pathway (Scheme 5).
Scheme 5. Effect of HOBt on Overall Catalytic Cycle.
2.1.4. Reaction Kinetics and Impact of Additives
Having identified, by 19F{1H} and 13C{1H} NMR, the major reactant-derived and catalyst-derived components present in the reaction mixture, the empirical rate equation was established by analysis of the decay in substrate 25a during the pseudosteady-state phase of the catalysis (Scheme 6).15 The standard conditions [25a (34 mM), (+)-BTM (6.8 mM), HOBt (6.8 mM), and iPr2NH (47 mM)], afforded a pseudo-first-order rate constant, kobs = 1.37 × 10–4 s–1.
Scheme 6. Empirical Rate Equation,
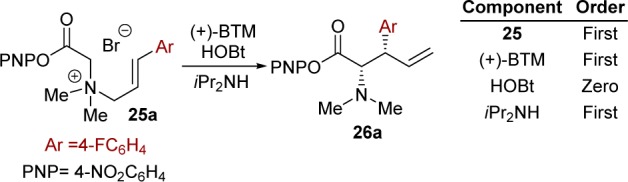
d[26a]/dt = kobs[25a]; kobs = k[BTM][NR3].
Conditions: 25a (17–68 mM), (+)-BTM (1.7–13.6 mM), HOBt (0–34 mM), iPr2NH (23.8–95.2 mM), d3-MeCN/d6-DMSO (9:1), 253 K, 4 h.
There was a linear relationship between the enantiopurity of (+)-BTM and product 26a, consistent with predominant or exclusive speciation of the catalyst in an active, monomeric, form.11 Systematic variation of the concentration of the reaction components afforded empirical first-order dependencies on (+)-BTM (1.7–13.6 mM) and diisopropylamine (23.8–95.2 mM), with no rate impact from the HOBt (0–34 mM). Addition of n-Bu4N 4-nitrophenoxide (27.2 mM) in the absence of HOBt resulted in a large increase in rate (kobs = 2.42 × 10–4 s–1) and reduced the accumulation of the [2,3]-rearrangement acyl ammonium intermediate 29c (≤2 mM). Control studies showed that n-Bu4NBr (27.2 mM) resulted in a similar rate enhancement (kobs = 2.35 × 10–4 s–1) but did not suppress the accumulation of 29c (5 mM). The observed increase in kobs may be rationalized by an increase in ionic strength of the medium.11 The n-Bu4N 4-nitrophenoxide thus promotes catalyst turnover of acyl ammonium 29c into product 26a and (+)-BTM. Addition of 4-nitrophenol (0–34 mM) resulted in a decrease in rate, with a negative first-order dependency. Overall, this suggests that 4-nitrophenoxide or benzotriazololate is required for efficient turnover of acyl ammonium 29c into products 26a/30 and (+)-BTM.
2.1.5. Secondary Kinetic Isotope Effect (SKIE)
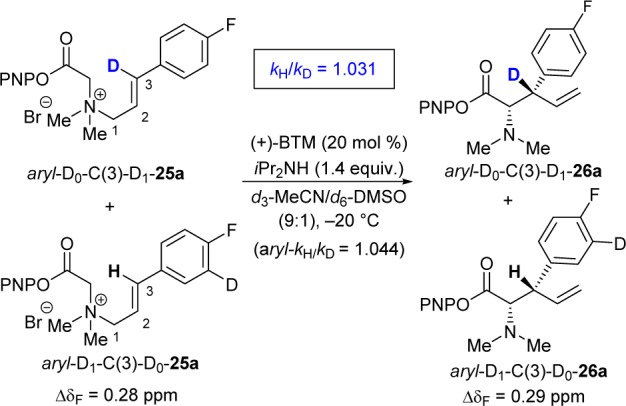
Although it is clear that the [2,3]-rearrangement to generate 29 is irreversible, it is not evident whether this is the product-determining step, i.e. the first irreversible step in the cycle. During the [2,3]-rearrangement step, the carbon that becomes C(3)–H in 29 undergoes rehybridization from sp2 to sp3. The process to generate a C(3)–D isotopologue of 29 would be thus expected to exhibit a 2H-SKIE. Alternatively, if deprotonation at C(2)–H to generate an allylic ammonium ylide (Scheme 3) was the product-determining step, there would not be any significant SKIE, as C(3)–D is remote.
