

Abstract
X-ray crystallography at X-ray free-electron laser (XFEL) sources is a powerful method for studying macromolecules at biologically relevant temperatures. Moreover, when combined with complementary techniques like X-ray emission spectroscopy (XES), both global structures and chemical properties of metalloenzymes can be obtained concurrently, providing new insights into the interplay between the protein structure/dynamics and chemistry at an active site. Implementing such a multimodal approach can be compromised by conflicting requirements to optimize each individual method. In particular, the method used for sample delivery greatly impacts the data quality. We present here a new, robust way of delivering controlled sample amounts on demand using acoustic droplet ejection coupled with a conveyor belt drive that is optimized for crystallography and spectroscopy measurements of photochemical and chemical reactions over a wide range of time scales. Studies with photosystem II, the phytochrome photoreceptor, and ribonucleotide reductase R2 illustrate the power and versatility of this method.
INTRODUCTION
X-ray scattering/diffraction and spectroscopy are valuable tools for studying biomolecular mechanisms because they yield direct and element-specific molecular geometry and electronic state information. X-ray signals generated by excitation with <50 femtosecond pulses from an X-ray Free Electron Laser (XFEL) are produced before radiation-induced changes occur (picosecond time-scale)1–4. Consequently, XFELs provide new opportunities to study biomolecules at ambient temperature and probe enzymatic reaction intermediates prepared in situ. To understand enzymatic mechanisms in detail, both electronic and molecular structural information are needed for reaction intermediates and in practice this requires that complementary time-resolved methods be used to probe the reaction. Time-resolved studies of biomolecules, however, are challenging at an XFEL because the focused XFEL pulses destroy the sample after each shot. This requires that the sample be continually replenished in a consistent state, with time-delays from reaction initiation to the X-ray probe in the picoseconds to seconds time scale5,6.
Recently, we reported an approach for simultaneously collecting X-ray crystal diffraction (XRD) data and X-ray emission spectroscopy (XES) data, with the goal of investigating catalysis in metalloenzymes4. XES directly probes the redox and spin state of the active site metal(s) and ensures that the atomic structure revealed by XRD is in the correct catalytic state. However, optimal sample delivery conditions (crystal size, sample path lengths for spectroscopy, overall sample consumption, etc.) for collecting XRD and XES datasets are not the same. Several continuous-flow sample delivery strategies for XFEL experiments have been established (Fig. 1a)7–10. Liquid jet systems like the gas dynamic virtual nozzle (GDVN)7 and the microfluidic electrokinetic sample holder (MESH)8 use small crystals to obtain the flow rates reported, as a consequence of the narrow feed capillaries they use, whereas lipidic cubic phase (LCP) or grease matrix injectors9,10 use highly viscous media to achieve low flow rates with medium size capillaries. Ejection from an ADE has a high upper bound for the crystal size, while keeping sample consumption at low levels, orders of magnitude lower than Rayleigh jets of the same pathlength. Fixed-target systems have also been successfully used at XFEL facilities11–14, but such systems have not yet been demonstrated for multi-modal data collection.
Figure 1. Experimental Setup.
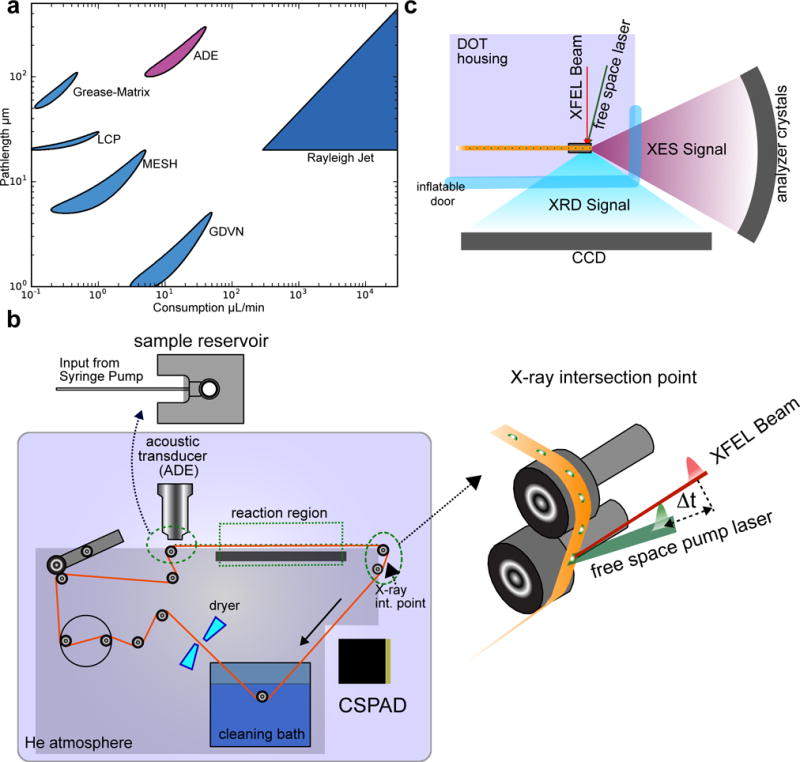
a. A comparison of sample consumption rate (assuming 120 Hz operation) and pathlength accessible with the replenishing methods used at XFELs. b. The experimental setup of DOT. The conveyor belt delivers droplets of sample at a high rate (up to 120 Hz). A large (283 liter) gas-tight chamber houses the entire instrument and is maintained at 95–97% Helium via a 30 cubic feet per hour purge flow. Droplets are deposited onto a polyimide belt using an ADE, and ejected from an open 2.5 mm diameter reservoir that is continuously resupplied via a capillary feed line attached to a syringe pump (top left b). In the interaction region, the belt was run at a small angle with respect to the z-axis (vertical) in the xz-plane (zoomed in view of interaction point). Positioning the droplet in the X-ray focus is accomplished by moving the entire system in the horizontal plane, while the droplet z-position (vertical) on the tape at the intersection is adjusted by changing the deposition delay. The belt is cleaned and dried in situ, enabling continuous use for days. The reaction initiation point for longer time delays is shown in the top right of panel b. Bottom right b. The XFEL beam passes parallel to the belt surface, striking the droplet atop the belt. c. The data collection geometry for XRD and XES. An inflatable (and X-ray transparent) plastic film door with a 160° aperture to the X-ray interaction region allows both XES and XRD to be collected simultaneously.
Acoustic Droplet Ejection (ADE) offers a sweet spot in sample consumption rate and available pathlength to accommodate both XES and XRD simultaneously for biomolecules15,16. To extend the ADE method to probe longer time delays (milliseconds to a few seconds) after the initiation of reactions and/or more complex reaction initiation schemes, we describe here a novel conveyor belt system. The conveyor belt continuously transports droplets containing crystal slurry or protein solution through the reaction region, consisting of an array of laser excitations for photochemical reactions, or gas-activation area for chemically-triggered reactions (Fig. 1b,c and Supplemental Fig. 1). The sample delivery parameters employed here were optimized for crystals of diameter 20–100 μm, solvent pathlengths of 200–300 μm, and sample consumption of 0.8–6 nL/shot with sample replacement rates of up to 120 Hz.
To demonstrate that this acoustic droplet ejection-drop on tape (ADE-DOT) method is versatile for capturing reaction intermediates, we show here two types of reactions triggered and observed in situ by Kβ1,3 or Kα XES using solution samples; (i) the water oxidation reaction of photosystem II (PS II), which requires multiple timed laser excitations, and (ii) an O2-activation reaction at the MnII/FeII cluster of the class Ic ribonucleotide reductase R2 (RNR) from Chlamydia trachomatis (Ct), which yields the active MnIV/FeIII cluster via a MnIV/FeIV intermediate17–19. The application of DOT to crystallography is further demonstrated by (iii) atomic-resolution XRD data of a bilin containing phytochrome photoreceptor and (iv) simultaneous XRD and XES of the Mn/Fe RNR.
