

Publisher's Note: There is an Inside Blood Commentary on this article in this issue.
Key Points
ASK1 regulates TxA2 generation through p38 MAPK-dependent phosphorylation of cPLA2.
Because of impaired platelet function, Ask1−/− mice are protected from arterial thrombosis and pulmonary thromboembolism.
Abstract
Mitogen-activated protein kinases (MAPKs) are expressed in platelets and are activated downstream of physiological agonists. Pharmacological and genetic evidence indicate that MAPKs play a significant role in hemostasis and thrombosis, but it is not well understood how MAPKs are activated upon platelet stimulation. Here, we show that apoptosis signal-regulating kinase 1 (ASK1), a member of the MAP3K family, is expressed in both human and murine platelets. ASK1 is rapidly and robustly activated upon platelet stimulation by physiological agonists. Disruption of Ask1 (Ask1−/−) resulted in a marked functional defect in platelets. Ask1−/− platelets showed an impaired agonist-induced integrin αIIbβ3 activation and platelet aggregation. Although there was no difference in Ca2+ rise, platelet granule secretion and thromboxane A2 (TxA2) generation were significantly attenuated in Ask1−/− platelets. The defective granule secretion observed in Ask1−/− platelets was a consequence of impaired TxA2 generation. Biochemical studies showed that platelet agonists failed to activate p38 MAPK in Ask1−/− platelets. On the contrary, activation of c-Jun N-terminal kinases and extracellular signal-regulated kinase 1/2 MAPKs was augmented in Ask1−/− platelets. The defect in p38 MAPK results in failed phosphorylation of cPLA2 in Ask1−/− platelets and impaired platelet aggregate formation under flow. The absence of Ask1 renders mice defective in hemostasis as assessed by prolonged tail-bleeding times. Deletion of Ask1 also reduces thrombosis as assessed by delayed vessel occlusion of carotid artery after FeCl3-induced injury and protects against collagen/epinephrine-induced pulmonary thromboembolism. These results suggest that the platelet Ask1 plays an important role in regulation of hemostasis and thrombosis.
Introduction
The mitogen-activated protein kinase (MAPK) signaling cascades are well conserved in eukaryotic cells.1 There are 3 MAPK families: extracellular signal-regulated kinases (ERKs) that are mainly activated downstream of growth factors, c-Jun N-terminal kinases (JNKs), and p38 MAPKs that are activated by cellular stress.2-4 Activation of each MAPK occurs through a sequential phosphorylation by 2 classes of serine/threonine kinases. MAPK is phosphorylated first by MAPK kinase (MAP2K/MEK/MKK), which is in turn phosphorylated by MAP2K kinase (MAP3K/MEKK/MKKK). In platelets all 3 MAPKs are expressed and are activated downstream of physiological agonists.5 Although ERK2 knockout mice are not viable,6 studies using pharmacological inhibitors showed prolonged arteriole occlusion time, uncovering an important role for these MAPK in regulating thrombosis.7 Genetic ablation of JNK1 results in defective thrombosis, primarily due to a defect in granule secretion.8 Although homozygous p38α knockout mice die in utero,9 heterozygous mice show defective thrombosis.10
Apoptosis signal-regulating kinase (ASK1) is a well-characterized member of the MAP3K family, which is activated by a number of cellular stressors, such as oxidative stress, endoplasmic reticulum (ER) stress, UV exposure, and inflammatory cytokines.11,12 ASK1 regulates the JNK and p38 MAPK pathways by phosphorylating MKK4/MKK7 and MKK3/MKK6, respectively, in nucleated cells.13 Activation of ASK1 during cellular stress leads to apoptosis.13 In nucleated cells, ASK1 is held in an inactive state through the association with one of several endogenous inhibitors, such as thioredoxin (Trx), 14-3-3, and calcium and integrin binding protein 1 (CIB1).14-16 Replacement of these inhibitors by activators, such as tumor necrosis factor-α receptor-activated factor (TRAF) 2 and TRAF6 results in the activation of ASK1 by autophosphorylation of a threonine residue (human T838; murine T845) in the activation loop.17 Several molecular mechanisms were reported to regulate ASK1 in the nucleated cells.18 Reactive oxygen species (ROS)-mediated dissociation of Trx and Ca2+-dependent dissociation of CIB1 activate ASK1,14,15 and TRAF2 and TRAF6 binding facilitate ASK1 autophosphorylation and activation.17,19,20 Conversely, 14-3-3ζ binding inhibits ASK1 activation.16
We have previously reported that CIB1, which plays an important role in platelet function,21 regulates ASK1 in cells.15 However, expression of ASK1 in platelets is not known. Here, we show for the first time that ASK1 is expressed in both human and murine platelets and is rapidly activated by physiological agonists. Disruption of Ask1 in mice results in impaired platelet functions in part due to failure of agonist-induced TxA2 generation, a consequence of impaired p38 MAPK–dependent activation of cPLA2, resulting in protection of mice from arterial thrombosis.
Materials and methods
Reagents
Adenosine 5′-diphosphate (ADP) and collagen were purchased from Chronolog. AYPGKF was purchased from AnaSpec. Human fibrinogen (Fg) and α-thrombin were purchased from Enzyme Research. Anti-ASK1, anti-HSC-70, anti-CD36, anti-GP1bα, anti-glycoprotein VI (GPVI), and anti-αIIb were obtained from Santa Cruz Biotechnology. Phosphospecific-p38, P-ERK1/2, P-ASK1 Thr845, P-MKK4, P-MKK7, P-MKK 3/6, P-JNK, anti-p38, anti-MKK3, anti-MKK4, anti-MKK7, and anti-JNK were from Cell Signaling. Fluorescein isothiocyanate (FITC)-Fg, FITC-anti-P-selectin, anti-mouse CD41, and anti-JAM-A were obtained from BD Pharmingen. Anti-mouse CLEC-2, clone 17D9, was obtained from Biolegend. Anti-β3 and anti-PAR4 were obtained from EMD Millipore. Convulxin was from Pentapharm. Phycoerythrin (PE)-conjugated JON/A, PE-conjugated GPIbα, and the control immunoglobulin G (IgG) were obtained from Emfret. U44619, 2MeSADP, and all other chemicals unless indicated were of analytical grade from Sigma. PE-conjugated anti-GPVI was from R&D Systems.
