

Abstract
Objective: Folic acid (FA) supplementation reduces the risk of atherosclerosis and stroke. Phenotypic change from differentiated to dedifferentiated vascular smooth muscle cells (VSMCs) plays an important role in atherosclerosis development; however, the exact mechanisms remain unknown. This study aimed to assess whether FA through mammalian target of rapamycin (mTOR)/P70S6K signaling inhibits platelet derived growth factor (PDGF-BB) induced VSMC dedifferentiation. Methods: VSMCs from primary cultures were identified by morphological observation and α-smooth muscle actin (α-SM-actin, α-SMA) immunocytochemistry. Then, VSMCs were induced by PDGF-BB and treated with varying FA concentrations. Rapamycin and MHY-1485 were used to inhibit or activate the mTOR/P70S6K pathway, respectively. Next, MTT, Transwell, and wound healing assays were employed to assess proliferation and migration of VSMCs. In addition, Western blotting was used to evaluate protein levels of α-SMA, calponin, osteopontin, mTOR, p-mTOR, P70S6K and p-P70S6K in VSMCs. Results: VSMCs showed phenotypic alteration from differentiated to dedifferentiated cells in response to PDGF-BB. MTT, Transwell and wound healing assays showed that FA markedly inhibited proliferation and migration in PDGF-BB-induced VSMCs, in a time and concentration-dependent manner. FA treatment increased the expression levels of the contractile phenotype marker proteins α-SMA and calponin compared with VSMCs stimulated by PDGF-BB alone. Furthermore, FA significantly suppressed mTOR and P70S6K phosphorylation compared with PDGF-BB alone. Similar to FA, downregulation of mTOR signaling by rapamycin inhibited VSMC dedifferentiation. In contrast, upregulation of mTOR signaling by MHY-1485 reversed the FA-induced inhibition of VSMC dedifferentiation. Conclusion: Folic acid inhibits dedifferentiation of PDGF-BB-induced VSMCs by suppressing mTOR/P70S6K signaling.
Keywords: Vascular smooth muscle cells, folic acid, dedifferentiation, mTOR/P70S6K signaling pathway
Introduction
Atherosclerosis is one of the most common and underlying causes of heart disease and stroke in Western societies and throughout the world [1]. Vascular smooth muscle cells (VSMCs) in mature, normal blood vessels are highly specialized cells with a differentiated, quiescent, and contractile phenotype; they exhibit elevated levels of contractile proteins such as α-smooth muscle actin (α-SMA) and calponin (Cal). In atherosclerosis and arterial restenosis, however, VSMCs switch to a dedifferentiated, proliferative, and migratory phenotype (synthetic phenotype), with decreased levels of contractile proteins and increased expression of osteopontin (OPN) [2,3]. Recent investigations have reported that VSMC phenotypic switching is widely observed in atherosclerosis, and established that inhibiting VSMC phenotypic switching may be beneficial in advanced atherosclerosis [2,4]. VSMC phenotypic switching plays a critical role in all stages of the atherosclerotic process [5].
Platelet-derived growth factors (PDGFs) represent a family of 4 isoforms (A, B, C, and D), and are strong mitogens for VSMCs in vessels. PDGFs regulate cell proliferation and migration, extracellular matrix accumulation, and the production of pro- and anti-inflammatory mediators, to maintain tissue permeability and hemodynamics. They are involved in various vascular pathophysiological processes, including atherosclerosis, restenosis, fibrosis and angiogenesis [6]. PDGF-BB, released primarily by vascular endothelial cells and platelets at the sites of vascular injury, is considered one of the most potent mitogens and chemoattractant stimulants for VSMC proliferation and migration, through modulation of several transcription factors and key molecular signalling pathways [7]. Proliferation, migration, protein production or secretion, and phenotypic modulation of VSMCs are activated by PDGF receptor signaling [8]. Therefore, in the present study, PDGF-BB was selected to induce VSMC dedifferentiation.
The mammalian target of rapamycin (mTOR) signaling pathway impacts most major cellular functions, with an outsized role in regulating protein synthesis, as well as cell cycle progression and proliferation [9]. Specifically, mTOR is a widely expressed protein kinase that regulates translation initiation of specific growth-related mRNA subsets through the effectors P70S6K and 4EBP1/eIF4E [10]. VSMCs can dedifferentiate from a quiescent/contractile phenotype to the proliferative/synthetic one, and these changes are associated with mTOR/P70S6K activation to promote protein translation and cell growth [11].