RESULTS
The Drop-on-Tape setup
The conveyor belt is the core component of our DOT sample delivery technique and is used to build a queue of droplets (Fig. 1b, top right) that arrive at the XFEL beam in a desired state of preparation. Synchronized with the XFEL, droplets are deposited from an open 2.5 mm diameter reservoir onto a polyimide belt using an ADE15. ADE can eject large crystals without complications such as orifice clogging, which commonly plagues delivery systems using narrow (<100 micron) capillaries. A wide (~250 micron) capillary supply line maintains constant reservoir levels (Fig. 1b top left), allowing ejection conditions to remain stable and maintenance-free for hours. A focused XFEL beam (<25 μm2) that passes parallel to the belt surface probes the droplet atop the belt. After X-rays probe the sample, the belt is cleaned before it loops back to the start.
DOT is well-suited to probe a wide range of photochemical reactions in enzymes and inorganic systems. Excitation periods ranging from seconds to hundreds of milliseconds are accessible by positioning an evenly spaced array of laser excitation points along the belt (Fig. 2a). Shorter delays (femtoseconds to hundreds of milliseconds) are accessed by a movable excitation point near the X-ray interaction region (Fig. 1b). The length of the time delay is the ratio of the distance (D) between the points of reaction initiation and X-ray interaction to the tangential velocity of the belt (ν). The precision of the belt velocity required to achieve a desired time delay is determined by the ratio of the droplet diameter to the delay, where longer delay times and smaller droplets require higher precision. Delays of at least 3 seconds were readily achieved with 270 μm droplets (see Fig. 2a). In liquid jet systems, by contrast, excitation pulses for delay times longer than 10 ms need to be coupled into the capillary that supplies the jet, as was implemented for previous PS II studies20. The current method, with discrete excitation in a droplet-based system, is more flexible for multi-step excitation experiments and excludes population mixing resulting from laminar flow in a capillary.
Figure 2. Photo-initiated XES of PS II.
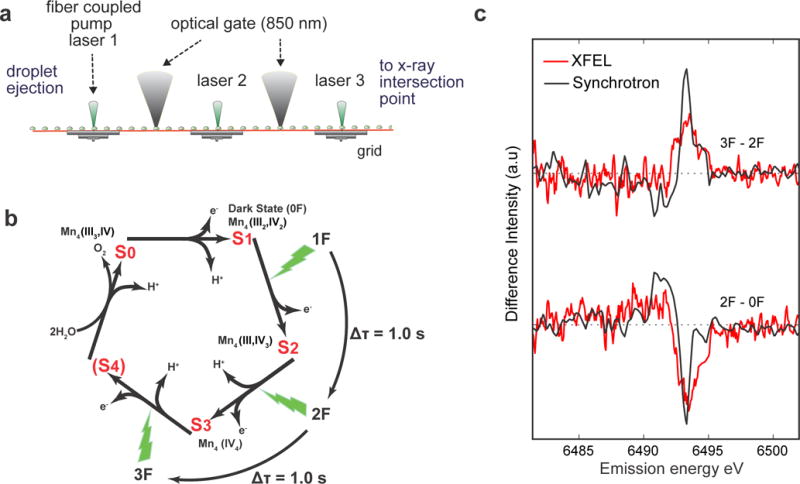
a. The photoexcitation setup employed for PS II, comprised of a precision-machined grid of fixed excitation positions with 60 mm spacing, is shown. Optical gates, which measure droplet arrival times on the grid, are realized by two low-power (<0.5 mW) continuous-wave near infrared (NIR, 850 nm) point sources, delivered via optical fiber. NIR light scatters from the droplets as they pass over the gates and is collected onto high-speed Si-photodiodes for readout. b. A schematic of the mechanism depicting S-state advancement using periodic laser flashes. The interval between laser flashes for both DOT and freeze-quench methods was 1.0 s. Flash states (denoted as 0F, 1F, 2F, or 3F) are highly enriched in the pure reaction intermediates S1, S2, S3, and S0, respectively, but are not completely pure due to small back reaction rates and photon misses (see ref. 23). c. A comparison of Mn Kβ1,3 XES difference spectra of PS II solution collected with the DOT instrument at room temperature using an XFEL (red) and the same state differences collected using a freeze-quench method at 8 K with a synchrotron source (black). The XFEL spectra contain about 120,000 shots per difference spectrum. For details of the collection conditions and difference analysis see Online Methods and Supplementary Fig. 7.
Photoactivation of photosystem II
Fig. 2a shows the DOT setup used to study PS II catalytic intermediates in solution sample. The setup was also used for recently reported studies of PS II crystal samples 21. The oxidation of water by PS II requires multiple laser flashes to complete the four-electron redox chemistry that is catalyzed by the Mn4CaO5 cluster22,23. This complex reaction progresses through four intermediates, called S-states (Fig. 2b). Long delay times between flashes (0.1–1.0 seconds) are required to ensure nearly complete production of each successive S-state due to a slow rate-limiting quinone movement24,25. To accommodate this intricate reaction mechanism, we implemented a feedback control system of the belt speed and deposition delay, which affected the flashing period and droplet phase, respectively. This was provided by a pair of optical gates that reported the arrival time of the droplet stream in the excitation grid (Fig. 2a and Supplementary Fig. 1).
Kβ1,3 emission (metal 3p to 1s transition) is sensitive to oxidation and spin state26,27 and serves as an indicator of the S-state advancement in PS II28. Fig. 2c shows the Mn Kβ1,3 XES difference spectra of PS II in solution collected with DOT. 270 μm diameter droplets (~5 nL) were injected onto the conveyor belt at room temperature in a helium atmosphere (Fig. 1b). The emission signal was collected with a 16 crystal wavelength-dispersive von Hamos spectrometer29,30, which collects the full Kβ1,3 energy range31 without scanning. These data are in good agreement with those collected at a synchrotron source using the freeze quench method for preparing PS II solution in different S-states26 (Fig. 2c). In the transition from the dark S1 state (0F, F: flash) to the most oxidized 2F (S3-rich) state, a negative difference on the high-energy side of the peak is seen, indicating oxidation. On the other hand, a clear positive difference relative to 2F at the high energy side is seen upon illumination to 3F (S0-rich), indicating transition of the major fraction of PS II from S3 to the most reduced S0 state. The purity of the states prepared, witnessed by the difference amplitude, on the belt is comparable with the best case S-state population acquired by freeze quench methods, while operating at room temperature and in high-throughput mode.
Chemical activation of ribonucleotide reductase by gas exposure
DOT can also be used to study chemically activated reactions. To demonstrate this, we examined O2 activation of Ct RNR by replacing the aforementioned laser excitation grid with an incubation chamber that exposes the drops to oxygen gas as they travel to the X-ray beam (Fig 3a,b). We built a chamber that exposes a 60 mm portion of the conveyor belt to a 4.7 L/min O2 flow. Leakage of O2 to the surrounding Helium environment was mitigated by using a flow-through exhaust design, guard baffles, and differential pumping. Tape speed through the chamber determines the gas exposure time, and we explored a range of exposures from 0.5 to 8 seconds.
Figure 3. XES of gas-activated RNR.
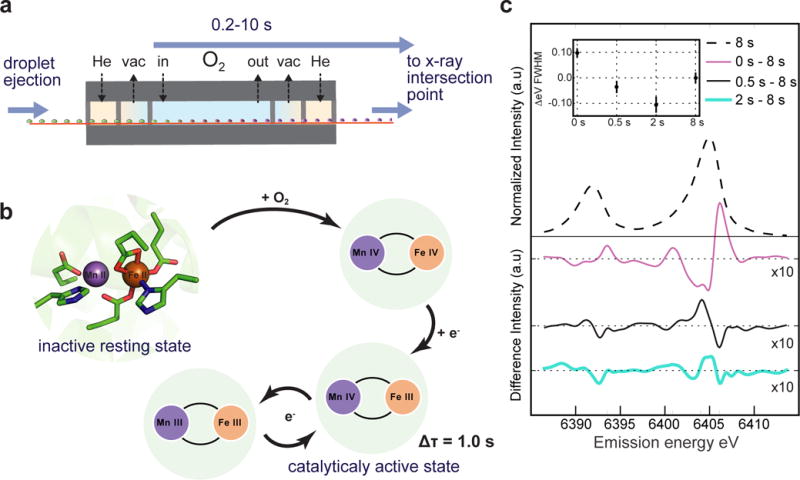
a. A schematic of the differentially pumped oxygen gas activation setup is shown. b. The known reaction scheme of Ct RNR17–19 is shown. Oxygen is activated at the MnII/FeII cluster to produce a high valent MnIVFeIV intermediate in a biomolecular reaction (kform = 13±3 mM−1sec−1 at 5 °C from the literature18), which then decays to the active MnIVFeIII state (first-order kdecay = 0.02 sec−1 at 5 °C also from literature18). c. RNR solution was monitored at 25 °C with Fe Kα emission for various O2 exposure times. The inset shows the Kα1 FWHM as a function of exposure time relative to an 8 s exposure. Error bars are computed via bootstrap residual sampling (1000 samples per data point). Difference spectra with respect to an 8 s exposure (smoothed by wavelet denoising) are shown in the main plot.