Mouse strains
Congenic Ask1−/− mice in C57BL/6 genetic background generated by backcrossing for more than 10 generations was described previously.22 Age (8-16 weeks) and gender-matched wild-type (WT) C57BL/6 mice were used as controls for all in vivo and in vitro experiments. Integrin β3−/− mice were obtained from Jackson Laboratories. Approval for animal experimental studies was received from the institutional animal care and use committees of the University of Delaware and Thomas Jefferson University.
Platelet preparation and aggregation
Whole blood was drawn by venipuncture from healthy adult volunteers (of both gender and race) with informed consent. Approval was obtained from the institutional review boards of the University of Delaware and Thomas Jefferson University, according to the Declaration of Helsinki. Blood was collected in acidified citrate dextrose as an anticoagulant. Human and murine platelet-rich plasma (PRP) and washed platelets were prepared, as has been previously described.23,24 Platelet aggregation was performed using PRP containing 2 × 108/mL platelets using a Chrono-Log Lumi-Aggregometer (Chrono-Log), as has been described.25 Aggregation traces were recorded using Aggrolink software (Chrono-Log).
Immunoblotting
An aliquot of 100 μL of washed platelet suspension (4 × 108 platelets/mL) in Tyrode’s buffer was resting or stimulated with thrombin (0.1 U/mL), ADP (20 μM), U46619 (5 μM), epinephrine (2 μM), or convulxin (10 ng/mL) at various time points, and the reaction was stopped by adding 5× reducing sample buffer. Immunoblotting was performed, as has been described.26
Flow cytometry
P-selectin surface expression, FITC-labeled Fg, and PE-labeled JON/A binding to washed murine platelets (0.6 × 108/mL) resting or activated with agonist were measured by flow cytometry (Accuri C6, BD), as has been described.25
Tail-bleeding assay
Tail bleeding of age- and gender-matched WT and Ask1−/− mice (4-6 weeks) was performed under anesthesia prior to genotyping, as has been described.25 Time (in seconds) required for cessation of blood flow was recorded.
Platelet transfusion
Platelet transfusion experiments were performed, as has been described.27 In recipient WT and Ask1−/− mice, platelets were depleted by injecting anti-CD41 antibody, as has been described.28 After 24 h the reduction in platelet counts was assessed by Hemavet. Donor platelets isolated from WT and Ask1−/− mice were transfused into the recipient mice, and the FeCl3-induced carotid artery thrombosis assay was performed as has been described.25
In vivo thrombosis models
FeCl3-induced carotid artery thrombosis assay was performed, as has been described.25 Briefly, the carotid arteries of anesthetized WT and Ask1−/− mice were exposed, and an injury was inflicted upon the arteries by placing a piece of Whatman No. 1 filter paper (1 mm × 1 mm) saturated with freshly prepared 10% FeCl3 (anhydrous) for 2 min. The time for complete occlusion (lack of detectable blood flow) was recorded.
Pulmonary thromboembolism assay was performed, as has been described.24 Briefly, a mixture of collagen (0.4 mg/kg; Chronolog) and epinephrine (60 mg/kg; Sigma) in 100 μL of phosphate-buffered saline (PBS) were administered through the tail-vein injection into anesthetized WT and Ask1−/− mice. Two minutes after the onset of respiratory arrest, but while the heart was still beating or at the completion of the 10-minute observation period, 0.5 mL of Evans blue solution (1% in saline) was injected into the heart. Lungs were excised, photographed with a Nikon Coolpix camera, formalin fixed, and embedded into paraffin. Lung sections were stained with hematoxylin and eosin (H&E).
14C-serotonin release
14C-serotonin release was performed, as has been described.29 PRP from WT and Ask1−/− mice was incubated with 0.1 μCi of 14C-serotonin for 1 h at 37°C. Incorporation was stopped by adding imipramine (1 mM). An aliquot of 100 μL (2.5 × 108 platelets/mL) was stimulated with or without various concentrations of thrombin for 5 minutes, and the reaction was stopped by adding equal volume of stopping solution (2 mL of 1× PBS, 0.25 mL 37% formaldehyde, 2.25 mL of 77-mM EDTA pH 7.5). Another set of unstimulated aliquots of 100 μL were lysed with an equal volume of 2% Triton-X (control for serotonin uptake in platelets). An aliquot (150 μL) of individual sample supernatants was counted in a liquid scintillation counter. The percentage of 14C-serotonin release was calculated using the following formula:
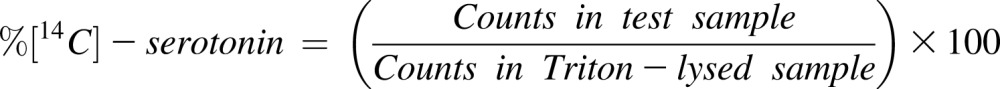 |
TxA2 generation
Thromboxane generation assay was performed by using an enzyme immunoassay kit (Enzo Life Sciences).26 Briefly, 100 μL of washed WT and Ask1−/− platelets (2.0 × 108/mL) were untreated or pretreated with pyrrophenone (1 μM) and stimulated with various agonists for 10 minutes. The level of thromboxane B2, the stable metabolites of TxA2, was determined from supernatant (1:10), according to the manufacturer’s instructions.
Microfluidic flow assay
Microfluidic flow assays were performed in a 4-channel device with channel dimensions of 100 μm in height and 500 μm in width over collagen (200 μg/mL) that was absorbed into the clean glass surface, as has been described.30 Briefly, blood from WT and Ask1−/− mice (9-16 weeks) was collected using heparin (2 U/mL) and 40 μM of d-phenylalanyl-l-prolyl-l-arginine chloromethyl ketone (PPACK; Calbiochem). Whole blood was diluted 1:2 in Tyrode’s buffer and incubated with DiOC6 (1 mg/mL; Sigma) for 10 minutes at 37°C. Blood was perfused through the flow channel for 3 minutes at a shear rate of 800 s−1. The chamber was rinsed with Tyrode’s buffer for 3 minutes and fixed with 5% paraformaldehyde for 30 minutes. Images were taken with a ×20 objective on an EVOS microscope (Thermo Scientific) and analyzed with Image J (National Institutes of Health); fluorescence intensity was calculated by using a NIES-Elements AR 3.2 (Nikon).
Intracellular calcium
Intracellular calcium levels were measured, as has been described.31 Washed platelets from WT and Ask1−/− mice (1.0 × 107/mL) were incubated with 2.5 μM Fluo-4-AM (Life Technologies) for 1 minute in calcium-free Tyrode’s buffer at 37°C. Platelets were stimulated in the presence of 1 mM of extracellular Ca2+ with various agonists, following 60-s baseline, and fluorescence intensity was measured by Accuri C6 flow cytometer (BD).
