

Abstract

The isomerization of estragole to trans-anethole is an important reaction and is industrially performed using an excess of NaOH or KOH in ethanol at high temperatures with very low selectivity. Simple Ru-based transition-metal complexes, under homogeneous, ionic liquid (IL)-supported (biphasic) and “solventless” conditions, can be used for this reaction. The selectivity of this reaction is more sensitive to the solvent/support used than the ligands associated with the metal catalyst. Thus, under the optimized reaction conditions, 100% conversion can be achieved in the estragole isomerization, using as little as 4 × 10–3 mol % (40 ppm) of [RuHCl(CO)(PPh3)3] in toluene, reflecting a total turnover number (TON) of 25 000 and turnover frequencies (TOFs) of up to 500 min–1 at 80 °C. Using a dimeric Ru precursor, [RuCl(μ-Cl)(η3:η3-C10H16)]2, in ethanol associated with P(OEt)3, a TON of 10 000 and a TOF of 125 min–1 are obtained with 100% conversion and 99% selectivity. These two Ru catalytic systems can be transposed to biphasic IL systems by using ionic-tagged P-ligands such as 1-(3-(diphenylphosphanyl)propyl)-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide immobilized in 1-(3-hydroxypropyl)-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl) imide with up to 99% selectivity and almost complete estragole conversion. However, the reaction is much slower than that performed under solventless or homogeneous conditions. The use of ionic-tagged ligands significantly reduces the Ru leaching to the organic phase, compared to that in reactions performed under homogeneous conditions, where the catalytic system loses catalytic performance after the second recycling. Detailed kinetic investigations of the reaction catalyzed by [RuHCl(CO)(PPh3)3] indicate that a simplified kinetic model (a monomolecular reversible first-order reaction) is adequate for fitting the homogeneous reaction at 80 °C and under biphasic conditions. However, the kinetics of the reaction are better described if all of the elementary steps are taken into consideration, especially at 40 °C.
1. Introduction
Trans-anethole is a naturally occurring alkylbenzene,1 which can be extracted from anise or fennel oils,2,3 albeit with variable proportions of the cis isomer as an impurity. Extraction of trans-anethole from natural sources cannot supply the growing market (food, drugs, and cosmetics),4−7 generating the need for a synthetic alternative. For industrial purposes, only trans-anethole is of use, as the cis isomer has a higher toxicity and unpleasant organoleptic properties.8−10 Currently, trans-anethole is obtained by estragole isomerization (Scheme 1), promoted by an excess of NaOH or KOH.11,12 However, there are a number of disadvantages for this process, including the requirement of high temperatures (>200 °C), low conversion in anethole (∼60%), a lack of stereoselectivity (trans/cis ratio 82:18), and the significant amounts of basic wastes that are generated.
Scheme 1. Isomerization of Estragole to Trans-Anethole.

To overcome these limitations, several protocols using homogeneous13−16 and heterogeneous17,18 metal-based catalysts have been developed.1 The best results were obtained using Ru(IV) catalysts in homogeneous systems (conversions and trans-anethole selectivities of up to 99%).16 Alternatively, heterogeneous systems show only moderate selectivities for the trans isomer (85–88%) with conversion of around 86–98%.17,18 However, the search for more efficient and selective systems, capable of attaining green and sustainable approaches to trans-anethole, is desirable19−21 and is an on-going challenge for the relevant scientific communities. Indeed, catalytic estragole to trans-anethole isomerization, using green reaction media, has already been reported using ethanol–glycerol and ethanol–H2O.16 Similarly, ionic liquids (ILs) have emerged as one of the most important and investigated solvents for several green chemical transformations.22−26 The combination of these liquids with charge-tagged ligands has proven to be a valuable tool in organometallic catalysis.27−30 The ionic modification of these ligands confers a particular solubility profile that makes catalyst/product recovery possible and often improves the activity of the catalytic species compared to that of the parent tag-free analogue.31 Moreover, new selective processes can be envisaged by changing the solubility and diffusion of substrates and products in the active phase of a reaction.32−34 However, little is known about the rate law and kinetics of this apparently simple isomerization reaction in which the desired product (trans isomer) is the thermodynamic product. An intimate knowledge of the reaction law and intrinsic kinetics is necessary to design more active and selective catalysts in both homogeneous and multiphase conditions. To address these issues, we have investigated, in detail, the kinetics of estragole isomerization under both homogenous and biphasic conditions using Ru, Rh, and Pd catalyst precursors dissolved in organic solvents, immobilized in ILs, and under “solventless” conditions.