Fig. 3c shows the Fe Kα1 and Kα2 XES difference spectra of Fe/Mn RNR solution in which we monitored the advancement of the Fe site from its reduced form (FeII) to the oxidized form by varying O2 exposure time. Both Kα1 and Kα2 exhibit spectral changes as a function of O2 exposure time. The full width half max (FWHM) of the Kα1 line, which serves as a proxy for oxidation state32,33, shows the reaction progressing towards peak oxidation at 2 s and then reduction again at 8 s exposure, which roughly matches expected changes for the formation of MnIV/FeIV intermediate and subsequent decay to MnIV/FeIII (Fig. 3b)17–19. The difference spectra with respect to the 8 s O2 exposure show that the Fe Kα spectra contain complex lineshape changes, with both shifts and width changes throughout the reaction cycle. We note that no significant signs of droplet dehydration were detected for these studies of RNR solution. In samples containing a significant amount of Polyethylene Glycol (PEG), like many protein crystal buffers, PEG diffraction appears if the belt droplets are exposed to Helium for too long. The lower bound belt speed is thus limited by sample dehydration and needs to be evaluated on a per-sample basis.
For X-ray spectroscopy studies as described above, the ability to control pathlengths (e.g. 1/e is 520 μm at 6500 eV for water) is important for efficient data collection, by balancing the sample consumption rate and the data acquisition time. The acoustic pulse in the ADE system can be tuned to provide a range of droplet sizes (50–400 μm diameter), with larger droplets simply requiring a lower central frequency and/or higher driving force. The ability to access larger droplets is particularly beneficial for collecting signals from weaker transitions like Kβ2,5/Kβ” that can uniquely probe changes in the metal-ligand environment while its intensity is ~60 times lower than that of Kβ1,327.
Femtosceond crystallography using the DOT setup
ADE’s ability to adjust droplet size also allows a wide range of crystal sizes to be analyzed, while the discrete nature of delivery keeps sample consumption at moderate levels. Crystals >20 μm in size are of general interest for Serial Femtosecond Crystallography (SFX)2–4, as their larger crystal volume, in comparison to <5 μm crystals used in many liquid jet systems, can translate to stronger diffraction signals and potentially higher resolution. Also, diffraction quality may be more easily optimized at synchrotron beamlines prior to XFEL experiments for crystals in this size range. For the XRD results presented in this study, we used 5.5 nL droplets (~270–290 μm hemisphere) with crystal sizes ranging from 20 to 100 μm. With this size a 100% droplet hit rate was achieved with favorable signal collection rates and reasonable sample consumption (3.3 μL/min at 10 Hz). XRD solvent background was present, but manageable with a beam focus of ~5 μm. Thus, the solvent pathlength that can be achieved with the DOT (100 to 300 μm) is practical, vide infra, for the study of crystals surveyed here. However, practical considerations of what droplet size to select for which crystal size must be made for balancing the diffractivity of crystals and the solvent background; there is a pragmatic trade-off between factors favoring smaller droplets (solvent background, sample consumption) and those favoring larger droplets (instrument precision and distance of the X-ray beam from the belt). A consideration for XRD, which is unique to this setup, is polymer scatter and absorption from the polyimide conveyor belt. Polymer scatter was typically lower in amplitude than solvent scatter, and absorption was successfully treated in post-analysis (see Supplementary Fig. 2).
To explore the applicability of DOT for different protein crystals, we examined four samples: two different phytochrome constructs (50 μm plates or 100 μm needles), RNR (20–30 μm, bipyramidal), and PS II (20–50 μm, bipyramidal) (Supplementary Fig. 3, Supplementary Table 1). To collect diffraction from crystals of various sizes without saturating the detector, we used a high dynamic range Rayonix MX 170 HS detector, operated at its maximum data rate of 10 Hz in the 2×2 binning mode. For all crystallography data presented here, an incident X-ray energy of 9.5–9.8 keV was used. High-resolution room temperature structures were obtained in all cases (Supplementary Table 2)21. Data collection time varied depending on the crystal number density, but complete data sets could be collected in ~ 40–80 min with between 10 and 50% of all shots giving indexed diffraction patterns (Supplementary Table 1 and Supplementary Fig. 4).
Room temperature structures of phytochromes
XRD data were collected for the chromophore-binding region of Deinococcus radiodurans bacterial phytochrome (BphP) containing only the PAS (Per/Arnt/Sim) and GAF (cGMP phosphodiesterase/adenylyl cyclase/FhlA) domains (residues 1–321) or the entire photosensory module (PSM, residues 1–501) in their dark-adapted (Pr) states (Supplementary Table 2). These data yielded novel room temperature structures of DrBphP dimers at 1.65 and 3.3 Å resolution, respectively (Fig. 4a,b). The chromophore binding pocket of the PAS-GAF bili-protein was highly congruent with the best resolved structure of the DrBphP-PSM to date34, but diverged from an alternate binding pocket orientation seen for recently reported monomeric crystal assemblies of DrBphP35,36, and the photochemically compromised D207A mutant (Supplementary Fig. 5)34. Whether both bilin configurations represent authentic structural microstates of the Pr photostate in BphP dimers is not yet known. Additionally, we detected a significant shift of the PHY domain within the crystal lattice of the PSM dimer as compared to models generated with diffraction data collected at 100 K, suggesting that cryopreservation and/or cryogenic temperatures perturb the structure and/or dynamics of the GAF and PHY domains (Fig. 4b).
Figure 4. XRD of various enzymes.
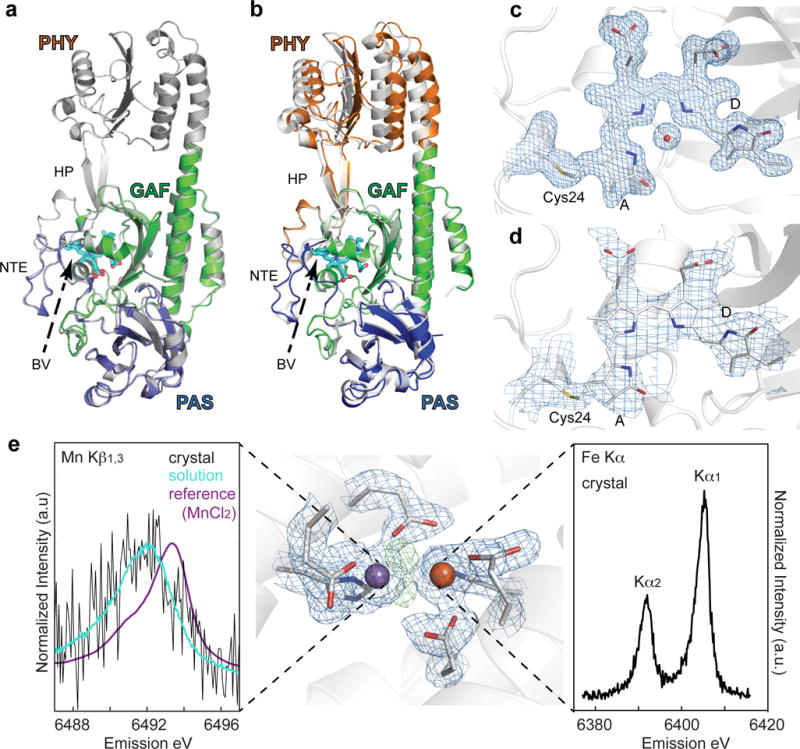
a. The PAS-GAF (blue and green, respectively) and the PSM (white) constructs from D. radiodurans BphP in their dark-adapted Pr states are shown superimposed. b. Superposition of atomic models of the PSM in the room-temperature Pr state (colored) with that derived from diffraction data collected at 100 K (white, PDB ID 4Q0J). The β-sheets of the GAF domains were superposed, allowing the respective positions of the PAS and PHY domains of the two models to be identified. The largest differences between the models were found at the PHY domain. For the room temperature model the domains were colored blue, green, and orange for the PAS, GAF, and PHY domains, respectively. Several important features are labeled: HP, hairpin; NTE, amino-terminal extension; BV, biliverdin. BV from the room temperature structure is shown for orientation. c and d. Composite simulated-annealing omit maps (2Fo-Fc) contoured at 1 σ were superimposed with the corresponding PAS-GAF (c) or PSM (d) model of DrBphP22. For clarity only electron density around Cys24 (gray), biliverdin (cyan) and for (c) the pyrrole water is shown. e. Metal site electron density (2Fo-Fc) at the heterodinuclear Mn/Fe site in aerobic class Ic RNR, metal ions and protein ligands are indicated (contoured to 1.3 σ) in blue. Residual positive difference electron density map (Fo-Fc) representing non-protein ligands is indicated in green (contoured to 3.5 σ). The manganese and iron atoms are depicted as purple and orange spheres, respectively. Kβ1,3 XES of oxidized RNR in crystal and in solution collected at room temperature with an XFEL is shown to the left. A Mn(II)Cl2 calibration standard is also shown to illustrate the absolute oxidation state of the solution and crystal spectra. Fe Kα XES Data collected from oxidized RNR crystals is shown at the right.