Hematologic analysis
Murine whole blood (50-75 μL) was drawn by retro-orbital bleed into a tube containing 1 μL (10 units) of heparin to prevent coagulation. The blood cell analysis was performed using a Hemavet 950 (Drew Scientific).
Detection of occult fecal blood
Occult fecal blood was detected using Hemoccult kit (Beckman Coulter), following the manufacturer’s instructions.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed, as has been described.32 Ultrathin counterstained sections were imaged by a Zeiss Libra 120 TEM, equipped with a CCD camera (Zeiss).
Statistical analysis
Statistical analysis of the data was performed using Prism 6 (GraphPad). Student’s t test (mean ± standard error of the mean) or Fisher’s exact test were used to determine statistical significance. P ≤ .05 was regarded as statistically significant. Each experiment was repeated independently at least 3 times.
Results
ASK1 is expressed in platelets and is activated by physiological agonists
A search of platelet proteomic databases showed that ASK1 (MAP3K5)—but not ASK2 (MAP3K6) and ASK3 (MAP3K15), two other members of the ASK1 family—is expressed in platelets.33,34 We found expression of ASK1 in both human and murine platelets (Figure 1A).35 To determine whether ASK1 is activated during human platelet stimulation by physiological agonists, we immunoblotted resting and activated washed human platelet lysates using antiphospho-T845 (P-T845) ASK1. P-T845ASK1 was undetectable in resting platelet lysates. ASK1 was rapidly phosphorylated by thrombin within 1 minute, which peaked at 3 minutes and then decreased (Figure 1B). ADP also activated ASK1 with kinetics similar to that of thrombin; however, the extent of activation was slightly lower (Figure 1C). Convulxin, a GPVI-specific agonist, robustly increased ASK1 phosphorylation with similar kinetics to that of thrombin and ADP (Figure 1D). U46619, a TxA2 mimetic, also strongly induced ASK1 phosphorylation. However, unlike ADP and thrombin, U46619-induced ASK1 phosphorylation persisted beyond 5 minutes (Figure 1E). Epinephrine on the other hand did not activate ASK1 (Figure 1F). These results strongly suggest that most platelet agonists are capable of activating ASK1 within a rapid time scale that is relevant to platelet activation.
Figure 1.

Platelet agonists induce ASK1 phosphorylation. (A) Western blot analysis of lysates from human and murine platelet (free from WBCs) blotted with anti-ASK1 (Cell Signaling; upper panel); reprobed with anti-HSC-70 to ensure equal loading (lower panel). (B-F) Washed human platelets (4 × 108) were stimulated with or without (resting) agonist at different time points, as indicated. Lysates were Western blotted using phosphospecific anti-T845ASK1, which recognizes both human and murine phospho-ASK1. The same blot was reprobed with anti-ASK1 to ensure equal protein loading in all the lanes. Western blot of lysates from resting and thrombin-stimulated platelets (Bi) and the quantitation of band intensity (Bii). Western blot of lysates from resting and ADP stimulated platelets (Ci) and the quantitation of band intensity(Cii). Western blot of lysates from resting and convulxin-stimulated platelets (Di) and the quantitation of band intensity (Dii). Western blot of lysates resting and U46619 stimulated platelet (Ei) and the quantitation of band intensity (Eii). Western blot of lysates from resting and epinephrine-stimulated platelets (Fi) and the quantitation of band intensity (Fii). Band density was calculated by using National Institutes of Health Image J software. All the experiments were repeated more than 3 times using platelets from different individual donors. ns, not significant; Thr, thrombin. ***P < .001.
Ask1 null platelets are defective to agonist stimulation
Because ASK1 is robustly and rapidly activated by platelet agonists, we evaluated platelet functions in Ask1−/− mice. As was expected, Ask1−/− platelets do not express Ask1 protein. However, we found no difference in expression levels of major platelet membrane proteins, nor in signaling proteins associated with MAPK signaling pathway between WT and Ask1−/− platelets (Figure 2A-B). The surface expression of CD41, GP1bα, CLEC-2, and GPVI was not affected because of the lack of Ask1 in platelets (Figure 2C). Platelet aggregations induced by a low dose of thrombin, U46619, collagen, and ADP were severely affected in Ask1−/− mice in comparison with WT mice (Figure 2D). However, the aggregation defect was overcome by a higher dose of agonists (Figure 2D). Furthermore, we found a significant attenuation of both JON/A- and Fg-binding induced by AYPGKF in Ask1−/− platelets in comparison with WT platelets, indicating a defect in integrin αIIbβ3 activation (Figure 2E-F). These results suggested that Ask1 is required for agonist-induced platelet activation.
Figure 2.

Platelet functions are defective in the absence of Ask1. (A-B) Representative Western blots of washed platelet lysates from WT and Ask1−/− mice blotted with antibodies specific to major platelet transmembrane proteins (A) and MAPK cascade proteins (B). (C) Quantitation of flow cytometric analysis of indicated surface protein expression from WT and Ask1−/− platelets. (D) Representative platelet aggregation tracings of platelet-rich plasma (PRP) from WT and Ask1−/− mice stimulated with 2 concentrations of agonists, as indicated. (E-F) Quantitation of flow cytometric analysis of PE-conjugated JON/A binding (E) and FITC-conjugated Fg binding (F) to WT and Ask1−/− mouse platelets stimulated with various concentrations of AYPGKF, as indicated. Data presented are from 3 independent experiments. CLEC2, C-type lectin-like receptor; MFI, mean fluorescence intensity; ns, not significant; WB, Western blot. *P < .05.
Ask1 is needed for platelet granule secretion
It is known that intracellular Ca2+ rise is one of the common and early steps of platelet activation. Platelet agonist-induced intracellular Ca2+ rise was unaffected in Ask1−/− platelets in comparison with WT platelets, suggesting that Ask1 functions downstream of Ca2+ release (Figure 3A-B). Thrombin induced a dose-dependent Ca2+ rise, which was similar in both WT and Ask1−/− platelets (Figure 3Bii). On the other hand, thrombin-induced dense granule secretion was significantly attenuated in Ask1−/− platelets in comparison with WT platelets (Figure 3C). Total 14C-serotonin incorporation was comparable in platelets of both genotype, suggesting that the defect is not in the granular content of Ask1−/− platelets. We also found that agonist-induced ATP release was also significantly lower in Ask1−/− platelets than in WT platelets (Figure 4H). Similar to dense granules, agonist-induced release of α-granules was also significantly attenuated in Ask1−/− platelets in comparison with WT platelets (Figure 3D). The platelet morphology and granule number were similar in Ask1−/− and WT platelets, suggesting that the attenuation of secretion was not caused by defective granule biogenesis (Figure 3E).