Herein, we present the development of homogeneous systems for estragole to trans-anethole isomerization and the direct transposition for biphasic systems containing ILs and charge-tagged ligands. Moreover, the kinetics aspects of this reaction, in homogeneous and biphasic systems, have also been investigated. Finally, we show that this simple catalytic reaction has complex kinetics.
2. Results and Discussion
2.1. Estragole Isomerization in Classical Organic Solvents
In our study, we first compared the efficiency of some of the most popular and most used alkene isomerization complexes,1 for the transformation of estragole to trans-anethole. [RuHCl(CO)(PPh3)3] 1, [RuH2(CO)(PPh3)3] 2, [RhCl(PPh3)3] 3, and [PdCl2(NCPh)2] 4 were evaluated for this transformation (entries 1–15, Table 1; for detailed information see Table S1). [RuHCl(CO)(PPh3)3] complex 1 is the most active among these catalysts and demonstrates 100% substrate conversion to anethole in 1 h and a trans selectivity of 95%. Using a ratio of estragole/1 of 100, the reaction could be performed in air without any changes to the conversion or selectivity (entries 1 and 2, Table 1). However, visual changes could be observed when the reaction was not performed in an inert medium, indicating partial decomposition of the catalyst (Figure S1). As previously reported, despite the high stability of precursor 1 in the solid state, in solution it quickly decomposes when exposed to air.35 In addition, the reaction could be performed under solventless conditions with no significant changes in the selectivity (entries 3 and 4, Table 1 and Figure S2). Indeed, in just 5 min, the reaction was completed, with a selectivity of 92% for the trans isomer at 80 °C. This result is best reported under solventless conditions using [RuCl3(AsPh3)2]·MeOH at very high temperatures (>200 °C) for 5 h.36
Table 1. Ru-Catalyst Precursors Used in Estragole Isomerizationa.

| entry | complex | estragole/[M] | medium | ligand | T (°C) | time (min) | conv. (%) | sel. (%)b | TONc | TOF (min–1)d |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 100 | toluene | 80 | 60 | 100 | 95 | 100 | 2 | |
| 2e | 1 | 100 | toluene | 80 | 60 | 100 | 95 | 100 | 2 | |
| 3 | 1 | 100 | none | 80 | 5 | 100 | 92 | 100 | 20 | |
| 4 | 1 | 100 | none | 80 | 60 | 100 | 95 | 100 | 2 | |
| 5f | 1 | 2500 | toluene | 80 | 10 | 100 | 95 | 2500 | 485h | |
| 6f | 1 | 10 000 | toluene | 80 | 30 | 100 | 96 | 10 000 | 1580h | |
| 7f | 1 | 25 000 | toluene | 80 | 1440 | 99 | 88 | 24 750 | 450i | |
| 8g | 1 | 25 000 | toluene | 80 | 1440 | 100 | 92 | 25 000 | 500i | |
| 9f | 1 | 10 000 | toluene | 40 | 1440 | 100 | 97 | 10 000 | 123j | |
| 10 | 2 | 100 | toluene | 80 | 240 | 96 | 78 | 96 | 1j | |
| 11 | 2 | 100 | toluene | 80 | 1440 | 100 | 96 | 100 | 1j | |
| 12 | 3 | 100 | toluene | 80 | 1440 | 99 | 78 | 99 | 0.5k | |
| 13 | 3 | 100 | ethanol | 80 | 240 | 99 | 84 | 99 | 1l | |
| 14 | 3/SnCl2 | 100 | ethanol | 80 | 60 | 99 | 97 | 99 | 2 | |
| 15 | 4 | 100 | toluene | 80 | 1440 | 97 | 96 | 97 | 2j | |
| 16f | 5 | 100 | ethanol | 80 | 240 | 91 | 97 | 91 | 1j | |