Folic acid (FA) is a cofactor in one-carbon metabolism and a crucial regulator of nucleotide synthesis and methylation reactions [12]. Low FA levels are tightly associated with an increased risk of human vascular diseases, including atherosclerosis and stroke. Conversely, high circulating FA concentrations are associated with reduced risk of primary coronary events [13]. Huo Y et al reported that enalapril-folic acid therapy is more effective in reducing first stroke than enalapril alone in Chinese adults with hypertension [14]. Guo H et al found that FA decreases homocysteine-induced MMP-2 secretion, suggesting a beneficial effect of FA on coronary artery disease pathogenesis [15]. However, FA’s effect on PDGF-BB-induced dedifferentiation in cultured VSMCs remains unknown. The purpose of this study was to assess whether FA directly inhibits the cell dedifferentiation effect of PDGF-BB through mTOR/P70S6K signaling.
Materials and methods
Materials
Male specific pathogen free (SPF) grade Sprague-Dawley (SD) rats (6 weeks old, 120-150 g) were provided by the experimental animal center of Zhejiang Academy of medical sciences. DMEM, fetal bovine serum (FBS), PBS, 0.25% trypsin-EDTA, penicillin, and streptomycin were obtained from Jinuo Biotech Company (Hangzhou, China). Rapamycin, MHY-1485, and DMSO were obtained from Sigma-Aldrich (USA). MTT was purchased from Emresco (USA). DAPI was from Roche (USA). Recombinant human PDGF-BB was obtained from R&D Systems (USA). Transwell chambers were purchased from Millipore (Germany). Primary antibodies against α-SMA, Cal, OPN, GAPDH, p-mTOR, mTOR, p-P70S6K, and P70S6K were from Abcam (Cambridge, MA, USA). Horseradish peroxidase-conjugated goat anti-rabbit IgG antibodies were obtained from Jackson ImmunoResearch Laboratories (USA).
Methods
Cell culture and identification
VSMCs were isolated from thoracic aortas of male SD rats by the improved tissue-sticking method developed in our previous studies, and cultured in DMEM containing 10% FBS at 37°C in a humidified atmosphere with 95% air and 5% CO2. Immunocytochemical identification of the contractile marker protein α-SMA was performed for primary VSMC identification. Primary VSMCs between passages 3 to 7 were used in subsequent experiments. VSMCs were cultured to 70-80% confluency, and serum-starved overnight before experiments. Rapamycin (20 nmol/L) or MHY1485 (10 µmol/L) was used to inhibit or activate the mTOR signaling pathway [16]. Before incubation PDGF-BB (20 ng/ml) for 24 h, VSMCs were pretreated with different concentrations of FA (1 μmol/l, 10 μmol/l, and 100 μmol/l, respectively) for 24 h.
MTT assay
VSMCs were cultured to 70% confluency and serum-starved overnight. In the experimental group, the cells were pretreated with different FA concentrations (1 μmol/l, 10 μmol/l, and 100 μmol/l, respectively) for 24 h then addition of PDGF-BB (20 ng/ml) for 12, 24 and 48 h, respectively. In the control group, cells were cultured without any treatment. PDGF-BB group cells were treated only with PDGF-BB (20 ng/ml). After treatment, MTT assay was used to examine the viability of cells in all groups. The MTT solution (20 µl) was added for 4 h at 37°C with 5% CO2. Then, the medium was carefully removed, and 150 µl DMSO were added to each well. The plate was gently rotated on an orbital shaker for 10 min at room temperature, and a microplate reader (Bio-Rad, Hercules, CA, USA) was used to determine absorbance at 490 nm.
Wound-healing assay
VSMCs were cultured until 90% confluency, serum-starved, and administered hydroxyurea overnight for synchronization and growth inhibition. A wound in VSMCs was created with a sterile 100 ul pipette tip. PBS was used to flush the 6-well plate, washing away the cell debris. After addition of various test articles, cells were analyzed at 0 and 24 h post-wounding under a Nikon microscope (Nikon Corporation, Tokyo, Japan) equipped with the Image-pro plus 6.0 software (Media Cybernetics Inc., Bethesda, MD, USA). The ratio of cell recovery area to whole wound area was used to evaluate cell migration.
Transwell migration assay
VSMCs (2×104 cells per well) were seeded in the upper chamber of the transwell system using an 8 µm aperture. Meanwhile, 250 µl of medium containing different concentrations of FA was added into the upper chamber. VSMCs were incubated for 24 h at 37°C for migration assays. Then, VSMCs in the upper chamber were washed and stained for 15 min with crystal violet. Migrated cells were counted in the lower chamber under a microscope, in 5 randomly selected high power fields.