Phytochromes undergo a cascade of conformational changes upon photoexcitation of the bilin chromophore, with dynamics that span from picoseconds to milliseconds37–39. However, the electronic state of the bilin chromophore is exceedingly sensitive to radiation-induced changes from synchrotron X-ray sources40, culminating in rupture of the thioether linkage between the Cys24 and the bilin at high dose. These complications were eliminated in our XFEL data (Fig. 4c,d), with both structures clearly showing continuous electron density along the thioether-bilin linkage, visible already in the initial Fo-Fc omit maps (Supplementary Fig. 6). The ability to collect RT structures of phytochrome that are free of radiation induced artifacts thus opens the possibility of elucidating bona fide structures of phytochrome photoconversion intermediates.
Combined X-ray emission and crystallography of ribonucleotide reductase
We also collected a high-resolution room temperature structure of S. erythraea Mn/Fe RNR (Fig. 4e). Ribonucleotide reductases are the only enzymes that catalyze the reduction of ribonucleoside-5′-diphosphates or triphosphates (NDP or NTP) to the 2′-deoxynucleotides (dNDP or dNTP). Class I RNRs generate an essential catalytic radical via a dinuclear metal site, involving redox state changes at the metal site41. XES can be collected simultaneously with XRD for many metalloproteins serving as a sensitive in-situ verification of state preparation. To demonstrate this, we collected a Mn Kβ1,3 and Fe Kα spectrum of Mn/Fe RNR, simultaneous with XRD, from crystal slurries (Fig. 4e). Mn XES from RNR protein crystals was overlaid with a solution spectrum, using the same dispersive X-ray emission spectrometer system. The results confirmed that the XRD data collected, and thus the electron density obtained, represent the metal site in its as-prepared oxidized form and that the Mn ion was not photo-reduced to the (II) state before probing the scattering data. Model building and refinement is currently underway and will be reported elsewhere.
DISCUSSION
Through the XRD and XES results reported here, we demonstrate that the ADE-DOT sample delivery method provides a foundation for multimodal data collection in both solution and crystalline samples. The conveyor belt in DOT is flexible and extensible, permitting either photochemical or chemical activation of samples in situ, and thus enables it to study a wide variety of transient reaction intermediates. For photochemical reactions, in particular, the ability to precisely illuminate a discrete volume eliminates potential flow-induced state-mixing prior to probing the sample. Here we carried out the gas activation studies of a solution sample, but the method can also be applied to crystalline samples; the influence of substrate transport speed on the observed kinetics would depend on crystal size.
Another extension of the current work that is envisioned is liquid mixing prior to probing the droplet. This would enable a much broader class of enzyme-substrate studies. For reactions with relatively slow kinetics (seconds timescale) where liquids can be mixed right before droplet ejection, the method as it is shown can already be applied. Faster reactions can be probed by incorporating a rapid mixing scheme directly either in droplet creation or shortly after deposition on to the belt.
ONLINE METHODS
Samples
(a) PS II
PS II dimer was extracted and purified from T. elongatus as reported previously42. PS II crystals ranging in size from 10–60 μm were then prepared using an established seeding protocol43. The crystals were transferred into a final buffer consisting of 0.1 M MES pH 6.5, 0.1 M ammonium chloride, 10% (w/v) ethylene glycol and 35% (w/v) polyethylene glycol (PEG) 5000, with ~0.5 mM chlorophyll concentration, before loading into the sample delivery syringe (Hamilton gastight syringe, 500 μl).
Dimeric PS II solution samples were extracted and purified as described previously44. The final chlorophyll concentration was adjusted to ~7.0 mM (70 mg/mL protein) in a final buffer comprising 0.1 M PIPES pH 7.0, 5 mM calcium chloride, 30% (v/v) ethylene glycol and 0.02% (w/v) detergent β-dodecyl maltoside (βDM).
(b) Phytochrome
D. radiodurans BphP apoproteins bearing an N-terminal T7 tag (MASMTGGQQMGRGS) and a C-terminal hexahistidine tag were expressed in Escherichia coli BL21(DE3) cells as the PSM (residues 1–501) or the PAS-GAF region (residues 1–321 and including the Y307S mutation45 using the pET21b plasmid (Novagen, Madison, WI). Cultures were grown at 16 °C in terrific broth and harvested by centrifugation 16 h after inducing apoprotein expression with isopropyl-D-1-thiogalactopyranoside. The cell pellets were frozen in liquid nitrogen and stored at −80 °C.
Frozen pellets were resuspended in lysis buffer (10% (w/v) glycerol, 20 mM HEPES-NaOH (pH 7.8), 500 mM NaCl, 0.05% Tween 20, 30 mM imidazole, 1 mM phenylmethanesulfonyl fluoride, and 1 mM 2-mercaptoethanol), sonicated, and clarified by centrifugation. DrBphP apoproteins were enriched from the lysates by nickel-nitrilotriacetic acid (Qiagen, Valencia, CA) chromatography, using the lysis buffer for washing and lysis buffer with 300 mM imidazole for elution. The DrBphP-containing eluates were incubated overnight with a molar excess of biliverdin (BV) (Frontier Scientific, Logan, UT), and the free BV was removed by filtration through a G-25 Sephadex column (GE Healthcare) equilibrated in the ion-exchange buffer (10% (w/v) glycerol, 20 mM HEPES-NaOH (pH 7.8), 20 mM NaCl, and 10 mM 2-mercaptoethanol). The chromoproteins were then purified by ion-exchange chromatography with a Q-Sepharose HP column (GE Healthcare) and employing a 20–1000 mM linear NaCl gradient for elution. The PAS-GAF chromoprotein was exchanged into 30 mM Trizma base-HCl (pH 8.0), and the PSM was exchanged into 10 mM HEPES-NaOH (pH 7.8) using a 50-mL G-25 Sephadex column (GE Healthcare), frozen as 30–50 μL droplets in liquid nitrogen, and stored at −80 °C.
Crystallization of DrBphP chromoproteins was conducted in batch mode. PAS-GAF chromoproteins at 40 mg/mL were mixed at a 1:1 volumetric ratio with 15–16% (w/v) PEG 3350, 19% (v/v) isopropanol, 5% (w/v) glycerol, and 100 mM tetrasodium citrate-citric acid (pH 5.6). 2 mL batches were then seeded with 10 μL of a finely crushed PAS-GAF crystal slurry. PSM crystals were produced in the same manner except that the crystallization solution contained 22–28% (w/v) PEG 3350, 2.5% (v/v) Tacsimate (pH 6.0), 2.5% (v/v) Tacsimate (pH 7.0), 2% (v/v) ethylene glycol, and 100 mM PIPES-NaOH (pH 6.5). Crystalline samples were mobilized by scraping with a pipet tip and suspended by aspiration. The crystal slurry was then loaded into a Hamilton syringe for sample delivery.