| 17 | 5 | 100 | ethanol | 80 | 1440 | 100 | 97 | 100 | 1j | |
| 18f | 5 | 100 | ethanol | P(OEt)3 | 80 | 60 | 100 | 99 | 100 | 2 |
| 19 | 5 | 100 | ethanol | P(OEt)3 | 80 | 1440 | 100 | 98 | 100 | 2j |
| 20f | 6 | 100 | ethanol | 80 | 60 | 99 | 99 | 99 | 2 | |
| 21 | 6 | 100 | ethanol | 80 | 1440 | 100 | 97 | 100 | 2j | |
| 22f | 6 | 100 | ethanol | P(OEt)3 | 80 | 10 | 99 | 100 | 99 | 18h |
| 23 | 6 | 100 | ethanol | P(OEt)3 | 80 | 1440 | 100 | 96 | 100 | 18h |
| 24f | 6 | 2500 | ethanol | P(OEt)3 | 80 | 60 | 99 | 99 | 2475 | 100l |
| 25f | 6 | 10 000 | ethanol | P(OEt)3 | 80 | 1440 | 98 | 96 | 9800 | 125l |
| 26f | 6 | 100 | toluene | P(OEt)3 | 80 | 180 | 100 | 91 | 100 | 1 |
| 27f | 6 | 100 | ethanol | PPh3 | 80 | 10 | 100 | 100 | 100 | 10 |
Reaction conditions: reaction in an argon atmosphere.
Selectivity for trans-anethole.
Cumulative TON values (mol products/mol Ru).
Calculated at the time indicated in each case unless otherwise stated [(mol products/mol Ru)/time].
Reaction in air.
Estragole filtered over aluminum oxide.
Estragole filtered over magnesol.
5 min.
10 min.
60 min.
120 min.
30 min.
Under the optimized reaction conditions (Tables S2 and S3), 100% conversion (92% selectivity in trans-anethole) can be achieved for the estragole isomerization using as little as 4 × 10–3 mol % (40 ppm) of 1, with a total turnover number (TON) of 25 000 and turnover frequencies (TOF) of up to 500 min–1 (entry 8, Table 1 and Figure S3). Furthermore, the removal of the peroxides (entries 5–8, Table 1) present in the substrate is essential in avoiding catalyst deactivation.37,38
The influence of temperature (from 40 to 80 °C) was evaluated for estragole isomerization using 1 (Figure 1 and Table S4). Hydride catalyst 1 has been demonstrated to be an efficient catalyst for estragole isomerization with a high potential for this reaction, due its high activity at low loadings of the catalyst and at low temperatures (40 °C) (entry 9, Table 1). It is important to note that at full estragole conversion, the formed cis-anethole is transformed into the trans isomer with the increasing reaction time (Figure 1), that is, the cis isomer is reduced from 7% after 2 h to 3% after 24 h at 60 °C. Further attempts to improve the selectivity of estragole isomerization were made using dimeric Ru precursors already described in the literature as being efficient for estragole isomerization.15,16 Ru precursors [RuCl(μ-Cl)(η6-C6H5OCH2CH2OH)]25 and [RuCl(μ-Cl)(η3:η3-C10H16)]26 were explored under homogeneous conditions (entries 16–27, Tables 1 and S7). As expected, dimeric Ru(II) complex 5 showed a conversion of 91% in 4 h and a trans-anethole selectivity of 97%, whereas dimeric Ru(IV) precursor 6 was more active and selective for this type of transformation, showing a conversion of 99% and a selectivity of 99% after a 1 h reaction (entries 16 and 20, Table 1). Second, we reasoned that the introduction of phosphorous ligands, phosphites and phosphines, with different electronic and steric properties, should exert some influence in terms of the activity and trans/cis selectivity of the reaction.