Figure 3.

Ask1 regulates granular secretion. (A) Representative tracings of intracellular Ca2+ of Fluo-4 loaded washed platelets from WT and Ask1−/− mice stimulated with ADP (10 μM) (i), convulxin (10 ng/mL) (ii), AYPGKF (100 μM) (iii), and U46619 (5 μM) (iv) in the presence of 1 mM of extracellular Ca2+ (left panels). Quantitation of data from more than 3 experiments (right panels). (B) Quantitation of intracellular Ca2+ from WT and Ask1−/− platelets treated with Ca2+ ionophore (A23187, 10 μM) (i) and various concentrations of thrombin (ii). (C) Quantitation of 14C-serotonin release from platelets (2.5 × 108/mL) from WT and Ask1−/− mice stimulated with various concentrations of thrombin and measured using a liquid scintillation counter. (D) Quantitation of P-selectin surface expression of WT and Ask1−/− platelets. (E) Representative TEM images of WT and Ask1−/− null platelets. ns, not significant. Scale bar, 1 μm. *P < .05; **P < .01.
Figure 4.

Thromboxane generation is defective in the absence of Ask1. (A) Quantitation of TxA2 generation measured as thromboxane B2 (TxB2) in washed platelets isolated from WT and Ask1−/− mice and stimulated with different agonists, as indicated. Data presented are from 3 independent experiments. (B) Quantitation of thrombin-induced 14C-serotonin secretion in WT and Ask1−/− platelets in the presence or absence of aspirin. (C) Quantitation of TxA2 generation induced by various agonists in WT and Ask1−/− platelets pretreated with pyrrophenone (1 μM) for 30 minutes. (D-E) Western blot of lysates of resting or thrombin-stimulated washed platelets (D) or indicated agonist-stimulated washed platelets (E) isolated from WT and Ask1−/− mice blotted using phosphospecific anti-cPLA2. The same blot was reprobed with anti-cPLA2 to ensure equal protein loading in all the lanes. A representative blot from 3 independent experiments is shown. (F) Quantitation of JON/A-binding in platelets from WT and Ask1−/− mice resting or treated with AA (100 μg/mL). (G) Quantitation of P-selectin expression in WT and Ask1−/− platelets resting or treated with U46619 (1 μM). (H) Quantitation of ATP secretion in platelets from WT and Ask1−/− mice resting or treated with U46619 (100 nM). ASA, acetylsalicylic acid. *P < .05; **P < .01; ***P < .001.
Ask1 regulates TxA2 generation
In addition to Ca2+, TxA2 generation is also important for the regulation of granule secretion.36 We found that thrombin, collagen, and convulxin caused robust generation of TxA2 in WT platelets, as was expected (Figure 4A). However, TxA2 generation induced by these agonists was severely attenuated in Ask1−/− platelets (Figure 4A). To determine whether the observed defect in granule secretion was in fact due to reduced TxA2 generation, we determined the thrombin-induced 14C-serotonin secretion in the presence of aspirin, which inhibits Cox1, a key enzyme required for TxA2 generation.37 As was expected, we found that aspirin significantly attenuated 14C-serotonin secretion in WT platelets; however, it had no effect on thrombin-induced 14C-serotonin secretion in Ask1−/− platelets (Figure 4B). Interestingly, the amount of 14C-serotonin secretion in the WT platelets in the presence of aspirin was the same as that in Ask1−/− platelets in the presence or absence of aspirin (Figure 4B). These data suggest that the granule secretion defect seen in Ask1−/− platelets was mainly dependent on TxA2 generation. Because cPLA2 is a key enzyme in the biogenesis of TxA2, we used pyrrophenone, a specific cPLA2 inhibitor, to determine the contribution of cPLA2 in agonist-induced TxA2 generation. We found that pretreatment of both WT and Ask1−/− platelets with 1 μM of pyrrophenone strongly inhibited thrombin-, collagen-, and convulxin-induced TxA2 generation, suggesting that cPLA2 was primarily responsible for the observed agonist-induced TxA2 generation in murine platelets (Figure 4C). It is known that cPLA2 activity is regulated by phosphorylation of its S505 residue.38 Although treatment of WT platelets with thrombin showed an increase in S505 phosphorylation of cPLA2 in a time-dependent manner, thrombin failed to induce any S505 phosphorylation of cPLA2 in Ask1−/− platelets (Figure 4D). ADP and collagen also failed to induce S505 phosphorylation of cPLA2 in Ask1−/− platelets (Figure 4E). These results suggest that agonist-induced cPLA2 S505 phosphorylation is entirely dependent on Ask1. If the sole function of Ask1 is activation of cPLA2, which is required for arachidonic acid (AA) release, then exogenous addition of AA should rescue functional defects seen in Ask1−/− platelets. We found that AA-induced integrin activation was significantly attenuated in Ask1−/− platelets (Figure 4F). Because TxA2 stimulates signaling by activating TPα in platelets, we also tested whether TxA2-induced granule secretion is rescued in Ask1−/− platelets. We found significant attenuation of α-granule as well as dense granule secretion induced by U46619, a TxA2 mimetic (Figure 4G-H). These results suggested that in addition to TxA2 generation, Ask1 also functions in regulating other pathways downstream of platelet agonists, including TxA2.