Immunofluorescence
VSMC culture medium was removed, and cells were washed three times with PBS and fixed in 4% paraformaldehyde. After cell permeabilization (0.1% Triton) and blocking (10% goat serum), VSMCs were incubated with anti-α-SMA antibodies overnight. Anti-mouse fluorescein isothiocyanate (FITC) conjugated secondary antibodies were added for 1 h at room temperature, before analysis under an inverted phase-contrast microscope.
Western blot
VSMCs culture medium was removed, and cells were washed three times with PBS. VSMCs were then lysed with lysis buffer (Beyotime, Haimen, China) on ice for 30 min.
Cell lysates were centrifuged at 12,000 g for 15 min at 4°C. Equal amounts of soluble protein were separated by 12% SDS-PAGE and protein bands were electro-transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% skim milk, followed by primary antibody incubation overnight. Secondary antibodies conjugated with horseradish peroxidase were used in these experiments. The ECL chemiluminescence detection kit was used to visualize the target proteins as well as the internal control. Protein bands were obtained by autoradiography and analyzed by Quantity One 4.4 (Bio-Rad, Hercules, CA, USA). Antibodies used included: rabbit polyclonal α-SMA, Cal, OPN, GAPDH, p-mTOR, mTOR, p-P70S6K, and P70S6K (1:1,000, Sigma-Aldrich).
Statistical analysis
Each experiment was repeated at least three times, with a fresh batch of cells. The SPSS 19.0 statistical software (SPSS Inc., Chicago, IL, USA) was used to perform statistical analyses. Measurement data are mean ± standard deviation (SD). Multiple groups were compared by one-way analysis of variance (ANOVA), followed by Bonferroni post-hoc tests. Unpaired Student’s t test was used for two-group comparisons. P<0.05 was considered statistically significant.
Results
VSMC identification
After primary culture of VSMCs for 7-9 days, morphological observations revealed that long spindle-shaped cells with strong cytoplasmic refractive index, oval nucleus, and rich cytoplasm migrated from the tissues. The typical VSMC ‘peak-to-valley’ growth pattern was observed after the cells overlapped (Figure 1A). Alpha smooth muscle actin (α-SMA) immunocytochemical staining revealed an abundance of green myonemes in the cytoplasm; nuclei were light blue after counterstaining with DAPI, demonstrating that the cells obtained were indeed VSMCs, with high purity (Figure 1B).
Figure 1.
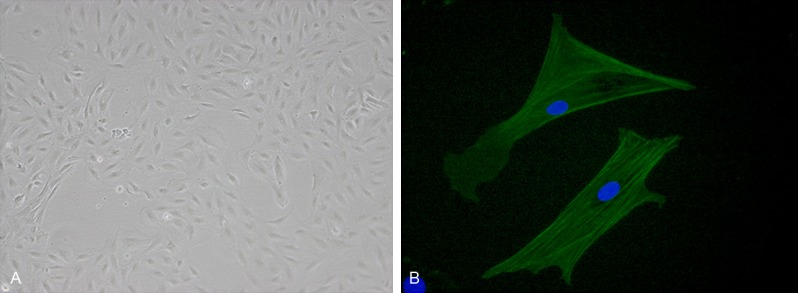
VSMC primary culture identification. A. VSMC “peak-to-valley” growth (magnification, 100×); B. VSMC α-SM-actin immunocytochemical staining (magnification, 400×). VSMC, vascular smooth muscle; SM, smooth muscle.
Folic acid inhibits PDGF-BB-induced VSMC dedifferentiation
VSMCs were pre-treated with various concentrations of folic acid followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. Folic acid inhibited the PDGF-BB-induced proliferation and migration of VSMCs, in a concentration-dependent manner (Figure 2A and 2B, P<0.05) as assessed by MTT and wound-healing assays. In addition, Transwell chamber assay revealed that folic acid could also attenuate PDGF-BB-induced VSMC invasion in a concentration-dependent manner (Figure 2C). What’s more, α-SMA immunocytochemical staining exhibited myoneme morphological changes in the cytoplasm after FA treatment (Figure 2D). Western blot demonstrated that PDGF-BB-induced VSMCs reduced α-SMA and Cal protein expression levels, and increased OPN protein expression, indicating dedifferentiation of VSMCs; these effects were exerted in a concentration-dependent manner (Figure 2E and 2F, P<0.05). These results indicated that folic acid inhibited PDGF-BB-induced VSMC dedifferentiation.
Figure 2.