(c) RNR
A crystallizable construct of the RNR R2 from S. erythraea was purified and prepared as described in ref46. Crystals suitable for this XFEL experiment were obtained at 20 °C under aerobic conditions using the batch crystallization method, in a 1:1 protein solution to crystallization buffer ratio. The crystallization buffer (10% (w/v) PEG3350, 1% (v/v) Tacsimate pH 4.0) was slowly released at the bottom of a 20 mg/mL protein solution containing tube such that phase separation occurred between the upper protein solution and the lower crystallization buffer. The tube was incubated upright without mixing for crystal growth. A pellet of up to 30 μm sized square bipyramidal shaped crystals appeared overnight. This crystal pellet was then resuspended in the crystallization buffer supplemented with 10% glycerol and loaded into the sample delivery syringe used for data collection.
For solution XES measurements, a histidine-tagged construct of Ct R2 was expressed and purified as previously described19,47; the resulting apo-protein was anaerobically reconstituted with 0.75 equivalents of Fe(II) and Mn(II) per monomer.
Representative images of crystals of all four samples used for the data collection are shown in Supplemental Fig. 3.
Cleaning and drying the belt for continuous use
During operation of the DOT, residual sample was removed continuously from the PI belt. After passing the X-ray probe point, the belt was immersed in a water tank where 4 high pressure water jets were used to remove sample debris. To remove waste and maintain a constant level in the tank, water was drained using a peristaltic pump (Masterflex L/S digital pump, HV-77921-75, Cole-Parmer Instrument Company, LLC, USA). Water jets were produced through 250 μm inner diameter PEEK tubing (Part # 1531, IDEX Health & Science LLC, USA), and were generated by an HPLC pump (Prep 100 pump, P40PFT01, Scientific Systems, Inc., USA) at 100 mL/min, which produced an effective pressure of 800 psi. The belt was then dried with He that was forced through small diameter nozzles (Atto super air nozzle, model 1108SS, EXAIR Corporation, USA). To minimize He consumption, the gas was continuously circulated using a compressor comprising a diaphragm pump (KNF Neuberger, Inc., type N145.1.2AN.18) and a small reservoir. This mechanism was necessary to have enough backing pressure to purge water from the belt surface. Effective drying is also important for the operation of the rollers to maintain a constant tape speed and was achieved by blowing He at a flow rate of 100 standard cubic feet (SCF) per hour with a backing pressure of 20 psi.
Sample delivery system
The tape drive instrumentation included a 15 MHz spot-focused piezoelectric immersion transducer (Part # V319, Olympus NDT) with a focal length of 1.0 in. A waveform generator (model 33612A, Keysight Technologies) triggered by the XFEL master clock was used to produce short bursts of RF energy which were amplified by a 500-W RF amplifier (Model 500A250C, Amplifier Research, Souderton, PA, USA) and used to drive the transducer for droplet ejection. The transducer was immersed in degassed water within a 3D-printed plastic sleeve with a sample reservoir at the end. A motorized stage was used to adjust the up and down position of the transducer such that the acoustic energy was focused at the liquid-gas interface. To change the position of the droplet deposition parallel to the beam, a second motorized stage was used to move the transducer and sleeve together. The sample reservoir was a 3D printed small plastic well (6 μL volume) with a thin polypropylene window to minimize the attenuation of the acoustic ejection pulse. Sample was continuously fed to the reservoir, using a syringe pump, through a 250 μm ID glass capillary. To avoid crystal settling, the syringe pump was mounted onto a shaker that rotated the syringe 180°. The 3D files/schematics of custom-built parts and more details of the sample delivery system are included in the Supplementary Protocol48.
Laser excitation system
A Coherent Evolution frequency doubled Nd:YLF laser provided 100 ns pulses of 527 nm light which were fiber coupled into the multi-flash excitation system depicted in Supplemental Fig. 1. An identical and independent Evolution provided the free-space excitation pulse near the X-ray interaction region for studies of PS II. For all excitations in PS II studies a fluence of 0.12 J/cm2 was used, with a focal diameter of 400 μm. The penetration depth of PS II at 527 nm is about 40 μm in PS II crystals and 400 μm in PS II solution at the concentrations we employ. Thus, we are close to the limit in crystal size that can be excited from a single side. For solution samples, the situation is better (as the concentration is about 10× lower).
Gas activation system
The oxygen incubation chamber (Fig. 3a) used for solution studies of RNR was 3D printed using ABS-like resin and has 5 internal chambers. The central compartment, where O2 gas is pumped, is a 60 mm long, 14 mm diameter cylinder. The flanking chambers, two on each side, are each 15 mm long and 14 mm in diameter. To mount the conveyor belt on the drive, the top half of the chamber was made to be removable and after mounting the belt the chamber top was sealed to the bottom half with the aid of vacuum grease. Entrance holes into the sides of the O2 activation chamber and between internal chambers measured 0.9 mm tall by 3 mm wide. The vacuum pressure on the chambers immediately flanking the central O2 chamber was set to −0.5 psig through a variable vacuum regulator. More details of the gas activation system are included in the Supplementary Protocol48.
Data collection
The DOT setup was employed at two end stations in Linac Coherent Light Source (LCLS): X-ray Pump Probe49 (XPP) and Macromolecular Femtosecond Crystallography (MFX). Photoactivated studies began at XPP and continued at MFX over several beamtimes. Chemically activated studies were performed at MFX. The X-ray beam with a pulse width of ~50 fs was focused at either end station to 5×5 μm2 (FWHM) using Be lenses. A pulse energy of 2.5 mJ at 9.5 keV, corresponding to ~1.6 × 1012 photons/pulse, was used at a repetition rate of 10 Hz for crystal samples. For XES, solution samples of PSII were excited at 7.5 keV and at 30 Hz rep rate. Fe XES of RNR solution was collected at 60 Hz using 9.5 keV excitation and 2.5 mJ pulse energy. To minimize the belt diffraction background, the tails of the beam were obscured with a pinhole lead collimator. XRD data were collected in the forward direction using a Rayonix MX 170 HS detector at a distance of 58 to 108 mm from the interaction point allowing for detection of a maximum inscribed resolution of 1.4 or 2.0 Å, respectively. To calibrate the detector distance, the XRD pattern of silver behenate, enclosed in a quartz capillary (Hampton Research, 10 μm wall thickness) was recorded with an attenuated beam (≤1%). Mn XES spectra were recorded perpendicular to the beam on a Cornell-SLAC pixel-array detector (CSPAD-140k)50 using a multi-crystal wavelength-dispersive hard X-ray spectrometer based on the von Hamos geometry29. Studies on Fe Kα XES employed the newly available ePIX-100 detector51 in the same geometry. Mn XES was analyzed by an array of 16 Si (440) crystal analyzers, mounted at a distance of 500 mm from the interaction point, with the center of the array at an angle of 81° with respect to the interaction point. The Bragg angle range covered by these analyzer crystals was 85.8–83.4° which corresponds to a Mn Kβ1,3 energy range from 6474.6–6499.4 eV. To calibrate the emission energy, the XES spectrum of MnCl2 solution (500 mM), in a quartz capillary, was collected using an unfocused beam. The emission energy was then calibrated to the published value of 6493.3 eV of MnCl2 spectrum peak position30. Fe Kα XES was analyzed by a single Ge (440) crystal placed 250 mm from the interaction point, with the center of the crystal at 75.41° with respect to the interaction point and resolved both Kα lines from 6370 to 6420 eV. Calibration of the Fe spectrometer was similar to the method for Mn, using as the reference Fe(III)(NO3)3 instead.