The SKIE was measured by competition using aryl-D1-C(3)-D0-25a and aryl-D0-C(3)-D1-25a, in a double-labeling method16 in which the C(3)–D/C(3)–H ratio as a function of fractional conversion is determined by 19F NMR (ΔδF = 0.28 ppm; aryl-D0/aryl-D1). After correction for the effect of aryl deuteration,17,18 a value of kH/kD = 1.031 was obtained. The presence of a small positive SKIE is consistent with a product-determining [2,3]-rearrangement transition state (Scheme 7). A linear free energy relationship analysis of a range of C(3)-aryl substrates, against standard Hammett sigma values, showed the C(3) position to be relatively insensitive to electronic substituent effects.11
Scheme 7. C(3) SKIE Competition Experiment.
2.1.6. Crossover and Reversibility Studies
We have previously demonstrated the [2,3]-rearrangement step to be intramolecular and irreversible.10 To distinguish which steps prior to the [2,3]-rearrangement are reversible, a crossover reaction between ammonium salts 25a and 31 (1:1) bearing two distinct activated ester groups (4-NO2C6H4 and 3,5-(CF3)2C6H3) and two distinct C(3)-aryl units (4-FC6H4 and 3-FC6H4) was monitored in situ under catalytic conditions (Scheme 8A). Complete equilibration with ammonium salts 32 and 33 was observed,11 consistent with reversible generation of nonrearranged (+)-BTM acyl ammonium intermediates. To examine reversibility at the deprotonation step, the [2,3]-rearrangement of α-dideuterio ammonium salt α-[D2]-25 (75% D2) was monitored in situ. The product was obtained with significantly lower deuterium incorporation (29% D), consistent with a reversible deprotonation step (Scheme 8B).19 Reaction of ammonium salt 33 in the presence of rearrangement product 26b (Scheme 8C) bearing distinct C(3)-aryl units and activated esters demonstrated no crossover, consistent with catalyst turnover being irreversible, confirming the conclusions deduced from isotopic entrainment.
Scheme 8. Crossover Studies.

(i) (+)-BTM (20 mol %), iPr2NH (1.4 equiv), 25a (0.5 equiv), 31 (0.5 equiv), d3-MeCN/d6-DMSO (9:1), 253 K, 4 h; (ii) (+)-BTM (20 mol %), HOBt (20 mol %), iPr2NH (1.4 equiv), 26b (0.5 equiv), 33 (0.5 equiv), MeCN, 253 K.
2.2. Computational Studies
2.2.1. Computed Catalytic Cycle
We computed all intermediates, transition structures (TSs), and possible salt complexes involved in the catalytic cycle shown in Scheme 9. Exhaustive searches were performed to locate all pertinent conformations. Geometries and thermodynamic corrections were computed at the M06-2X20/6-31G(d)21 level of theory.22 Vibrational frequencies and thermal corrections to the Gibbs free energy were calculated at −20 °C and 1 atm to match the experimental conditions. Further energy refinements were completed using M06-2X/6-311++G(2df,p).23 Implicit solvation corrections were applied using the polarized continuum model (PCM)24 with UFF radii for acetonitrile in both the geometry optimizations and the single-point energy refinements. The hybrid meta-GGA functional M06-2X is generally more robust than B3LYP at accounting for dispersion and nonbonding interactions routinely found in organocatalytic reactions.25 Kinetic isotope effects were calculated using the theory of Bigeleisen and Mayer26 along with the rigid-rotor harmonic oscillator approach (ΔHΔS).27 Quantum mechanical tunneling effects were also calculated for both methods using the one-dimensional parabolic approximation.28 The calculation of the KIE was automated by use of the Onyx isotope effect program.29 The catalytic cycle and the computed reaction coordinate are summarized in Scheme 9.
Scheme 9. Proposed Catalytic Cycle and Computed Reaction Coordinate.

Direct acylation begins with BTM attack on allylic ammonium activated substrate (TS-II, ΔG‡ = 14.8 kcal·mol–1) to form tetrahedral intermediate III. Release of PNPO– (TS-IV, ΔG‡ = 12.0 kcal·mol–1) gives dication V. Indirect acylation through formation of the ammonium ketene III′ was ruled an unlikely reactive intermediate based on its unfavorable thermodynamics (ΔG‡ = 22.0 kcal·mol–1). The endergonicity of dication V (ΔG‡ = 3.3 kcal·mol–1) confirms the observed reversibility of catalyst acylation. Dication V is in equilibrium with ylide VII (ΔG = 1.6 kcal·mol–1) through deprotonation of the α-proton of V by PNPO– (ΔG‡ = 11.4 kcal·mol–1), also in agreement with the experimentally observed reversibility of the deprotonation step.30,31
NBO analyses reveal significant enolate character of ylide intermediate VII. Intermediate VII subsequently undergoes stereoselective and turnover-rate limiting [2,3]-rearrangement (TS-VIII-(2S,3S)-Major, ΔG‡ = 17.3 kcal·mol–1) to yield enantio- and diastereoenriched acyl ammonium product–catalyst complex IX. Catalyst turnover is found to be stepwise, begins with PNPO– attack (TS-X) and ends with catalyst and product release (TS-XII). The barrier for PNPO– attack as calculated from intermediate IX (ΔG‡ = 16.9 kcal·mol–1) indicates that, in the absence of HOBt, this step is highly competitive with rearrangement as turnover-rate limiting.