Ask1 regulates TxA2 generation through p38 MAPK-dependent cPLA2 phosphorylation
The kinase responsible for the phosphorylation and activation of cPLA2 is controversial.5 It was reported that both p38 and ERK MAPKs regulate cPLA2 activity by phosphorylation of S505 downstream of agonist activation.39,40 Because we found a complete lack of phosphorylation of cPLA2 in Ask1−/− platelets, we investigated which MAPK is actually responsible for cPLA2 phosphorylation in murine platelets. We found that thrombin, ADP, and collagen robustly induced ERK1/2 as well as JNK phosphorylation in both WT and Ask1−/− platelets, suggesting that the activation of JNK and ERK1/2 is not dependent on Ask1 and that they are not responsible for cPLA2 phosphorylation (Figure 5A-B). On the contrary, agonists failed to induce p38 MAPK phosphorylation in Ask1−/− platelets (Figure 5C-D), suggesting that Ask1 is required for p38 MAPK phosphorylation and p38 MAPK is responsible for cPLA2 activation in murine platelets. To determine which MKK is activated by Ask1, we evaluated thrombin-induced phosphorylation of MKK3, MKK4, MKK6, and MKK7. We found robust time-dependent activation of all of these MKKs in WT platelets (Figure 5E-G). However, thrombin failed to phosphorylate MKK3, MKK4, and MKK6 at any time point tested (Figure 5E-F). On the other hand, phosphorylation of MKK7 occurred at the 1-minute time point but was rapidly attenuated in Ask1−/− platelets in comparison with WT platelets (Figure 5G). Interestingly, ADP- and collagen-induced phosphorylation was significantly attenuated in Ask1−/− platelets, suggesting that MKK7, a kinase responsible for activation of JNKs, is not under control of Ask1 (Figure 5H).
Figure 5.

Ask1 regulates TxA2generation through activation of p38 MAPK. (A-B) Western blots of lysates of WT and Ask1−/− platelets stimulated with thrombin (Thr), ADP, or collagen and blotted using phosphospecific anti-ERK1/2 (Ai) or phosphospecific anti-JNK1/2 (Bi). The same blots were reprobed with anti-ERK1/2 or anti-JNK1/2 to ensure equal protein loading in all the lanes. (Aii, Bii) Quantitation of data. (C) Western blots of lysates of WT and Ask1−/− platelets stimulated with AYPGKF for various time points and blotted using phosphospecific anti-p38. The same blot was reprobed with anti-p38 to ensure equal protein loading in all the lanes. Shown is a representative blot from 3 independent experiments. (D) Western blots of lysates from panel A blotted using phosphospecific anti-p38. The same blot was reprobed with anti-p38 to ensure equal protein loading in all the lanes. (E-G) Western blots of lysates of WT and Ask1−/− platelets stimulated with thrombin for indicated time points and blotted using phosphospecific anti-MKK3/6 (E), phosphospecific anti-MKK4 (F), or phosphospecific anti-MKK7 (Gi). The same blot was reprobed with anti-MKK3 (E), anti-MKK4 (F), or anti-MKK7 (Gi) to ensure equal protein loading in all the lanes. Shown is a representative blot from 3 independent experiments. (Gii) Quantitation of band density from panel Gi. (H) Quantitation of band density of Western blot of lysates from panel A, blotted using phosphospecific anti-MKK7 normalized to total MKK7. ns, not significant. *P< .05; **P< .01.
Ablation of Ask1 impairs thrombosis
Ask1−/− mice are fertile and healthy with no visible abnormalities. While no statistically significant differences were observed in the hematological parameters between Ask1−/− and WT mice, the reduced white blood cell (WBC) count was very close to significance (Table 1). Interestingly, however, we found blood in the feces of 70% of adult Ask1−/− mice in comparison with 20% of WT mice. Integrin β3−/− mice, which are known to have internal bleeding, were 100% positive for fecal blood (Figure 6A).41 These results suggested that Ask1−/− mice have bleeding diathesis. To determine whether the ablation of Ask1 affected hemostasis, we compared the duration of tail bleeding in WT and Ask1−/− mice. WT mice had a mean tail-bleeding time of 100 ± 9.6 s, whereas Ask1−/− mice had a mean tail-bleeding time of 430 ± 35.3 s (Figure 6B). These results suggest that Ask1 regulates hemostatic function in mice. To determine the role of ASK1 in thrombosis, we performed FeCl3-induced carotid artery thrombosis assay. We found that carotid occlusion was significantly delayed in Ask1−/− mice in comparison with WT mice (Figure 6C). In WT mice the average time to occlude was 7.16 ± 0.48 minutes; it was 10.39 ± 0.57 minutes in Ask1−/− mice (Figure 6D). The majority of Ask1−/− mice (57%) either did not occlude at all or had an unstable occlusion during the 30 minutes of the assay, in comparison with 22% of WT mice (Figure 6E). These results strongly suggest that Ask1 plays an important role in regulating thrombosis.
Table 1.
Whole blood profile of Ask1−/− mice
| Parameter (units) | WT (n = 11) | Ask1−/− (n = 9) | P value |
|---|---|---|---|
| Leukocytes | |||
| White blood cells (K/µL) | 9.88 ± 2.68 | 7.15 ± 3.48 | .063 |
| Erythrocytes | |||
| Red blood cells (M/µL) | 8.55 ± 0.91 | 9.07 ± 0.79 | .197 |
| Hematocrit (%) | 41.97 ± 4.40 | 45.31 ± 4.90 | .126 |
| Thrombocytes | |||
| Platelets (K/µL) | 783 ± 136 | 862 ± 146 | .226 |
| Mean platelet volume (fL) | 5.12 ± 0.13 | 5.03 ± 0.10 | .131 |
WT, wild-type.
Figure 6.

Ask1 regulates in vivo thrombosis. (A) Percentage of adult mice testing positive for fecal occult blood. (B) Tail-bleeding times performed by amputating the terminal 3-mm segment of the tail of anesthetized 4- to 6-week-old WT (n = 17) and Ask1−/− (n = 17) mice and immersing them into warm saline solution prior to genotyping. Bleeding time was measured as the time from the start of bleeding to cessation of bleeding. The bleeding was manually stopped for those for which bleeding continued for more than 10 minutes. (C) Tracings of Doppler flow of carotid artery after 10% FeCl3-induced injury in anesthetized 8- to 12-week-old WT (n = 13) and Ask1−/− (n = 14) mouse right common carotid artery. Duration of the injury is denoted by 1; time to occlusion is denoted by 2; cessation of flow is denoted by 3. (D) Quantitation of occlusion time from panel C. (E) Quantitation of percentage of mice from panel C with stable or unstable occlusion or no occlusion. *P < .05; **P < .01; ***P < .001.