Folic acid inhibits PDGF-BB-induced VSMC dedifferentiation. Control, VSMCs were cultured without any treatment; PDGF-BB, VSMCs were treated only with PDGF-BB (20 ng/mL); Folic acid, VSMCs were pretreated with various concentrations (1, 10 and 100 µmol/L) of folic acid followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. A. MTT assay was used to assess the proliferation of VSMCs. B. Wound-healing assay was employed to evaluate migration in VSMCs. C. Transwell assay was used to assess the invasion ability of VSMCs. D. α-SMA immunocytochemical staining was used to assess myonemes and cell morphology changes. E, F. Western blot was employed to quantitate the expression levels of contractile proteins (*P<0.05 vs control group; #P<0.05 vs treatment with PDGF-BB alone; n=3).
Folic acid inhibits PDGF-BB-induced VSMC dedifferentiation via mTOR/P70S6K signaling
To further explore the cellular mechanisms by which FA inhibits PDGF-BB-induced VSMC dedifferentiation, VSMCs were pre-treated with various concentrations (1, 10, and 100 µmol/L) of folic acid before stimulation with PDGF-BB (20 ng/mL) for 24 h. As shown in Figure 3A and 3B, p-mTOR and p-P70S6K protein levels in PDGF-BB-induced VSMCs were higher than those in the control group; meanwhile, p-mTOR and p-P70S6K protein levels in PDGF-BB-induced VSMCs treated with various concentrations of folic acid for 24 h were significantly lower than PDGF-BB group values; this effect was concentration-dependent. In addition, p-mTOR and p-P70S6K protein amounts were decreased at 2 h to 6 h after treatment with 10 µmol/L folic acid and stimulation with PDGF-BB (Figure 3C and 3D, P<0.05). These results indicated that folic acid inhibited PDGF-BB-induced VSMC dedifferentiation, suppressing the mTOR/P70S6K signaling pathway.
Figure 3.
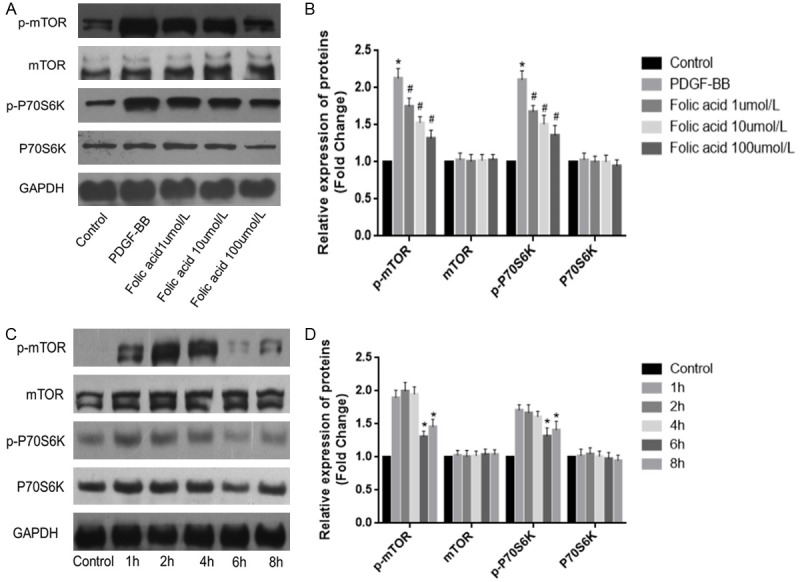
Folic acid inhibits PDGF-BB-induced VSMC dedifferentiation via mTOR/P70S6K signaling. Control, VSMCs were cultured without any treatment; PDGF-BB, VSMCs were treated only with PDGF-BB (20 ng/mL); Folic acid, VSMCs were pretreated with various concentrations (1, 10 and 100 µmol/L) of folic acid followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. A, B. Western blotting was applied to assess p-mTOR and p-P70S6K protein levels in VSMCs treated with various concentrations of folic acid followed by stimulation with PDGF-BB. Folic acid treatment resulted in p-mTOR and p-P70S6K protein levels significantly lower compared with PDGF-BB group values, in a concentration-dependent manner. C, D. Western blotting was applied to evaluate p-mTOR and p-P70S6K protein levels in VSMCs treated with 10 µmol/L folic acid followed by stimulation with PDGF-BB; expression levels of p-mTOR and p-P70S6K were decreased at 2 h to 6 h (*P<0.05 vs control group; #P<0.05 vs treatment with PDGF-BB alone; n=3).
Rapamycin inhibits mTOR/P70S6K signaling pathway suppressed VSMC dedifferentiation
To further examine the role of mTOR/P70S6K signaling in PDGF-BB-induced VSMC dedifferentiation, rapamycin (20 nmol/L) was applied to block the mTOR/P70S6K signaling pathway. VSMCs were treated with PDGF-BB (20 ng/mL) for 24 h. MTT and wound-healing assays showed that rapamycin obviously decreased VSMC proliferation and migration (Figure 4A and 4B). Transwell chamber assay revealed that rapamycin markedly attenuated PDGF-BB-induced invasion ability in VSMCs (Figure 4C). What’s more, Western blot showed that rapamycin starkly increased α-SMA and Cal protein expression levels and decreased OPN protein amounts (Figure 4D and 4E, P<0.05). These results demonstrated that rapamycin suppressed mTOR/P70S6K signaling pathway inhibited VSMC dedifferentiation.