2.2.2. Effect of Counterions on the Theoretical KIE
The presence of counterions posed a challenge to the accuracy of DFT and significantly increased the complexity of the conformational search and the number of relevant structures to consider.32 Almost all species present in the catalytic cycle prior to catalyst turnover bear a positive charge, with intermediate V being dicationic. Species indicated to include a counterion in Scheme 9 were optimized with the explicit ion shown. Given the charged nature of the species present, the identification of the structures that compose the free energy span33 resulted from considering all possible counterion coordination combinations for all conformations of each charged species in the catalytic cycle. This exhaustive process led to the identification of acyl substrate I and TS-VIII as the two states contributing to the free energy span. Computed barriers show the rearrangement step as the first irreversible step of the mechanism, thereby allowing kinetic isotopic fractionation to occur. The computed KIE then depends on the vibrational frequencies of I and TS-VIII, and could then be utilized to corroborate the computed thermodynamics and barriers of this free energy span.34
In a multistep reaction with highly charged and zwitterionic species, leveraging KIE prescribes a means to identify not only the structures that compose the free energy span33 but also which ions coordinate, the specific binding site of the counterion,35 and the conformation.27b,36 We sought to identify the coordination state of TS-VIII by leveraging both the KIE and computed barriers for TS-VIII-F, i.e. bearing the 4-fluoro substituent used for KIE determination (Scheme 7). Coordination to TS-VIII-F, formation of byproduct salt complexes, and conformations all affect the barrier in going from I-F to TS-VIII-F. Two possible counterions, PNPO– and Br–, were considered as TS counterions, while iPr2NH2+ was evaluated as a component of the possible remaining complexes (Figure 6). No coordination to the TS (Figure 6, left) leaves H-bond complex PNPOH···iPr2NH as the lowest-energy remaining complex, giving an overall ΔG‡ = 18.3 kcal·mol–1. Bromide ion binding to the TS (TS-VIII-F-(2S,3S)···Br, Figure 6, middle) also leaves complex PNPOH···iPr2NH (ΔG‡ = 18.7 kcal·mol–1). PNPO– binding and the complexation of iPr2NH2+ and Br– gives an even higher barrier (TS-VIII-F-(2S,3S)···PNPO, ΔG‡ = 22.8 kcal·mol–1). With no counterion coordination to the TS (TS-VIII-F-(2S,3S)-Major), the KIEcomp of 1.028 matches well with experiment (KIEexp = 1.031). Bromide complexation, which is 0.4 kcal·mol–1 higher, also matches fairly closely, giving a KIEcomp of 1.041.38 PNPO– complexation leads to an erroneously large magnitude of rate difference between kH/kD, yielding a KIEcomp of 1.050.
Figure 6.

Computed TSs, ions, complexes, and KIEs involving the 4-fluoro substituted substrate I-F. The computed KIE depends on the coordination state of substrate I-F and TS-VIII-F. The violet highlighted atom is the isotopic proton (H/D).37 All energies in kcal·mol–1. Shaded gray lines represent forming/breaking bonds. Green lines represent C–H electrostatic interactions and hydrogen bonds.
2.2.3. Stereocontrol Model
The computed diastereomeric [2,3]-rearrangement TSs are shown in Figure 8. All TSs feature concerted C–C bond formation and ammonium N–C bond cleavage.4,6 The four main elements that control the stereochemical outcome of the reaction are as follows (Figure 8):
Figure 8.
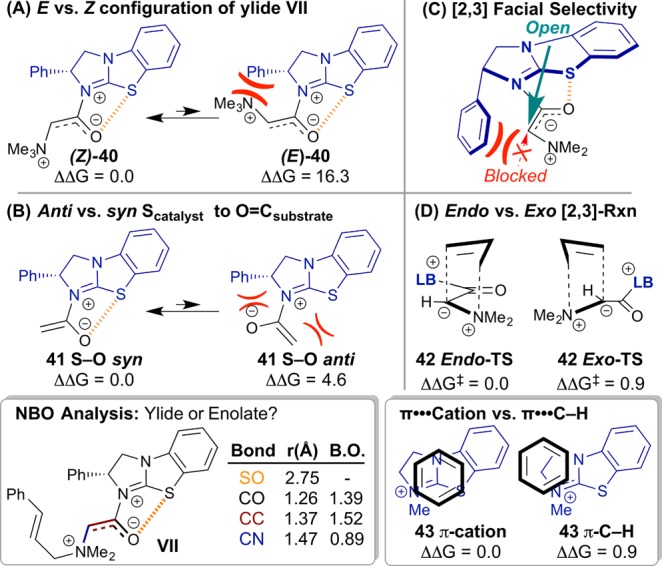
Computed model systems (all energies in kcal·mol–1 and distances in Å). (A) Preference for Z over E enolate. Enolate-like character indicated by bond orders (B. O.) estimated from the Wiberg bond indices (bottom left inset). (B) Effect of S···O interaction on acylated catalyst conformation. (C) With the acylated catalyst conformation held rigid (S···O), the BTM stereodirecting Ph sterically biases open enolate face. (D) Endo rearrangement is favored. In the major TS, this preference is reinforced by a π–cation interaction (bottom right inset).