Processing of Data of Raw XRD images
Analysis of the diffraction data was performed using cctbx.xfel52,53. Diffraction images were indexed using LABELIT54 with a target cell to guide the choice of basis vectors. For PS II samples, two populations of unit cells in the P212121 space group were identified (isoform A: a=117.5 Å, b=223.6 Å, c=329.5 Å; isoform B: a=117.7 Å, b=223.2 Å, c=310.5 Å). PS II diffraction images were re-indexed twice, imposing either isoform A or isoform B unit cell parameters as constraints during unit cell refinement, to generate two disjoint sub-datasets for each illuminated state. For other samples, the single predominant unit cell identified in the first indexing step was imposed as a basis set target and a unit cell constraint in a second round of indexing. For all samples, resolution limits were determined independently for each image in order to ensure that the weak signal at high resolution in the highest-resolution images was not diluted by noise contributions from lower-resolution images. To determine resolution limits on each image, reflections were sorted into bins of equal size by resolution, and all bins beyond and including the lowest-resolution bin in which the average signal to uncertainty ratio <I/σ(I)> was below 0.1 were discarded. This approach permits the inclusion of a small number of negative intensities while in practice eliminating most nonphysical negative intensity reflections. Signal was integrated to these resolution limits and the resulting data were merged using cxi.merge. The resolution cut-off for the final merged data was determined based on a combination of several criteria, including where the data falls below 10 fold multiplicity, where the CC1/2 becomes ‘unstable’, meaning it begins to oscillate at higher resolution instead of uniformly decreasing, and where the values of I/σ(I) do not uniformly decrease any more.
Roughly half of the forward scattering signal passes through the PI belt, slightly lowering the Bragg spot intensities in this region. We developed a ray-tracing model to predict the absorption by the belt at a given point on the detector and used this to correct Bragg intensities during integration. Our model uses three fit parameters: distance of the crystal from the edge of the belt f, crystal height h, and the angle Θ between the vertical axis of the detector and the conveyor belt. Belt thickness and material absorption cross section are treated as known constants. These various parameters are illustrated in Supplemental Fig. 2c with the overall problem depicted in Supplemental Fig. 2b. Parameters f, h, and Θ are obtained indirectly through geometric relations to two boundary lines that are observed on the detector. The boundary lines (corresponding to maximum and minimum belt absorption) are highlighted in Supplemental Fig. 2a and c in red and blue, respectively. An estimate for the uncertainty of the Bragg spot intensity is needed in subsequent processing steps to weight observations. The uncertainty of the absorption correction to the spot intensity is estimated by the change in spot intensity upon perturbing the fit parameters. We typically find that the perturbations in f produce the largest effect and that perturbations on the order of a droplet diameter dominate other uncertainty estimates. Consequently, Bragg spots, which are shadowed by the belt, are down-weighted relative to those that are un-shadowed during merging. Separate resolution cut-offs are used for Bragg spots that are affected and unaffected by Kapton belt absorption.
XES data reduction and analysis
Supplemental Fig. 6 panel a shows the raw data of the averaged XES spectrum collected with the pixel array detector. To subtract the background signal, a fifth order two-dimensional polynomial background was fit to the image using information outside of the Region of Interest (ROI), shown in bounded by red lines. The fit of the background was extrapolated into the ROI (panel b, with the intensity multiplied by 4) to estimate its intensity there. The estimated background is shown as the red line in panel Supplemental Fig. 6 c. The final XES spectrum is obtained by integrating a tight ROI around the background subtracted signal (depicted in Supplemental Fig. 6 d). The resulting spectrum is shown in panel e. Difference spectra are computed normalizing the component spectra by their respective sums and then subtracting the components.
Synchrotron XES data collection
Room temperature difference Mn Kβ1,3 XES shown in Figure 2 are shown in reference to cryogenic difference spectra of PS II S-states collected at BL 6–2 in SSRL (Stanford Synchrotron Radiation Lightsource). For these synchrotron measurements, the incidence energy was set to 10.4 keV with the beamline monochromator, which uses two cryogenically cooled Si crystals in (111) reflection. The X-ray beam was focused to 0.45 (V) × 0.45 (H) mm (fwhm) by means of vertical and horizontal focusing mirrors. The X-ray flux at 10.4 keV was ~ 1 × 1013 photons/s/mm2. Samples were prepared in 0, 2, and 3 flash states by a freeze quench method, wherein a sample holder containing a 300 µm thick layer of PS II solution from T. elongatus (8.5 mM chlorophyll) is flashed from two sides with a Spectra-Physics Quanta Ray Pro 230-10 Nd:YAG laser (0.3 J/cm2, 9 ns pulse duration) with a 1 s flashing interval and subsequently plunged (within 1 second) into liquid nitrogen. During the measurement, samples were kept at 10 K in a continuous flow liquid helium cryostat (Oxford Instruments CF1208) under helium exchange gas atmosphere. Emission spectra were recorded by means of a high-resolution crystal-array spectrometer, using the 440 reflection of 7 spherically bent Si(110) crystals (100 mm diameter, 1 m radius of curvature), aligned on intersecting Rowland circles. An energy-resolving Si drift detector (Vortex) was positioned at the focus of the 7 diffracting elements. A helium-filled polyethylene bag was placed between the cryostat and the spectrometer to minimize signal attenuation due to air absorption. Each energy point in the spectra was collected at a fresh sample spot. The maximum exposure time at each spot was 2.5 s and the signal was read out in bins of 50 ms duration. To determine an appropriate exposure time to avoid radiation-induced effects, the signal intensity was measured as a function of exposure time at a single emission energy for each S-state. No significant changes were observed within the first 1.5 s of exposure, and so 20 time bins (equivalent to 1 s) were averaged for the final spectra. The signal intensity from each sample spot was normalized by the emission signal intensity recorded at 6491.5 eV within 7 s from the same sample spot, after going through all the fresh spots.
Phytochrome Structure determination, model building and refinement
Initial phases for the PAS-GAF and PSM datasets were calculated by PHASER55 (using the 1.45-Å DrBphP PAS-GAF structure (PDB ID code 2O9C) generated by Wagner et al45 as the search model for PAS and GAF domains, and residues 325–501 of PDB ID code 4Q0J as the PHY domain search model for the PSM34. To minimize model bias at the chromophore, the biliverdin and side chains of the following residues were omitted during molecular replacement and initial structural refinements: Cys24, Ile29, Arg70, Met174, Tyr176, Glu193, His196, Phe198, Phe203, Asp207, Ile208, Pro209, Tyr216, Arg222, Arg254, Ser257, Met259, His260, Tyr263, Leu264, Met267, Ser272, Ser274, Leu286, His290, and Arg310. Manual model building was conducted with COOT56, and structures were refined and simulated-annealing omit maps were calculated with PHENIX57. The initial refinement for each structural model included simulated annealing to reduce model bias. Model validation was conducted with MOLPROBITY58, and superpositions and figure preparation were conducted with the PyMOL Molecular Graphics System.
RNR Structure determination, model building and refinement
The RNR structure was solved using PHASER55 by molecular replacement using the atomic coordinates of the R2 subunit of ribonucleotide reductase from Chlamydia trachomatis (PDB ID code: 1SYY59) as starting model. A well-contrasted solution was obtained with 1 molecule per asymmetric unit (space group P41212). A preliminary crystallographic refinement was performed using PHENIX57 and the three-dimensional model was examined and modified using the program COOT56. Supplemental Table 2 lists the crystallographic statistics in which the test set represents 5% of the reflections. The current Ramachandran plot indicates that 93.0% and 0.3% of the residues are in the favored and disallowed regions, respectively. Figure 4e was prepared using the PyMOL Molecular Graphics System.
Data availability
Accession codes
Coordinates and structure factors of the phytochrome PAS-GAF and PSM structures have been deposited in the RCSB Protein Data Bank under accession codes 5MG0 (doi:10.2210/pdb5mg0/pdb)60 and 5MG1 (doi:10.2210/pdb5mg1/pdb)61, respectively.
Code Availability
Source code for the cctbx.xfel software package, used for diffraction data processing is freely available and is distributed via the Phenix software package (http://phenix-online.org/). Further information can be obtained via the cctbx.xfel homepage (http://cci.lbl.gov/xfel/).