Figure 4.
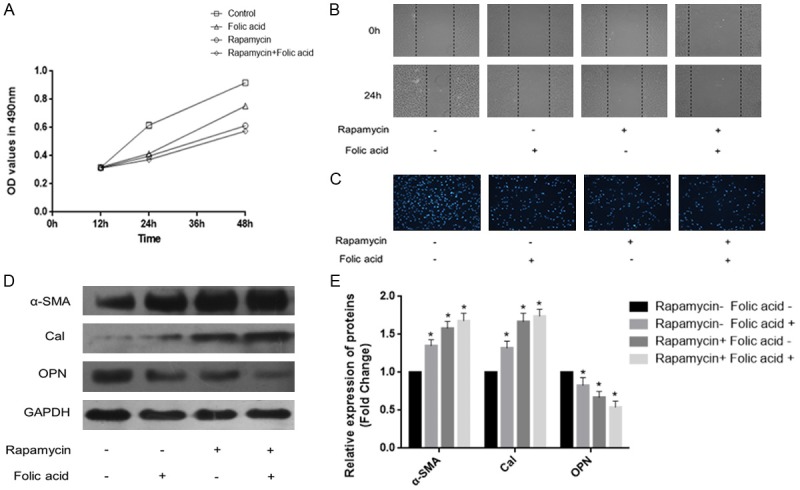
Rapamycin inhibits mTOR/P70S6K signaling suppressed VSMC dedifferentiation. VSMCs were treated with PDGF-BB (20 ng/mL) for 24 h. A. MTT assay was used to assess the proliferation of VSMCs. B. Wound-healing assay was performed to evaluate migration in VSMCs. C. Transwell assay was carried out to assess the invasion ability of VSMCs. D, E. Western blotting was applied to quantitate the expression of contractile proteins (*P<0.05 vs treatment with PDGF-BB alone; n=3).
MHY-1485 attenuates folic acid inhibited PDGF-BB-induced VSMC dedifferentiation
We next assessed the role of mTOR/P70S6K signaling in PDGF-BB-induced VSMC dedifferentiation. MHY-1485 (10 µmol/L) was applied to activate the mTOR/P70S6K signaling pathway, and the effects of folic acid on VSMC dedifferentiation were examined. VSMCs were treated with PDGF-BB (20 ng/mL) for 24 h. MTT and wound-healing assays showed that MHY-1485 significantly attenuated the ability of folic acid to inhibit VSMC proliferation and migration (Figure 5A and 5B). In addition, Transwell assay revealed that MHY-1485 overtly attenuated the ability of folic acid to inhibit VSMC invasion (Figure 5C). Western blot showed that MHY-1485 remarkably suppressed the folic acid related increased α-SMA and Cal protein levels and decreased OPN protein amounts (Figure 5D and 5E, P<0.05). What’s more, MHY-1485 treatment resulted in notably increased p-mTOR and p-P70S6K protein expression levels (Figure 5F and 5G, P<0.05). These findings demonstrated that activated mTOR/P70S6K signaling attenuated folic acid dependent inhibition of PDGF-BB-induced VSMC dedifferentiation.
Figure 5.