2.2.3.1. E vs Z Configuration of the Enolate in Ylide VII
NBO analysis indicates that both ylide VII and TS-VIII-(2S,3S)-Major have significant enolate character.39 Ylide VII displays a C–O bond order of 1.39 and a C–C bond order of 1.52 (Figure 8, bottom left inset), while TS-VIII-(2S,3S)-Major displays a C–O bond order of 1.54 and a C–C bond order of 1.21.11 The computed bond order of 1.52 for ylide VII suggests partial C–C double bond character leading to distinct isomeric E and Z enolate configurations prior to rearrangement with the configurations set in place by the deprotonation step. The Z-configuration is heavily favored over the E, as shown in the model system Z/E-35 where the Z is favored by >16 kcal·mol–1. All stable [2,3]-rearrangement transition structures feature the Z-enolate.
2.2.3.2. Anti vs syn Scatalyst to Osubstrate Orientation
In all the lowest energy conformations of the ylide-VII and rearrangement TS-VIII, the S–O relationship is syn. The syn distances (∼2.7–2.8 Å) are significantly below the sum of the van der Waals radii (3.4 Å), indicating close-contact S···O interactions (Figures 7 and 8, orange lines).40 Computed model systems show >4 kcal·mol–1 preference for the conformation which contains the 1,5-S···O interaction (anti/syn-36, Figure 8). All [2,3]-rearrangement TSs that do not bear the S···O interaction are higher by >6 kcal·mol–1.11 The conformational bias toward the S–Osyn arrangement is proposed to result from nO to σ*C–S delocalization coupled with electrostatic attraction of the partially positive sulfur atom and partially negative oxygen atom.41
Figure 7.
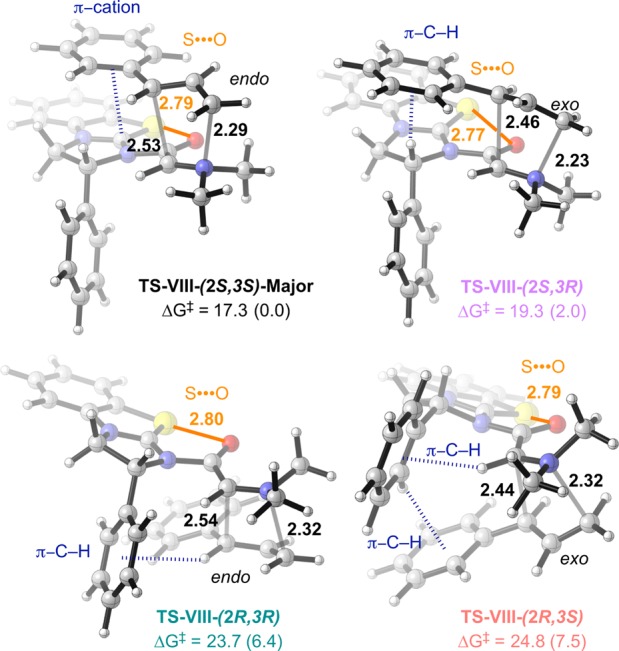
Stereodetermining [2,3]-rearrangement TSs.34 All energies in kcal·mol–1, and distances in Å. Shaded gray lines represent forming/breaking bonds. Solid orange lines represent nonbonding S···O interactions. Dashed blue lines represent aromatic interactions.
2.2.3.3. Facial Selectivity of Rearrangement
The S···O interaction significantly rigidifies the ylide-VII structure, leaving conformational freedom only to the substrate cinnamyl group. With rearrangement possible from either face of the catalyst isothiourea plane, the facial selectivity is controlled by the catalyst Ph stereodirecting group. The most favorable [2,3]-TSs favor approach opposite to this group (TS-VIII-(2S,3S)-Major and TS-VIII-(2S,3R), Figure 7). Approach on the same side as the stereodirecting Ph is disfavored by >6 kcal·mol–1 (TS-VIII-(2R,3R) and TS-VIII-(2R,3S)).