Supplementary Material
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences (CSGB) of the Department of Energy (DOE) under contract DE-AC02-05CH11231 (J.Y. and V.K.Y.) for X-ray methodology and instrumentation, by National Institutes of Health (NIH) Grants GM110501 (J.Y.) for instrumentation development for XFEL experiments, GM102520 and GM117126 (N.K.S.) for development of computational protocols for XFEL data, and GM055302 (V.K.Y.) for PS II biochemistry, structure and mechanism, the Ruth L. Kirschstein National Research Service Award (5 F32 GM116423-02, F.D.F.), the Human Frontiers Science Project Award No. RGP0063/2013 310 (J.Y., U.B. and A.Z.), and the Science and Technology Center program of the National Sciences Foundation through BioXFEL under agreement No. 1231306 (J.A.C., M.D.M. and G.N.P.). U.S. National Science Foundation grant MCB-1329956 supports R.D.V. J.A.C. was supported by a training fellowship from the Gulf Coast Consortia on the Houston Area Molecular Biophysics Program (NIHGMS Grant No. T32GM008280). C.J.P. was supported by NIH NRSA (GM113389-01). Portions of this work were supported by a Brookhaven National Laboratory/US DOE, Laboratory Directed Research and Development grant 11-008 (C.G.R., M.A., A.M.O.); the National Institutes of Health (NIH)/NCRR Grant 2-P41-RR012408, NIH/National Institute of General Medical Sciences (NIGMS) Grant 8P41GM103473-16 and the US DOE, Office of Biological and Environmental Research (OBER) Grant FWP BO-70 (A.M.O. and B.A.); NIH NIGMS grant Y1GM008003 (M.A.). A.M.O., P.A. and P.T.D. were supported in part by Diamond Light Source and A.M.O. acknowledges support from a Strategic Award from the Wellcome Trust and the Biotechnology and Biological Sciences Research Council (grant 102593). P.B. was supported by a Wellcome Trust DPhil studentship. The Knut and Alice Wallenberg Foundation, the Swedish Cancer Society, The Wenner-Gren foundations and the Swedish Research Council (grants 2013-541 and 2013-5884) provided support to M.H. C.A.S. acknowledges support from the US Department of Energy, Office of Science, Chemical Sciences, Geosciences, and Biosciences Division. The DFG-Cluster of Excellence “UniCat” coordinated by the Technische Universität Berlin and Sfb1078 (Humboldt Universität Berlin), TP A5 (A.Z.); the Solar Fuels Strong Research Environment (Umeå University), the Artificial Leaf Project (K&A Wallenberg Foundation 2011.0055) and Energimyndigheten (36648-1) (J.M.) are acknowledged for supporting this project. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science, DOE, under Contract No. DE-AC02-05CH11231. Testing of crystals and various parts of the setup were carried out at synchrotron facilities that were provided by the Advanced Light Source (ALS) in Berkeley and the Stanford Synchrotron Radiation Lightsource (SSRL) in Stanford, funded by DOE OBES. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH (P41GM103393). Use of the Linac Coherent Light Source (LCLS) and SSRL, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, OBES under Contract No. DE-AC02-76SF00515. Brookhaven National Laboratory’s (BNL) contribution to data collection at the LCLS is supported by the Life Science and Biomedical Technology Research (LSBR) program at the National Synchrotron Light Source II (NSLS-II) which operates under a DOE BER contract DE-SC0012704 and DOE BES contract DE-AC02-98CH10886 and is supported by an NIH-NIGMS grant P41GM111244. We thank Theodore Rendahl from LCLS for help with controls and Bruno Martins, Stuart Myers, Matthew Cowan, Grace Shea-McCarthy, and Cornelius Whalen from Brookhaven National Laboratory for help with engineering and controls. We are grateful to Rich Ellison, Joe Olechno, Rick Stearns, and Babur Hadimioglu from LABCYTE for sharing their experience with the acoustic transducers and related issues, Claudio Saracini from LBNL for his help with the preliminary testing of the droplet on tape system, Pieter Glatzel (ESRF) for discussion of the Fe Kα emission and Frances Houle of LBNL for sharing her reaction-diffusion simulation software Kinetiscope and useful discussions.
Footnotes
Author Contributions
A.M.O., J.K., V.K.Y. and J.Y. conceived the experiment, which was designed by F.D.F., S.G., J.K., V.Y., and J.Y.
Phytochrome, PSII and RNR samples were prepared, characterized, and provided by E.S.B., R.C., C.J.P., J.A.C., M.I., R.H., A.Z., H.L., V.S., M.Z., S.K., J.M., A.K.B., J.M.B., C.K., M.H., G.N.P., and R.D.V.
The acoustic injectors were designed by F.D.F., J.K., S.G., C.G.R. and A.M.O.
SFX and XES experiments were carried out by F.D.F., S.G., B.A., E.P., C.d.L., C.A.S., C.G.R., R.G.S., T.K., M.K., S.K., P.T.D.; U.B., G.P., J.K., V.K.Y, A.M.O., and J.Y.
Beamline setup was done by J.M.G., C.A.S., S.N., J.E.K., D.Z., M.C., S.S., H.L., D.S., M.L. and R.A.-M.
XRD and XES data were analyzed by A.S.B., I.D.Y., T.M.C., P.A., P.B., L.L., M.D.M., T.K., M.A., F.D.F., J.K. E.S.B, T.F., C.W., and N.K.S.
F.D.F., J.K, E.S.B., A.M.O., V.K.Y, and J.Y. wrote the paper with contributions from all authors.
References
- 1.Emma P, et al. First lasing and operation of an ångstrom-wavelength free-electron laser. Nat Photon. 2010;4:641–647. [Google Scholar]
- 2.Chapman HN, et al. Femtosecond X-ray protein nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boutet S, et al. High-resolution protein structure determination by serial femtosecond crystallography. Science. 2012;337:362–364. doi: 10.1126/science.1217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kern J, et al. Simultaneous Femtosecond X-ray Spectroscopy and Diffraction of Photosystem II at Room Temperature. Science. 2013;340:491–495. doi: 10.1126/science.1234273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 6.Levantino M, Yorke BA, Monteiro DC, Cammarata M, Pearson AR. Using synchrotrons and XFELs for time-resolved X-ray crystallography and solution scattering experiments on biomolecules. Curr Opin Struct Biol. 2015;35:41–48. doi: 10.1016/j.sbi.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 7.DePonte DP, et al. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J Phys D: Appl Phys. 2008;41:195505. [Google Scholar]
- 8.Sierra RG, et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr Sect D: Biol Crystallogr. 2012;68:1584–1587. doi: 10.1107/S0907444912038152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weierstall U, et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun. 2014;5:3309. doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugahara M, et al. Grease matrix as a versatile carrier of proteins for serial crystallography. Nat Methods. 2015;12:61–63. doi: 10.1038/nmeth.3172. [DOI] [PubMed] [Google Scholar]
- 11.Hunter MS, et al. Fixed-target protein serial microcrystallography with an x-ray free electron laser. Sci Rep. 2014;4:6026. doi: 10.1038/srep06026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baxter EL, et al. High-density grids for efficient data collection from multiple crystals. Acta Crystallogr D Struct Biol. 2016;72:2–11. doi: 10.1107/S2059798315020847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roedig P, et al. Room-temperature macromolecular crystallography using a micro-patterned silicon chip with minimal background scattering. J Appl Crystallogr. 2016;49:968–975. doi: 10.1107/S1600576716006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oghbaey S, et al. Fixed target combined with spectral mapping: approaching 100% hit rates for serial crystallography. Acta Crystallogr D Struct Biol. 2016;72:944–955. doi: 10.1107/S2059798316010834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roessler CG, et al. Acoustic Injectors for Drop-On-Demand Serial Femtosecond Crystallography. Structure. 2016;24:631–640. doi: 10.1016/j.str.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roessler CG, et al. Acoustic methods for high-throughput protein crystal mounting at next-generation macromolecular crystallographic beamlines. J Synchtron Radiat. 2013;20:805–808. doi: 10.1107/S0909049513020372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griese JJ, Srinivas V, Högbom M. Assembly of nonheme Mn/Fe active sites in heterodinuclear metalloproteins. J Biol Inorg Chem. 2014;19:759–774. doi: 10.1007/s00775-014-1140-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang W, Hoffart LM, Krebs C, Bollinger JM. A manganese (IV)/iron (IV) intermediate in assembly of the manganese (IV)/iron (III) cofactor of Chlamydia trachomatis ribonucleotide reductase. Biochemistry. 2007;46:8709–8716. doi: 10.1021/bi700906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang W, et al. A manganese (IV)/iron (III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science. 2007;316:1188–1191. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 20.Kern J, et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat Commun. 2014;5:4371. doi: 10.1038/ncomms5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young ID, et al. Structure of photosystem II and substrate binding at room temperature. Nature. 2016;540:453–457. doi: 10.1038/nature20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kok B, Forbush B, McGloin M. Cooperation of charges in photosynthetic O2 evolution–I. A linear four step mechanism. Photochem Photobiol. 1970;11:457–475. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- 23.Renger G. Primary Processes of Photosynthesis: Principles and Apparatus. Royal Society of Chemistry; Cambridge, UK: 2008. [Google Scholar]