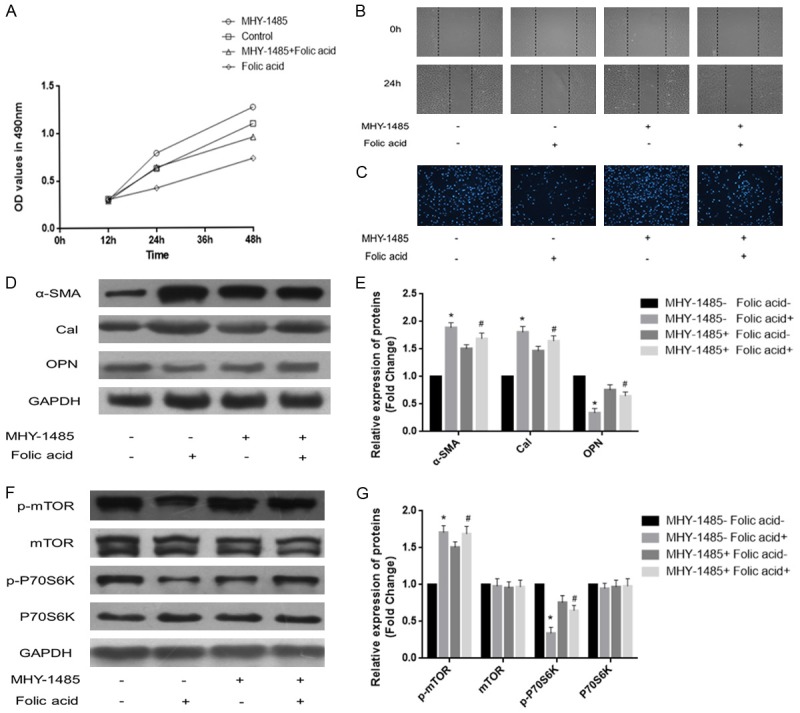
MHY-1485 attenuates folic acid mediated inhibition of PDGF-BB-induced VSMC dedifferentiation. VSMCs were treated with PDGF-BB (20 ng/mL) for 24 h. A. MTT assay was performed to assess the proliferation of VSMCs. B. Wound-healing assay was carried out to evaluate migration in VSMCs. C. Transwell assay was used to determine the invasion ability of VSMCs. D, E. Western blotting was applied to assess the expression levels of contractile proteins. F, G. Western blotting was applied to assess p-mTOR and p-P70S6K protein levels in VSMCs; MHY-1485 attenuated folic acid inhibited mTOR/P70S6K signaling pathway (*P<0.05 vs treatment with PDGF-BB alone; #P<0.05 vs treatment with 10 µmol/L folic acid; n=3).
Discussion
Atherosclerosis is the major underlying cause of myocardial infarction, heart failure, stroke, and peripheral vascular diseases. Therefore, deep understanding of molecular and cellular mechanisms driving atherosclerosis development and progression are necessary for formulating adequate treatment strategies. VSMCs are the main cell type in blood vessels; their chief function through vessel wall contraction and relaxation is to control blood flow and pressure. Unlike many other mature cell types in the adult body, VSMCs do not terminally differentiate but retain remarkable plasticity. They have the unique ability to switch between a differentiated and quiescent “contractile” state and the highly proliferative and migratory “synthetic” one, in response to certain environmental cues [17]. Dedifferentiation of VSMCs are key events in the development of atherosclerosis, and inhibition of VSMC phenotype switching counts among the mechanisms by which drugs play their cardiovascular protective role [18].
PDGF-BB is known to stimulate phenotype modulation in VSMCs from differentiated to dedifferentiated phenotype [19]. In the present study, we also found that PDGF-BB induced proliferation and migration in smooth muscle cells, increasing protein expression of the dedifferentiation associated OPN and reducing the levels of the differentiation specific contractile proteins α-SMA and Cal. Consistent with previous reports, this study showed that PDGF-BB successfully induced VSMC dedifferentiation.
It was suggested that digoxin through ILK signaling exerts an inhibitory effect on PDGF-BB-induced proliferation, migration and phenotypic modulation of VSMCs [20]. Meanwhile, TORRES G et al indicated that glucagon-like peptide-1 (GLP1) stimulates mitochondrial fusion, increases mitochondrial activity, and decreases PDGF-BB-induced VSMC dedifferentiation via PKA/Drp signaling [21]. Furthermore, GAN J et al reported that rosuvastatin suppresses MAPK signaling, inhibiting PDGF-BB-induced VSMCs proliferation and migration [22]. Consistent with these reports, the present study indicated that folic acid could decrease the proliferation and migration ability of VSMCs induced by PDGF-BB. Moreover, folic acid could also reduce the expression of the dedifferentiation related protein OPN, while increasing the amounts of the differentiation specific contractile proteins α-SMA and Cal. These findings indicate that folic acid could inhibit VSMC dedifferentiation.
Folic acid is a water-soluble vitamin involved in many critical cellular pathways, including DNA, RNA, and protein methylation as well as DNA synthesis and maintenance; folic acid can be a limiting factor in all these reactions [12]. HOU T C et al reported that folic acid inhibits endothelial cell migration by reducing RhoA activity mediated by induced FR/cSrc/p190RhoGAP-signaling [23]. Additionally, CHOU Y et al showed that folic acid inhibits homocysteine-mediated increases of VSMC proliferation and migration by inactivating AKT1 [24]. The present study confirmed that folic acid inhibits PDGF-BB induced VSMC differentiation, further demonstrating that folic acid exerts this effect by suppressing the mTOR/P70S6K signaling pathway.