2.2.3.4. Endo vs exo [2,3]-TS
Rearrangement can occur either endo or exo with respect to the substrate C=O (Figure 8). In the simple allyl model TS, the endo/exo preference is ∼1 kcal·mol–1. This preference is ∼2 kcal·mol–1 between TS-VIII-(2S,3S)-Major and TS-VIII-(2S,3R), and additional interactions contribute to the diastereoselectivity. In the major TS there is a π–cation interaction,42 which is favored over the π-C–H interaction found in the minor.43 Truncated fully optimized model systems probing the difference in energy between these interactions in the context of cationic BTM reveal a ∼1 kcal·mol–1 preference for 38 π-cation over 38 π-C–H (Figure 8, bottom right inset).11 These two factors contribute to the computed 2 kcal·mol–1 preference for TS-VIII-(2S,3S)-Major over TS-VIII-(2S,3R), in good agreement with the experimental selectivity of 1.5 kcal·mol–1.
3. Conclusions
The experimental and computational investigation reported herein has provided mechanistic and stereochemical insight into the enantioselective isothiourea-catalyzed [2,3]-rearrangement of allylic ammonium ylides. Analysis by in situ19F NMR has allowed reaction profiles to be established and has identified an intermediate species.
Isotopic labeling of catalyst (15N) and substrate (13C) has confirmed the constitution of the catalytic intermediate as 29/IX by 13C NMR. Isotopic entrainment has shown 29/IX to be an irreversibly generated intermediate that is productive toward catalysis. A series of crossover experiments have provided detailed information regarding the reversibility of each individual step of the catalytic cycle. The turnover-rate limiting step of the process varies between product release and [2,3]-rearrangement, depending on substrate conversion. The effect of excess HOBt upon the reaction is to accelerate product release, thus generating a greater proportion of the free BTM catalyst (Figure 5). This may then result in more effective interception of the background racemic reaction, and thus greater diversion onto the enantioselective pathway (Scheme 5). Computational analysis has provided finer detail for the fundamental steps in the catalytic cycle as well as the key interactions that control the stereochemical outcome of the process. The insight gained into this process will have implications in a wider context, especially in the use of activated esters in Lewis base catalysis, which is currently under investigation in our laboratories.44
Acknowledgments
We thank Dr. Carl Poree, Dr. Lorna Murray, Mr. Juraj Bella, Dr. Dušan Uhrín (all Edinburgh), Mrs. Melanja Smith and Dr. Tomas Lebel (both St Andrews) for assistance with NMR experiments. The research leading to these results (T.H.W., J.E.T., G.C.L.-J., and A.D.S.) has received funding from the ERC under the European Union’s Seventh Framework Programme (FP7/2007-2013)/E.R.C. Grant Agreement Nos. 279850 and 340163. A.D.S. thanks the Royal Society for a Wolfson Research Merit Award. We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University. P.H.-Y.C. is the Bert and Emelyn Christensen Professor and gratefully acknowledges financial support from the Stone Family of OSU. Financial support from the National Science Foundation (NSF) (CHE-1352663) is acknowledged. D.M.W. acknowledges the Bruce Graham and Johnson Fellowships of OSU. A.C.B. acknowledges the Johnson Fellowship of OSU. D.M.W., A.C.B., R.C.J., and P.H.-Y.C. also acknowledge computing infrastructure in part provided by the NSF Phase-2 CCI, Center for Sustainable Materials Chemistry (CHE-1102637).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b11851.