Because the Ask1−/− mice used in this study were created through a global deletion,22 it is possible that in addition to the lack of platelet Ask1, an absence of endothelial Ask1 also contributes to the observed thrombosis defect. To achieve thrombosis without major involvement of endothelial cells, we performed a collagen/epinephrine-induced thromboembolism assay.24 We found that the majority of the WT mice died from asphyxia, and only <20% survived beyond 3 minutes after the administration of the agonist mixture (Figure 7A). In contrast, >80% of Ask1−/− mice survived beyond 3 minutes, suggesting that they were protected from thromboembolism (Figure 7A). The inability of the Evan’s blue dye to penetrate the lungs of WT mice was visible by their pink color; on the contrary, Ask1−/− mouse lungs were blue because of free passage of the dye, indicating lack of obstruction (Figure 7B). Furthermore, large thrombi obstructing the pulmonary vasculature were visible in the histological sections of the WT lungs, whereas no such thrombi obstructing the pulmonary vasculature were detected in Ask1−/− lungs (Figure 7C). These results suggest that defective platelet thrombus formation protects the Ask1−/− mice from pulmonary thromboembolism. To further evaluate the thrombus growth in the absence of the influence of the endothelium, we assayed in vitro thrombus formation by perfusing whole blood through a microfluidic device coated with fibrillar collagen. We found that 47% fewer Ask1−/− platelets adhered to collagen in comparison with WT platelets (Figure 7Di,ii). Interestingly, the number of platelets in a thrombus (a measure of thrombus size) as indicated by fluorescence intensity of labeled platelets was significantly lower (4%) in Ask1−/− mouse blood than in WT mouse blood (Figure 7Di,iii). These data suggest that platelet Ask1 contributes to thrombus growth. In order to conclusively rule out the contribution of endothelial and leukocyte Ask1 in this process, we performed FeCl3-induced thrombosis in mice transfused with platelets after immunodepletion (Figure 7E). WT mice receiving WT platelets showed a mean occlusion time of 9 minutes, and Ask1−/− mice receiving Ask1−/− platelets showed a mean occlusion time of 21 minutes. WT mice receiving Ask1−/− platelets also showed an increased mean occlusion time (24 minutes). Interestingly, Ask1−/− mice receiving WT platelets showed a mean occlusion time of 7 minutes, which is similar to that of the WT recipient mice (Figure 7E). These results suggest that the thrombotic defect observed in Ask1−/− mice is due to impaired platelet functions. This was further supported by studies using platelet-specific Ask1 deletion.42
Figure 7.

Deletion of Ask1 protects mice from pulmonary thromboembolism. (A) Survival curve of WT (n = 10) and Ask1−/− (n = 15) mice after tail vein injection of a mixture of collagen (0.4 mg/kg) and epinephrine (60 mg/kg; Sigma) in 100 μL of PBS. Time of cessation of respiration (time needed to the onset of respiratory arrest that lasted at least 2-3 minutes) was recorded. (B) Pictures of mouse lungs from panel A, injected with 0.5 mL of Evans blue solution (1% in saline) into the heart 2 minutes after the onset of respiratory arrest, but while the heart was still beating or at the completion of the 10-minute observation period. Lungs were excised and photographed with a Nikon Coolpix camera. (C) Photographs of hematoxylin and eosin (H&E)–stained paraffin embedded sections of lung from panel B. Scale bar, 10 μm. (Di) Representative images of microfluidic chambers through which anticoagulated (heparin/PPACK) blood from WT and Ask1−/− mice labeled with DiOC6 was passed over on immobilized collagen (shear rate 800 s−1) for 3 minutes. Representative images were taken under ×20 magnification with an EVOS microscope; phase contrast (upper panel) and DiOC6 (green stained; lower panel). Percentage of surface area coverage (Dii) and fluorescence intensity (Diii) were analyzed with Image J (National Institutes of Health). Scale bar, 50 μm. (E) Quantitation of carotid occlusion time after FeCl3 injury in platelet-depleted recipient mice receiving platelets from indicated donor mice. (F) Schematic representation of the Ask1-dependent MAPK pathway that is activated downstream of agonist receptors and partly responsible for granular secretion and integrin αIIbβ3 activation. Epi, epinephrine; PARs, protease-activated receptors. *P < .05; **P < .01.
Discussion
Platelet activation during vascular injury could be viewed as a stress response. It is therefore not surprising to find that a stress response pathway is triggered during platelet activation, a terminal event analogous to apoptosis. MAPKs were shown to be activated at various steps of platelet activation and to play an important part in this process.5 However, how agonist-induced signaling events result in the activation of these kinases is not well understood. We found that ASK1, a MAP3K that relays signals to p38 and JNK MAPKs, is expressed in platelets and is robustly activated during platelet activation by physiological agonists. Furthermore, we show that in murine platelets, Ask1 is indispensable for p38 activation but is not required for activation of the ERK1/2 and JNK MAPKs. The upstream MAP3K that activates ERK1/2 in platelets was shown to be protein kinase C (PKC) and not Raf.43 The MAP3K responsible for platelet JNK activation remains to be determined.
It is not clear how ASK1 is activated during platelet activation. In nucleated cells, the activation mechanisms of ASK1 have been well characterized.44 ASK1 is maintained in a resting state by association with a number of proteins such as Trx and CIB1.14,15 During oxidative stress, ROS oxidizes Trx, causing its dissociation from ASK1 and permitting ASK1 activation by autophosphorylation.14 In certain nucleated cells, CIB1, a Ca2+-binding protein,21 binds to ASK1 at low Ca2+ levels, keeping it inactive. When Ca2+ levels rise, CIB1 dissociates from ASK1, allowing its activation.15 CAMKII, a Ca2+-dependent kinase, has also been reported to activate ASK1 in nucleated cells and could be responsible for inducing the activation of ASK1 in platelets.45,46 We find that ASK1 is activated downstream of most GPCRs in platelets (Figure 1). It is therefore possible that Gαq-dependent intracellular Ca2+ rise induced by G-protein-coupled receptors (GPCRs) could be important for ASK1 activation. However, other pathways could not be ruled out at this time, because some of the platelet GPCRs are also coupled to Gα12/13, and Gα12/13 has been reported to be involved in ASK1 activation in nucleated cells.47
ASK1 has been implicated to directly activate MKK3/6 as well as MKK4/7. MKK3/6 specifically activates p38, whereas MKK7 is specific to JNK1/2, and MKK4 can activate both p38 and JNK. The complete failure of activation of MKK3, MKK4, and MKK6 in Ask1−/− platelets suggests that Ask1 is only responsible for activation of these MKKs and that it is not needed for MKK7 phosphorylation, although absence of Ask1 results in rapid deactivation of MKK7, probably through activation of a phosphatase. The lack of a significant defect on JNK activation in Ask1−/− mice could be because of the fact that MKK7 is not activated by Ask1 in platelets.