In cardiovascular and metabolic systems, mTOR and its multi-protein complexes TORC1 and TORC2 regulate various cell processes, including proliferation, migration, growth and differentiation [25]. The serine/threonine kinase ribosomal protein P70S6K is a downstream target of mTORC1 [26]. Enhanced activation of mTOR/P70S6K is associated with cardiac hypertrophy and impaired endothelial function; rapamycin treatment to reduce this activation corrects some of the abnormalities [11]. LEE K Y et al observed that mesoglycan induced AMPK activation suppresses VSMC proliferation via an mTOR dependent mechanism [27]. In addition, CIDAD P et al found that Kv1.3 channels modulate human vascular smooth muscle cell proliferation independently of the mTOR signaling pathway [28]. Furthermore, OSMAN I et al revealed that pioglitazone inhibits mTOR/P70S6K and ERK signaling, and attenuates PDGF-induced VSMC proliferation [29]. Consistent with these reports, the current study showed that folic acid inhibited PDGF-BB induced p-mTOR and p-P70S6K expression. Rapamycin was further used to inhibit the mTOR signaling pathway; as shown above, the outcome was similar to that obtained after folic acid treatment. Interestingly, MHY-1485, a novel activator of mTOR [16,30], restored folic acid-induced inhibition of VSMC dedifferentiation. These findings suggest that folic acid inhibits PDGF-induced VSMC dedifferentiation by suppressing the mTOR/P70S6K signaling pathway.
In conclusion, the present data suggest that folic acid inhibits the dedifferentiation effect of PDGF-BB through mTOR/P70S6K signaling, indicating that it may have beneficial effects on vascular proliferative disorders such as atherosclerosis. These findings provide a basis for clinical use of folic acid in AS treatment. However, this study has some limitations, e.g., whether folic acid inhibits VSMC phenotype switching through mTOR/4EBP1 signaling was not assessed.
Acknowledgements
This research was supported by a grant from the Public Welfare project of Zhejiang Province (No. 2016C33227).
Disclosure of conflict of interest
None.
References
- 1.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi N, Chen SY. Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J Biomed Res. 2014;28:40–46. doi: 10.7555/JBR.28.20130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45(Suppl A):A25–32. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, Iwao H. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol. 2003;23:795–801. doi: 10.1161/01.ATV.0000066132.32063.F2. [DOI] [PubMed] [Google Scholar]
- 8.Satoh K, Kikuchi N, Kurosawa R, Shimokawa H. PDE1C negatively regulates growth factor receptor degradation and promotes VSMC proliferation. Circ Res. 2015;116:1098–1100. doi: 10.1161/CIRCRESAHA.115.306139. [DOI] [PubMed] [Google Scholar]
- 9.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin KA, Rzucidlo EM, Merenick BL, Fingar DC, Brown DJ, Wagner RJ, Powell RJ. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol. 2004;286:C507–517. doi: 10.1152/ajpcell.00201.2003. [DOI] [PubMed] [Google Scholar]
- 11.Jia G, Aroor AR, Martinez-Lemus LA, Sowers JR. Overnutrition, mTOR signaling, and cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1198–1206. doi: 10.1152/ajpregu.00262.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr. 2012;3:21–38. doi: 10.3945/an.111.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duthie SJ, Beattie JH, Gordon MJ, Pirie LP, Nicol F, Reid MD, Duncan GJ, Cantlay L, Horgan G, McNeil CJ. Nutritional B vitamin deficiency alters the expression of key proteins associated with vascular smooth muscle cell proliferation and migration in the aorta of atherosclerotic apolipoprotein E null mice. Genes Nutr. 2015;10:446. doi: 10.1007/s12263-014-0446-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huo Y, Li J, Qin X, Huang Y, Wang X, Gottesman RF, Tang G, Wang B, Chen D, He M, Fu J, Cai Y, Shi X, Zhang Y, Cui Y, Sun N, Li X, Cheng X, Wang J, Yang X, Yang T, Xiao C, Zhao G, Dong Q, Zhu D, Wang X, Ge J, Zhao L, Hu D, Liu L, Hou FF CSPPT Investigators. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: the CSPPT randomized clinical trial. JAMA. 2015;313:1325–1335. doi: 10.1001/jama.2015.2274. [DOI] [PubMed] [Google Scholar]