Additional discussion, kinetic data, experimental procedures, characterization data, NMR spectra and HPLC chromatograms, computed geometries, energies, and vibrational frequencies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Sweeney J. B. Chem. Soc. Rev. 2009, 38, 1027–1038. 10.1039/b604828p. [DOI] [PubMed] [Google Scholar]; b West T. H.; Spoehrle S. S. M.; Kasten K.; Taylor J. E.; Smith A. D. ACS Catal. 2015, 5, 7446–7479. 10.1021/acscatal.5b02070. [DOI] [Google Scholar]
- Clark J. S.Nitrogen, Oxygen, and Sulfur Ylide Chemistry: A Practical Approach in Chemistry; Oxford University Press: New York, 2002. [Google Scholar]
- Kennedy C. R.; Guidera J. A.; Jacobsen E. N. ACS Cent. Sci. 2016, 2, 416–423. 10.1021/acscentsci.6b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas B.; Collins S. C.; Singleton D. A. J. Am. Chem. Soc. 2014, 136, 3740–3743. 10.1021/ja4128289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Soheili A.; Tambar U. K. J. Am. Chem. Soc. 2011, 133, 12956–12959. 10.1021/ja204717b. [DOI] [PubMed] [Google Scholar]; b Nash A.; Soheili A.; Tambar U. K. Org. Lett. 2013, 15, 4770–4773. 10.1021/ol402129h. [DOI] [PubMed] [Google Scholar]; c Soheili A.; Tambar U. K. Org. Lett. 2013, 15, 5138–5141. 10.1021/ol4025937. [DOI] [PubMed] [Google Scholar]
- Wu Y. D.; Houk K. N.; Marshall J. A. J. Org. Chem. 1990, 55, 1421–1423. 10.1021/jo00292a007. [DOI] [Google Scholar]
- Workman J. A.; Garrido N. P.; Sançon J.; Roberts E.; Wessel H. P.; Sweeney J. B. J. Am. Chem. Soc. 2005, 127, 1066–1067. 10.1021/ja043768i. [DOI] [PubMed] [Google Scholar]
- a Blid J.; Panknin O.; Somfai P. J. Am. Chem. Soc. 2005, 127, 9352–9353. 10.1021/ja0510562. [DOI] [PubMed] [Google Scholar]; b Blid J.; Panknin O.; Tuzina P.; Somfai P. J. Org. Chem. 2007, 72, 1294–1300. 10.1021/jo062178v. [DOI] [PubMed] [Google Scholar]
- Isothioureas have been widely explored as Lewis base catalysts across a range of organocatalytic processes. For a seminal report, see:; Birman V. B.; Guo L. Org. Lett. 2006, 8, 4859–4861. 10.1021/ol061906y. [DOI] [PubMed] [Google Scholar]; For reviews on isothiourea catalysis, see:; a Taylor J. E.; Bull S. D.; Williams J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. 10.1039/c2cs15288f. [DOI] [PubMed] [Google Scholar]; b Merad J.; Pons J.-M.; Chuzel O.; Bressy C. Eur. J. Org. Chem. 2016, 2016, 5589–5610. 10.1002/ejoc.201600399. [DOI] [Google Scholar]
- West T. H.; Daniels D. S. B.; Slawin A. M. Z.; Smith A. D. J. Am. Chem. Soc. 2014, 136, 4476–4479. 10.1021/ja500758n. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for details.
- (+)-F-BTM 27 (20 mol %), HOBt (20 mol %) gave comparable reactivity and stereocontrol (68% yield, 92:8 dr, 97% ee) to the reaction using (+)-BTM 20 under the same conditions.
- Typical range for one bond carbon–carbon coupling constants: sp2–sp2 1JCC 75–80 Hz, sp2–sp3 1JCC 35–60 Hz, sp3–sp3 1JCC 25–55 Hz:; Hansen P. E.; Wray V. Org. Magn. Reson. 1981, 15, 102–103. 10.1002/mrc.1270150122. [DOI] [Google Scholar]; The magnitude of observed 1JCN and 2JCN coupling constants are consistent with typical coupling constants (1,2JCN) reported in the literature:; Freyer V. W. Z. Chem. 1981, 21, 47–58. 10.1002/zfch.19810210203. [DOI] [Google Scholar]; See Supporting Information for select examples. A 2JCH value of ∼1–2 Hz would be expected for coupling between C(2) and C(3)H in the intermediate if the [2,3]-rearrangement has taken place. The presence or absence of this coupling could not be unambiguously established.
- The maximum 13C2 population attainable in the intermediate is dictated by the relative rate of build-up and decay of the intermediate. The simulation used in Figure 4 employs a rate ratio for step 1 vs step 2 (k1/k2) of 0.407 leading to a maximum 6 mM concentration of the intermediate, analogous to that found experimentally under catalytic conditions.
- An initial drop in substrate concentration is observed and after ∼3000 s. 25a decays with good pseudo-first-order kinetics.
- A direct competition experiment analyzed by 1H NMR was not possible due to siginifcant line broadening. To allow 19F NMR to be used, a double-labeling experiment was employed.
- Gonzalez J. A.; Ogba O. M.; Morehouse G. F.; Rosson N.; Houk K. N.; Leach A. G.; Cheong P. H. Y.; Burke M. D.; Lloyd-Jones G. C. Nat. Chem. 2016, 8, 1067–1075. 10.1038/nchem.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Perrin C. L.; Dong Y. J. Am. Chem. Soc. 2007, 129, 4490–4497. 10.1021/ja069103t. [DOI] [PubMed] [Google Scholar]; b Pehk T.; Kiirend E.; Lippmaa E.; Ragnarsson U.; Grehn L. J. Chem. Soc., Perkin Trans. 2 1997, 445–450. 10.1039/a606790e. [DOI] [Google Scholar]
- Retreatment of the PNP ester products under the reaction conditions resulted in no epimerization of the products.
- Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]
- Frisch M. J.et al. Gaussian 09; Gaussian, Inc.: Wallingford CT, 2009. See Supporting Information for full citation.
- Hariharan P. C.; Pople J. A. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]
- Miertuš S.; Scrocco E.; Tomasi J. Chem. Phys. 1981, 55, 117–129. 10.1016/0301-0104(81)85090-2. [DOI] [Google Scholar]
- a Wheeler S. E.; Seguin T. J.; Guan Y.; Doney A. C. Acc. Chem. Res. 2016, 49, 1061–1069. 10.1021/acs.accounts.6b00096. [DOI] [PubMed] [Google Scholar]; b Sunoj R. B. Acc. Chem. Res. 2016, 49, 1019–1028. 10.1021/acs.accounts.6b00053. [DOI] [PubMed] [Google Scholar]; c Walden D. M.; Ogba O. M.; Johnston R. C.; Cheong P. H.-Y. Acc. Chem. Res. 2016, 49, 1279–1291. 10.1021/acs.accounts.6b00204. [DOI] [PubMed] [Google Scholar]; d Gould E.; Walden D. M.; Kasten K.; Johnston R. C.; Wu J.; Slawin A. M. Z.; Mustard T. J. L.; Johnston B.; Davies T.; Cheong P. H.-Y.; Smith A. D. Chem. Sci. 2014, 5, 3651–3658. 10.1039/C4SC00879K. [DOI] [Google Scholar]
- Bigeleisen J.; Mayer M. G. J. Chem. Phys. 1947, 15, 261–267. 10.1063/1.1746492. [DOI] [Google Scholar]
- a Fong A.; Meyer M. P.; O’Leary D. J. Molecules 2013, 18, 2281–2296. 10.3390/molecules18022281. [DOI] [PMC free article] [PubMed] [Google Scholar]; b O’Leary D. J.; Rablen P. R.; Meyer M. P. Angew. Chem., Int. Ed. 2011, 50, 2564–2567. 10.1002/anie.201007322. [DOI] [PubMed] [Google Scholar]
- a Northrop D. B. J. Am. Chem. Soc. 1999, 121, 3521–3524. 10.1021/ja984177t. [DOI] [Google Scholar]; b Bell R. P.The Tunnel Effect in Chemistry; Chapman and Hall: New York, 1980. [Google Scholar]; c Bell R. P. Chem. Soc. Rev. 1974, 3, 513–544. 10.1039/cs9740300513. [DOI] [Google Scholar]
- Kinetic isotope effects calculated using the Onyx program:Brueckner A. C.; Cevallos S. L.; Ogba O. M.; Walden D. M.; Meyer M. P.; O’Leary D. J.; Cheong P. H.-Y.. Onyx, version 1.0; Oregon State University: Corvallis, OR, USA, 2016.
- Deprotonation by iPr2NH was also computed, but gave a much higher barrier of 20.5 kcal·mol–1. See Supporting Information for structural details.
- The presence of an ylide intermediate has been confirmed in the closely related [1,2]-Stevens rearrangement of ammonium acetophenones:Capobianco A.; Caruso T.; Palombi L.; Peluso A. Electrochim. Acta 2013, 92, 446–451. 10.1016/j.electacta.2013.01.079. [DOI] [Google Scholar]
- Bedin M.; Karim A.; Reitti M.; Carlsson A-C. C.; Topić F.; Cetina M.; Pan F.; Havel V.; Al-Ameri F.; Sindelar V.; Rissanen K.; Gräfenstein J.; Erdélyi M. Chem. Sci. 2015, 6, 3746–3756. 10.1039/C5SC01053E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free energy span is the energetic counterpart of turnover frequency:; a Rohmann K.; Hölscher M.; Leitner W. J. Am. Chem. Soc. 2016, 138, 433–443. 10.1021/jacs.5b11997. [DOI] [PubMed] [Google Scholar]; b Yu Z.-X.; Cheong P. H.-Y.; Liu P.; Legault C. Y.; Wender P. A.; Houk K. N. J. Am. Chem. Soc. 2008, 130, 2378–2379. 10.1021/ja076444d. [DOI] [PubMed] [Google Scholar]; c Kozuch S.; Shaik S. J. Am. Chem. Soc. 2006, 128, 3355–3365. 10.1021/ja0559146. [DOI] [PubMed] [Google Scholar]
- Plata R. E.; Singleton D. A. J. Am. Chem. Soc. 2015, 137, 3811–3826. 10.1021/ja5111392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Świderek K.; Paneth P. Chem. Rev. 2013, 113, 7851–7879. 10.1021/cr300515x. [DOI] [PubMed] [Google Scholar]
- Hirschi J. S.; Takeya T.; Hang C.; Singleton D. A. J. Am. Chem. Soc. 2009, 131, 2397–2403. 10.1021/ja8088636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structure images generated using the CylView molecular visualization program: Legault C. Y.CYLview, version 1.0b; Université de Sherbrooke: Quebec, Canada, 2009; http://www.cylview.org.