In platelets, cPLA2 is primarily responsible for the release of arachidonic acid during activation. cPLA2 translocation to the plasma membrane is dependent on intracellular Ca2+.48 However, its activity is regulated by the phosphorylation of S505 by MAPKs.38 Both p38 and ERK1/2 were reported to phosphorylate S505 of cPLA2.39,40 JNK1 does not appear to affect cPLA2 activation because Jnk1−/− platelets have no defect in TxA2 generation.8 Our finding that cPLA2 phosphorylation is abolished and that p38, but not ERK1/2, activation is eliminated in Ask1−/− platelets indicates that in murine platelets, p38 is solely responsible for cPLA2 phosphorylation and hence its activation. ERK1/2 has been shown to be negatively regulated by p38 in nucleated cells through protein phosphatase PP2A.49 Our observation of enhanced activation of ERK1/2 in Ask1−/− platelets could be because of the absence of p38 activation in these platelets.
Our in vivo experiments suggest that ASK1 plays a central role in regulating thrombosis by activating the downstream effectors of the MAPK cascade. This is supported by the finding that thrombosis is defective in Jnk1−/− mice as well as in p38+/− mice.10,50 We attribute the defective thrombosis in Ask1−/− mice to the absence of platelet Ask1, although the contribution of endothelial and leukocyte Ask1 to the severity of the phenotype cannot be ruled out at this time. In part, the defective thrombosis in Ask1−/− mice could result from the lower TxA2-dependent granule secretion. Our finding that U46619-induced platelet aggregation and granule secretion was strongly inhibited in Ask1−/− platelets suggests that Ask1 may also be required for integrin αIIbβ3 activation and for aggregation in addition to dense granule secretion. As is depicted in Figure 7F, our results suggest that the increased intracellular Ca2+ levels downstream of the agonist receptors causes activation of ASK1 through an as yet unknown mechanism. ASK1 activates MKK3/4/6, which in turn activates p38 MAPK. cPLA2 is then actived by p38 MAPK by phosphorylating S505 residue, resulting in AA release leading to TxA2 production. Newly produced TxA2 induces release of a population of dense granules. TxA2 also activates TPα, inducing platelet activation. Activation of integrin αIIbβ3 may be regulated by p38 MAPK, leading to platelet aggregation. Increased Ca2+ can independently induce the major population of granule secretion and integrin αIIbβ3 activation, probably through PKC and Ca2+ and by DAG-regulated guanine nucleotide exchange factor I, respectively.32,51
In summary, we show here for the first time that ASK1, an upstream kinase of the stress-induced MAPK pathway, is activated in platelets. Most platelet agonists activate the ASK1 signaling pathway. This pathway appears to play a central role in TxA2 generation and plays a significant role in granule secretion and integrin inside-out signaling. Together, these distinct functions of ASK1 strongly influence the thrombotic outcome.
Acknowledgments
The authors thank Brendan Bachman, Jeffrey Caplan, Natalie Barns, and Sharmila Chatterjee for technical assistance and Paul Bray and Arie Horowitz for helpful suggestions on the manuscript.
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL113188 (U.P.N.) and R01 HL 120728 (K.B.N.).
Footnotes
Presented in part in abstract form at the 52nd annual meeting of the American Society of Hematology, Orlando, FL, 6 December 2010, and at the International Society of Thrombosis and Hemostasis, Kyoto, Japan, 27 July 2011.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.U.N. designed the experiments, performed in vivo thrombosis assays and activation of MAPK cascade, interpreted data, and wrote the paper; P.P. performed activation of Ask1 effectors and TxA2 generation in murine platelets; R.D. performed ASK1 activation in human platelets; R.T. performed platelet aggregation, granular secretion, and flow cytometry; X.C. performed microfluidic flow experiments; K.G. performed calcium experiments; K.B.N. designed and constructed the microfluidic flow chamber; H.I. developed the Ask1 KO mouse; U.P.N. conceived the research, designed the experiments, interpreted data, and wrote and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for R.T. is the College of Doctoral Studies, Grand Canyon University, Phoenix, AZ.
Correspondence: Ulhas P. Naik, 1020 Locust St, Jefferson Alumni Hall, Suite 394, Philadelphia PA 19107; e-mail: ulhas.naik@jefferson.edu.
References
- 1.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79(1):143-180. [DOI] [PubMed] [Google Scholar]
- 2.Avruch J, Khokhlatchev A, Kyriakis JM, et al. . Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127-156. [DOI] [PubMed] [Google Scholar]
- 3.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18(45):6087-6093. [DOI] [PubMed] [Google Scholar]
- 4.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81(2):807-869. [DOI] [PubMed] [Google Scholar]
- 5.Adam F, Kauskot A, Rosa JP, Bryckaert M. Mitogen-activated protein kinases in hemostasis and thrombosis. J Thromb Haemost. 2008;6(12):2007-2016. [DOI] [PubMed] [Google Scholar]
- 6.Saba-El-Leil MK, Vella FD, Vernay B, et al. . An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003;4(10):964-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oury C, Daenens K, Hu H, Toth-Zsamboki E, Bryckaert M, Hoylaerts MF. ERK2 activation in arteriolar and venular murine thrombosis: platelet receptor GPIb vs. P2X. J Thromb Haemost. 2006;4(2):443-452. [DOI] [PubMed] [Google Scholar]
- 8.Adam F, Kauskot A, Nurden P, et al. . Platelet JNK1 is involved in secretion and thrombus formation. Blood. 2010;115(20):4083-4092. [DOI] [PubMed] [Google Scholar]
- 9.Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA. Deficiency of the stress kinase p38alpha results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med. 2000;191(5):859-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakurai K, Matsuo Y, Sudo T, Takuwa Y, Kimura S, Kasuya Y. Role of p38 mitogen-activated protein kinase in thrombus formation. J Recept Signal Transduct Res. 2004;24(4):283-296. [DOI] [PubMed] [Google Scholar]
- 11.Takeda K, Matsuzawa A, Nishitoh H, Ichijo H. Roles of MAPKKK ASK1 in stress-induced cell death. Cell Struct Funct. 2003;28(1):23-29. [DOI] [PubMed] [Google Scholar]
- 12.Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55(10):1230-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichijo H, Nishida E, Irie K, et al. . Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90-94. [DOI] [PubMed] [Google Scholar]