Figure 1.

Selectivity vs conversion at ■ 40 °C, ▲ 60 °C, and ○ 80 °C. Reaction conditions: estragole 2.0 mmol, estragole/1 ratio of 10 000, toluene 0.5 mL, and estragole filtered over aluminum oxide.
As expected, bimetallic compound 5, in the presence of the ligand, P(OEt)3, showed a greater reaction speed and selectivity, compared to that of the system in the absence of phosphite, reaching a conversion of 100% in 1 h and a trans selectivity of 99% (entry 18, Table 1). When P(OEt)3 was added to the system containing precursor 6, no changes were observed in the conversion and selectivity, but the reaction was faster than in the system in the absence of phosphite (entries 20 and 22, Table 1). The same behavior was observed when PPh3 was used (entry 27, Table 1). Reactions were also performed using toluene as the solvent, and the activity and selectivity decreased dramatically when compared to that of the system with ethanol (compare entries 22 and 26, Table 1). It is certain that phosphorous ligands play a key role on the catalytic activity of these bimetallic precursors. However, we cannot discard the fact that the good activities and selectivities occur mainly as a result of the nature of the solvent used, probably due to the easier hydride formation, which constitutes the first step of the process.15
We also investigated how the process is affected by changing the catalyst loading (for detailed information see Supporting Information (SI), Table S8). We observed that decreasing the quantity of Ru from 1 to 0.04 mol % did not modify the ratio of trans/cis. Alternatively, lower metal loadings (0.01 mol %) resulted in lower selectivities for the trans isomer. Under the optimized reaction conditions, a total TON of 9800 and TOFs of up to 125 min–1 were obtained (entry 25, Table 1 and Figure S7). Nevertheless, small amounts of 6 were sufficient for performing the selective estragole to trans-anethole isomerization in shorter reaction times with selectivities of up to 99%.
2.2. Estragole Isomerization in ILs
To obtain a biphasic system for estragole isomerization, experiments were conducted in a two-phase homogeneous system containing ILs by the direct transposition of the homogeneous system for the biphasic one. Initially, we evaluated the solubility of estragole and anethole in three imidazolium-based ILs at 80 °C (Scheme 2; Table S5). The highest solubility of anethole and estragole was found in BMI·NTf2 and the lowest in nPr(OH)MI·NTf2. BMI·PF6 (Scheme 2) showed an intermediate solubility and was chosen for preliminary isomerization biphasic tests.
Scheme 2. Estragole Solubility (S80 °C) in ILs.

Initially, we performed tests in biphasic systems containing the IL BMI·PF6 (Table S6). After 20 min, the system reached 100% conversion with a selectivity for trans-anethole of 96%. The biphasic isomerization of estragole in the presence of BMI·PF6 was straightforward, and essentially the same conversion and selectivities were obtained compared to those by the homogeneous test. The product phase was collected by simple decantation at the end of the reaction, followed by subjection to ICP-AES analysis (Table 2). Although, a high Ru content was measured (160 ppm), the IL phase could retain part of the Ru, as a total of 216 ppm of Ru was present in the reaction mixture (0.0052 mmol Ru/2.43 g). As expected, the reaction rate of the biphasic system was slower than in the homogeneous one, probably due to the mass transfer limitations that will be discussed in the Section 2.3. As the anethole is recovered by phase separation, and the main contaminant is Ru, we decided to verify whether the addition of the ionophilic phosphine ligands (Figure 2) would improve the immobilization of the Ru complex in the IL phase (Table S6).