- Bromide coordination to the TS is likely weak, as evidenced by the minor difference in energy between the bromide coordinated TS and uncoordinated TS (0.4 kcal·mol–1 favoring no counterion, Figure 6). This suggests a Boltzmann-weighted distribution of KIEs contributes to the experimentally oberserved KIE. The Boltzmann averaged KIE from the static TSs matches the observed KIE. However, the flexibility and energetic accessibility of conformers and complexes indicate that explicit solvation and dynamics also play a role.
- Computed bond orders using NBO analysis indicated enolate character for ylide VII. TS-VIII lies in between dication V and ylide VII in terms of enolate-like character. See Supporting Information for details.
- For S···O interactions as control elements in isothiourea catalysis, see:; a Robinson E. R. T.; Walden D. M.; Fallan C.; Greenhalgh M. D.; Cheong P. H.-Y.; Smith A. D. Chem. Sci. 2016, 7, 6919–6927. 10.1039/C6SC00940A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Abbasov M. E.; Hudson B. M.; Tantillo D. J.; Romo D. J. Am. Chem. Soc. 2014, 136, 4492–4495. 10.1021/ja501005g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu P.; Yang X.; Birman V. B.; Houk K. N. Org. Lett. 2012, 14, 3288–3291. 10.1021/ol301243f. [DOI] [PubMed] [Google Scholar]; d Birman V. B.; Li X.; Han Z. Org. Lett. 2007, 9, 37–40. 10.1021/ol0623419. [DOI] [PubMed] [Google Scholar]; For an alternative use of nonbonded S···O interactions in asymmetric synthesis, see:; Nagao Y.; Miyamoto S.; Miyamoto M.; Takeshige H.; Hayashi K.; Sano S.; Shiro M.; Yamaguchi K.; Sei Y. J. Am. Chem. Soc. 2006, 128, 9722–9729. 10.1021/ja056649r. [DOI] [PubMed] [Google Scholar]
- NBO calculation by Romo and Tantillo suggest a number of orbital interactions responsible for the conformational preference. See ref (40b). For additional discussions on the origin of the S···O interaction, see:; a Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. J. Med. Chem. 2015, 58, 4383–4438. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]; b Zhang X.; Gong Z.; Li J.; Lu T. J. Chem. Inf. Model. 2015, 55, 2138–2153. 10.1021/acs.jcim.5b00177. [DOI] [PubMed] [Google Scholar]; Fairlie has suggested electrostatic attraction and dipole minimization. See:; c Reid R. C.; Yau M.-K.; Singh R.; Lim J.; Fairlie D. P. J. Am. Chem. Soc. 2014, 136, 11914–11917. 10.1021/ja506518t. [DOI] [PubMed] [Google Scholar]
- a Wheeler S. E.; Bloom J. W. G. J. Phys. Chem. A 2014, 118, 6133–6147. 10.1021/jp504415p. [DOI] [PubMed] [Google Scholar]; b Yang X.; Liu P.; Houk K. N.; Birman V. B. Angew. Chem., Int. Ed. 2012, 51, 9638–9642. 10.1002/anie.201203327. [DOI] [PubMed] [Google Scholar]; c Yang X.; Bumbu V. D.; Birman V. B. Org. Lett. 2011, 13, 4755–4757. 10.1021/ol201911z. [DOI] [PubMed] [Google Scholar]; d Yamada S.; Fossey J. S. Org. Biomol. Chem. 2011, 9, 7275–7281. 10.1039/c1ob05228d. [DOI] [PubMed] [Google Scholar]; e Wheeler S. E.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 3126–3127. 10.1021/ja809097r. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhao Y.; Cotelle Y.; Sakai N.; Matile S. J. Am. Chem. Soc. 2016, 138, 4270–4277. 10.1021/jacs.5b13006. [DOI] [PubMed] [Google Scholar]; g Kennedy C. R.; Lin S.; Jacobsen E. N. Angew. Chem., Int. Ed. 2016, 55, 12596–12624. 10.1002/anie.201600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenske E. H.; Houk K. N. Acc. Chem. Res. 2013, 46, 979–989. 10.1021/ar3000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The research data underpinning this publication can be found at DOI: http://dx.doi.org/10.17630/0dbeff98-db91-4051-803c-24e46c073f22.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.