- 14.Saitoh M, Nishitoh H, Fujii M, et al. . Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596-2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon KW, Cho JH, Lee JK, et al. . CIB1 functions as a Ca(2+)-sensitive modulator of stress-induced signaling by targeting ASK1. Proc Natl Acad Sci USA. 2009;106(41):17389-17394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, Shao Z, Kerkela R, et al. . Serine 58 of 14-3-3zeta is a molecular switch regulating ASK1 and oxidant stress-induced cell death. Mol Cell Biol. 2009;29(15):4167-4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujino G, Noguchi T, Matsuzawa A, et al. . Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol Cell Biol. 2007;27(23):8152-8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiizaki S, Naguro I, Ichijo H. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv Biol Regul. 2013;53(1):135-144. [DOI] [PubMed] [Google Scholar]
- 19.Matsuzawa A, Saegusa K, Noguchi T, et al. . ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6(6):587-592. [DOI] [PubMed] [Google Scholar]
- 20.Nishitoh H, Saitoh M, Mochida Y, et al. . ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2(3):389-395. [DOI] [PubMed] [Google Scholar]
- 21.Naik UP, Patel PM, Parise LV. Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J Biol Chem. 1997;272(8):4651-4654. [DOI] [PubMed] [Google Scholar]
- 22.Tobiume K, Matsuzawa A, Takahashi T, et al. . ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2(3):222-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naik UP, Naik MU. Association of CIB with GPIIb/IIIa during outside-in signaling is required for platelet spreading on fibrinogen. Blood. 2003;102(4):1355-1362. [DOI] [PubMed] [Google Scholar]
- 24.Naik MU, Stalker TJ, Brass LF, Naik UP. JAM-A protects from thrombosis by suppressing integrin αIIbβ3-dependent outside-in signaling in platelets. Blood. 2012;119(14):3352-3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naik MU, Nigam A, Manrai P, et al. . CIB1 deficiency results in impaired thrombosis: the potential role of CIB1 in outside-in signaling through integrin alpha IIb beta 3. J Thromb Haemost. 2009;7(11):1906-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naik MU, Caplan JL, Naik UP. Junctional adhesion molecule-A suppresses platelet integrin αIIbβ3 signaling by recruiting Csk to the integrin-c-Src complex. Blood. 2014;123(9):1393-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decouture B, Dreano E, Belleville-Rolland T, et al. . Impaired platelet activation and cAMP homeostasis in MRP4-deficient mice. Blood. 2015;126(15):1823-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kostyak JC, Naik MU, Naik UP. Calcium- and integrin-binding protein 1 regulates megakaryocyte ploidy, adhesion, and migration. Blood. 2012;119(3):838-846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grabarek J, Ware JA. Protein kinase C activation without membrane contact in platelets stimulated by bryostatin. J Biol Chem. 1993;268(8):5543-5549. [PubMed] [Google Scholar]
- 30.Neeves KB, Onasoga AA, Hansen RR, et al. . Sources of variability in platelet accumulation on type 1 fibrillar collagen in microfluidic flow assays. PLoS One. 2013;8(1):e54680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeung J, Tourdot BE, Fernandez-Perez P, et al. . Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood. 2014;124(14):2271-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konopatskaya O, Gilio K, Harper MT, et al. . PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119(2):399-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burkhart JM, Vaudel M, Gambaryan S, et al. . The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73-e82. [DOI] [PubMed] [Google Scholar]
- 34.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naik MU, Ichijo H, Naik UP. Apoptosis signal-regulating kinase 1 regulates platelet function through integrin alpha IIb beta 3 outside-in signaling [abstract].Blood. 2010;116(21). Abstract 328. [Google Scholar]
- 36.Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278(33):30725-30731. [DOI] [PubMed] [Google Scholar]
- 37.Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J Immunol. 2001;167(5):2831-2838. [DOI] [PubMed] [Google Scholar]
- 38.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72(2):269-278. [DOI] [PubMed] [Google Scholar]
- 39.Kramer RM, Roberts EF, Um SL, et al. . p38 mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J Biol Chem. 1996;271(44):27723-27729. [DOI] [PubMed] [Google Scholar]
- 40.Shankar H, Garcia A, Prabhakar J, Kim S, Kunapuli SP. P2Y12 receptor-mediated potentiation of thrombin-induced thromboxane A2 generation in platelets occurs through regulation of Erk1/2 activation. J Thromb Haemost. 2006;4(3):638-647. [DOI] [PubMed] [Google Scholar]
- 41.Hodivala-Dilke KM, McHugh KP, Tsakiris DA, et al. . Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103(2):229-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamiyama M, Naguro I, Ichijo H. PO-16 - ASK1 regulates tumor lung metastasis and platelet functions. Thromb Res. 2016;140(Suppl 1):S182. [DOI] [PubMed] [Google Scholar]
- 43.Nadal-Wollbold F, Pawlowski M, Lévy-Toledano S, Berrou E, Rosa JP, Bryckaert M. Platelet ERK2 activation by thrombin is dependent on calcium and conventional protein kinases C but not Raf-1 or B-Raf. FEBS Lett. 2002;531(3):475-482. [DOI] [PubMed] [Google Scholar]
- 44.Hattori K, Naguro I, Runchel C, Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal. 2009;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kashiwase K, Higuchi Y, Hirotani S, et al. . CaMKII activates ASK1 and NF-kappaB to induce cardiomyocyte hypertrophy. Biochem Biophys Res Commun. 2005;327(1):136-142. [DOI] [PubMed] [Google Scholar]
- 46.Takeda K, Matsuzawa A, Nishitoh H, et al. . Involvement of ASK1 in Ca2+-induced p38 MAP kinase activation. EMBO Rep. 2004;5(2):161-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berestetskaya YV, Faure MP, Ichijo H, Voyno-Yasenetskaya TA. Regulation of apoptosis by alpha-subunits of G12 and G13 proteins via apoptosis signal-regulating kinase-1. J Biol Chem. 1998;273(43):27816-27823. [DOI] [PubMed] [Google Scholar]
- 48.Clark JD, Lin LL, Kriz RW, et al. . A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65(6):1043-1051. [DOI] [PubMed] [Google Scholar]
- 49.Liu Q, Hofmann PA. Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;286(6):H2204-H2212. [DOI] [PubMed] [Google Scholar]
- 50.Kauskot A, Adam F, Mazharian A, et al. . Involvement of the mitogen-activated protein kinase c-Jun NH2-terminal kinase 1 in thrombus formation. J Biol Chem. 2007;282(44):31990-31999. [DOI] [PubMed] [Google Scholar]
- 51.Stefanini L, Roden RC, Bergmeier W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood. 2009;114(12):2506-2514. [DOI] [PubMed] [Google Scholar]