- 24.de Wijn R, van Gorkom HJ. Kinetics of electron transfer from QA to QB in photosystem II. Biochemistry. 2001;40:11912–11922. doi: 10.1021/bi010852r. [DOI] [PubMed] [Google Scholar]
- 25.Alonso-Mori R, et al. Towards characterization of photo-excited electron transfer and catalysis in natural and artificial systems using XFELs. Faraday Discuss. 2016;194:621–638. doi: 10.1039/c6fd00084c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Messinger J, et al. Absence of Mn-centered oxidation in the S2 -> S3 Transition: Implications for the mechanism of photosynthetic water oxidation. J Am Chem Soc. 2001;123:7804–7820. doi: 10.1021/ja004307+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glatzel P, Bergmann U. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes - electronic and structural information. Coord Chem Rev. 2005;249:65–95. [Google Scholar]
- 28.Yano J, Yachandra V. Mn4Ca Cluster in Photosynthesis: Where and How Water is Oxidized to Dioxygen. Chem Rev. 2014;114:4175–4205. doi: 10.1021/cr4004874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alonso-Mori R, et al. A multi-crystal wavelength dispersive x-ray spectrometer. Rev Sci Instrum. 2012;83:073114. doi: 10.1063/1.4737630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alonso-Mori R, et al. Energy-dispersive X-ray emission spectroscopy using an X-ray free-electron laser in a shot-by-shot mode. Proc Natl Acad Sci U S A. 2012;109:19103–19107. doi: 10.1073/pnas.1211384109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alonso-Mori R, et al. Photon-in photon-out hard X-ray spectroscopy at the Linac Coherent Light Source. J Synchtron Radiat. 2015;22:612–620. doi: 10.1107/S1600577515004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vankó G, et al. Probing the 3d Spin Momentum with X-ray Emission Spectroscopy: The Case of Molecular-Spin Transitions. J Phys Chem B. 2006;110:11647–11653. doi: 10.1021/jp0615961. [DOI] [PubMed] [Google Scholar]
- 33.Svyazhin A, Kurmaev E, Shreder E, Shamin S, Sahle CJ. Local moments and electronic correlations in Fe-based Heusler alloys: Kα x-ray emission spectra measurements. J Alloys Compd. 2016;679:268–276. [Google Scholar]
- 34.Burgie ES, et al. Crystallographic and electron microscopic analyses of a bacterial phytochrome reveal local and global rearrangements during photoconversion. J Biol Chem. 2014;289:24573–24587. doi: 10.1074/jbc.M114.571661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edlund P, et al. The room temperature crystal structure of a bacterial phytochrome determined by serial femtosecond crystallography. Sci Rep. 2016;6:35279. doi: 10.1038/srep35279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhattacharya S, Auldridge ME, Lehtivuori H, Ihalainen JA, Forest KT. Origins of fluorescence in evolved bacteriophytochromes. J Biol Chem. 2014;289:32144–32152. doi: 10.1074/jbc.M114.589739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang CF, Farrens DL, Bjorling SC, Song PS, Kliger DS. Time-Resolved Absorption Studies of Native Etiolated Oat Phytochrome. J Am Chem Soc. 1992;114:4569–4580. [Google Scholar]
- 38.Mroginski MA, Murgida DH, Hildebrandt P. The chromophore structural changes during the photocycle of phytochrome: A combined resonance raman and quantum chemical approach. Acc Chem Res. 2007;40:258–266. doi: 10.1021/ar6000523. [DOI] [PubMed] [Google Scholar]
- 39.Braslavsky SE, Gärtner W, Schaffner K. Phytochrome photoconversion. Plant Cell Environ. 1997;20:700–706. [Google Scholar]
- 40.Li F, et al. X-ray Radiation Induces Deprotonation of the Bilin Chromophore in Crystalline D. radiodurans Phytochrome. J Am Chem Soc. 2015;137:2792–2795. doi: 10.1021/ja510923m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundin D, Berggren G, Logan DT, Sjoberg BM. The origin and evolution of ribonucleotide reduction. Life (Basel) 2015;5:604–636. doi: 10.3390/life5010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hellmich J, et al. Native-like Photosystem II Superstructure at 2.44 Å Resolution through Detergent Extraction from the Protein Crystal. Structure. 2014;22:1607–1615. doi: 10.1016/j.str.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 43.Ibrahim M, et al. Improvements in serial femtosecond crystallography of photosystem II by optimizing crystal uniformity using microseeding procedures. Struct Dyn. 2015;2:041705. doi: 10.1063/1.4919741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kern J, et al. Purification, characterisation and crystallisation of photosystem II from Thermosynechococcus elongatus cultivated in a new type of photobioreactor. Biochim Biophys Acta Bioenerg. 2005;1706:147–157. doi: 10.1016/j.bbabio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Wagner JR, Zhang J, Brunzelle JS, Vierstra RD, Forest KT. High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J Biol Chem. 2007;282:12298–12309. doi: 10.1074/jbc.M611824200. [DOI] [PubMed] [Google Scholar]
- 46.Kutin Y, et al. Divergent assembly mechanisms of the manganese/iron cofactors in R2lox and R2c proteins. J Inorg Biochem. 2016;162:164–177. doi: 10.1016/j.jinorgbio.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 47.Dassama LMK, et al. O2-Evolving Chlorite Dismutase as a Tool for Studying O2-Utilizing Enzymes. Biochemistry. 2012;51:1607–1616. doi: 10.1021/bi201906x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuller FD, et al. Droplet On Tape: Protocol. Nat Protoc. 2017 http://www.nature.com/protocolexchange/protocols/5489/
- 49.Chollet M, et al. The x-ray pump–probe instrument at the linac coherent light source. J Synchtron Radiat. 2015;22:503–507. doi: 10.1107/S1600577515005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herrmann S, et al. CSPAD-140k: A versatile detector for LCLS experiments. Nucl Instr Meth Phys Res. 2013;718:550–553. [Google Scholar]
- 51.Carini G, et al. 2014 IEEE Nuclear Science Symposium and Medical Imaging Conference (NSS/MIC) IEEE; Seattle, WA USA: 2014. pp. 1–3. [Google Scholar]
- 52.Sauter NK, Hattne J, Grosse-Kunstleve RW, Echols N. New Python-based methods for data processing. Acta Crystallogr Sect D: Biol Crystallogr. 2013;69:1274–1282. doi: 10.1107/S0907444913000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hattne J, et al. Accurate macromolecular structures using minimal measurements from X-ray free-electron lasers. Nat Methods. 2014;11:545–548. doi: 10.1038/nmeth.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sauter NK, Grosse-Kunstleve RW, Adams PD. Robust indexing for automatic data collection. J Appl Crystallogr. 2004;37:399–409. doi: 10.1107/S0021889804005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Högbom M, et al. The radical site in chlamydial ribonucleotide reductase defines a new R2 subclass. Science. 2004;305:245–248. doi: 10.1126/science.1098419. [DOI] [PubMed] [Google Scholar]
- 60.Burgie ES, et al. Structure of PAS-GAF fragment of Deinococcus phytochrome by serial femtosecond crystallography. RCSB Protein Data Bank. 2017 doi: 10.2210/pdb5mg0/pdb. [DOI] [Google Scholar]
- 61.Burgie ES, et al. Structure of the photosensory module of Deinococcus phytochrome by serial femtosecond X-ray crystallography. RCSB Protein Data Bank. 2017 doi: 10.2210/pdb5mg1/pdb. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession codes
Coordinates and structure factors of the phytochrome PAS-GAF and PSM structures have been deposited in the RCSB Protein Data Bank under accession codes 5MG0 (doi:10.2210/pdb5mg0/pdb)60 and 5MG1 (doi:10.2210/pdb5mg1/pdb)61, respectively.
Code Availability
Source code for the cctbx.xfel software package, used for diffraction data processing is freely available and is distributed via the Phenix software package (http://phenix-online.org/). Further information can be obtained via the cctbx.xfel homepage (http://cci.lbl.gov/xfel/).