
Fig. 5.
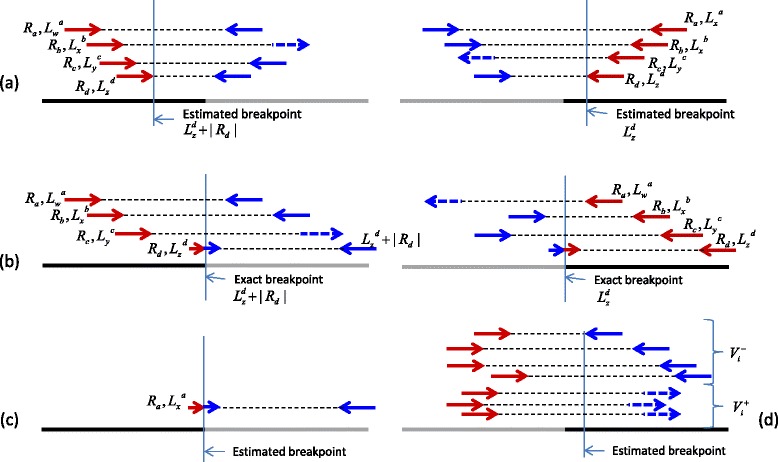
The figure shows how the breakpoints are estimated from a cluster of reads. The red segments of a read aligns to human genome (shown as a black line), and the blue segments belong to the viral genome (shown as a gray line). The solid arrows show properly aligned reads and dashed arrows indicate reads that are aligned incorrectly. For a read cluster (or ) we take the 3’-most(5’-most) aligned position of the read cluster as the estimated human breakpoint. In (a), there is no read passing through the actual breakpoint so the estimation can be off to the 3’ side (or 5’ side). This can be as much as the maximum insert size span of the library. However, if there is a split read R d (b), the exact human breakpoint can be recovered. To find the viral co-ordinate of the integration following procedure can be used. If a split read is available close to the estimated human breakpoint, the exact viral breakpoint can be found out c. Otherwise, the viral mappings of the cluster (or ) will be further sub-divided into two clusters based on the strand of the mapping. The cluster containing the largest number of reads will be considered as correct. Then, the viral breakpoints can be estimated using similar method as that for the human breakpoints d