- 15.Guo H, Lee JD, Uzui H, Yue H, Wang J, Toyoda K, Geshi T, Ueda T. Effects of folic acid and magnesium on the production of homocysteine-induced extracellular matrix metalloproteinase-2 in cultured rat vascular smooth muscle cells. Circ J. 2006;70:141–146. doi: 10.1253/circj.70.141. [DOI] [PubMed] [Google Scholar]
- 16.Cheng Y, Kim J, Li XX, Hsueh AJ. Promotion of ovarian follicle growth following mTOR activation: synergistic effects of AKT stimulators. PLoS One. 2015;10:e0117769. doi: 10.1371/journal.pone.0117769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu R, Leslie KL, Martin KA. Epigenetic regulation of smooth muscle cell plasticity. Biochim Biophys Acta. 2015;1849:448–453. doi: 10.1016/j.bbagrm.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 19.Chen S, Liu B, Kong D, Li S, Li C, Wang H, Sun Y. Atorvastatin calcium inhibits phenotypic modulation of PDGF-BB-induced VSMCs via down-regulation the Akt signaling pathway. PLoS One. 2015;10:e0122577. doi: 10.1371/journal.pone.0122577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan G, Wang Q, Hu S, Wang D, Qiao Y, Ma G, Tang C, Gu Y. Digoxin inhibits PDGF-BB-induced VSMC proliferation and migration through an increase in ILK signaling and attenuates neointima formation following carotid injury. Int J Mol Med. 2015;36:1001–1011. doi: 10.3892/ijmm.2015.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torres G, Morales PE, Garcia-Miguel M, Norambuena-Soto I, Cartes-Saavedra B, Vidal-Pena G, Moncada-Ruff D, Sanhueza-Olivares F, San Martin A, Chiong M. Glucagon-like peptide-1 inhibits vascular smooth muscle cell dedifferentiation through mitochondrial dynamics regulation. Biochem Pharmacol. 2016;104:52–61. doi: 10.1016/j.bcp.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gan J, Li P, Wang Z, Chen J, Liang X, Liu M, Xie W, Yin R, Huang F. Rosuvastatin suppresses platelet-derived growth factor-BB-induced vascular smooth muscle cell proliferation and migration via the MAPK signaling pathway. Exp Ther Med. 2013;6:899–903. doi: 10.3892/etm.2013.1265. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Hou TC, Lin JJ, Wen HC, Chen LC, Hsu SP, Lee WS. Folic acid inhibits endothelial cell migration through inhibiting the RhoA activity mediated by activating the folic acid receptor/cSrc/p190RhoGAP-signaling pathway. Biochem Pharmacol. 2013;85:376–384. doi: 10.1016/j.bcp.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Chou Y, Lin HC, Chen KC, Chang CC, Lee WS, Juan SH. Molecular mechanisms underlying the anti-proliferative and anti-migratory effects of folate on homocysteine-challenged rat aortic smooth muscle cells. Br J Pharmacol. 2013;169:1447–1460. doi: 10.1111/bph.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ha JM, Yun SJ, Kim YW, Jin SY, Lee HS, Song SH, Shin HK, Bae SS. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem Biophys Res Commun. 2015;464:57–62. doi: 10.1016/j.bbrc.2015.05.097. [DOI] [PubMed] [Google Scholar]
- 26.Chong ZZ, Maiese K. Mammalian target of rapamycin signaling in diabetic cardiovascular disease. Cardiovasc Diabetol. 2012;11:45. doi: 10.1186/1475-2840-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee KY, Lee DH, Choi HC. Mesoglycan attenuates VSMC proliferation through activation of AMP-activated protein kinase and mTOR. Clin Hypertens. 2015;22:2. doi: 10.1186/s40885-016-0037-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cidad P, Miguel-Velado E, Ruiz-McDavitt C, Alonso E, Jimenez-Perez L, Asuaje A, Carmona Y, Garcia-Arribas D, Lopez J, Marroquin Y, Fernandez M, Roque M, Perez-Garcia MT, Lopez-Lopez JR. Kv1.3 channels modulate human vascular smooth muscle cells proliferation independently of mTOR signaling pathway. Pflugers Arch. 2015;467:1711–1722. doi: 10.1007/s00424-014-1607-y. [DOI] [PubMed] [Google Scholar]
- 29.Osman I, Segar L. Pioglitazone, a PPARgamma agonist, attenuates PDGF-induced vascular smooth muscle cell proliferation through AMPK-dependent and AMPK-independent inhibition of mTOR/p70S6K and ERK signaling. Biochem Pharmacol. 2016;101:54–70. doi: 10.1016/j.bcp.2015.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi YJ, Park YJ, Park JY, Jeong HO, Kim DH, Ha YM, Kim JM, Song YM, Heo HS, Yu BP, Chun P, Moon HR, Chung HY. Inhibitory effect of mTOR activator MHY1485 on autophagy: suppression of lysosomal fusion. PLoS One. 2012;7:e43418. doi: 10.1371/journal.pone.0043418. [DOI] [PMC free article] [PubMed] [Google Scholar]