Table 2. Estragole Isomerization in Biphasic Systems Containing ILs and Charge-Tagged Ligandsa.
| entry | ligand | conversion (%) | selectivity (%)b | Ru content (mg/kg)c |
|---|---|---|---|---|
| 1 | 100 | 96 | 160 ± 4 | |
| first recharge | 39 | 82 | ||
| second recharge | 7 | 86 | ||
| 2 | L1 | 100 | 96 | 61.1 ± 2.1 |
| first recharge | 88 | 77 | ||
| second recharge | 17 | 82 |
Reaction conditions: estragole 13.0 mmol, estragole/1 = 2500, BMI·PF6 0.5 g, t = 20 min and 1 h, T = 80 °C, estragole filtered over aluminum oxide, and products separated by decantation.
Selectivity for trans-anethole.
Ru content in the organic phase.
Figure 2.

Ionophilic ligands used.
The addition of four equivalents of phosphine L1 relative to Ru completely inhibited the catalytic activity, which is expected as an excess of phosphine ligands has been shown to have detrimental effects on the isomerization activity.39 Indeed, an excess of P-containing ligands inhibits the formation of the 16-electron Ru–H catalytic active species (see below). When one or two equivalents of the ligand were used, good results could be obtained, whereas larger amounts led to long reaction times. Changing the nature of the ionophilic phosphine resulted in no observable changes in the catalytic results. ESI(+)-MS experiments indicated the substitution of the phosphine, present in the precursor 1, by the ionophilic phosphine, through the observation of species [RuHCl(CO)(PPh3)L1] (m/z = 751.0842) and [RuHCl(CO)(L1)2] (m/z = 1092.0302) in the reaction solution (Figure S6). This indicates that this ligand increases the immobilization of this catalyst in IL systems, reducing the lixiviation and contamination of the organic phase.
Attempts were made to recycle the ionic phase containing the catalyst. A new charge of the fresh substrate was added, and the reaction proceeded for 1 h; remarkably, changes in catalytic activity could be observed (entry 1, Table 2). The ionic phase completely lost its catalytic activity with a second charge of the substrate. Recycling experiments performed with the ionophilic ligand, L1, presented the same behavior, although the Ru content in the oil phase was lower, resulting in a small drop in the catalytic activity after the first cycle. The isomerization of estragole with BMI·NTf2, promoted by 6/P(OEt)3, gave a conversion of 83% in 3 h and a trans selectivity of 98% (entry 1, Table 3). To improve the catalytic activity, we decided to use an IL with similar characteristics to ethanol.
Table 3. Influence of IL in Estragole Isomerization Using 6/Phosphorous ligand systemsa.
| entry | ligand | IL (amount) | time | conv. (%) | sel. (%)b |
|---|---|---|---|---|---|
| 1 | P(OEt)3 | BMI·NTf2 (0.5 g) | 3 h | 83 | 98 |
| 2 | P(OEt)3 | nPr(OH)MI·NTf2 (0.5 g) | 30 min | 99 | 99 |
| 3 | P(OEt)3 | nPr(OH)MI·NTf2 (0.1 g) | 1 h | 99 | 99 |
| 4 | PPh3 | nPr(OH)MI·NTf2 (0.5 g) | 30 min | 99 | 99 |
| 5 | PPh3 | nPr(OH)MI·NTf2 (0.1 g) | 1 h | 99 | 99 |
Reaction conditions: estragole 2.0 mmol, estragole/6 ratio of 100, P(OEt)3 0.04 mmol, IL 0.5 g, T = 80 °C, and estragole filtered over aluminum oxide.
Selectivity for trans-anethole.
An experiment using the IL nPr(OH)MI·NTf2 was performed, and the reaction reached 99% conversion in 30 min with a selectivity of 99% for trans-anethole (entry 2, Table 2). In this case, the estragole was only partially miscible with nPr(OH)MI·NTf2, and a biphasic system was obtained. By comparing the reaction performed with BMI·NTf2 and that performed with nPr(OH)MI·NTf2, it became clear that even in a biphasic system, the catalyst was much more active and selective if an OH-functionalized IL is used. Interestingly, the amount of IL could be reduced 5-fold and used as a “liquid support” (entries 2 and 4, Table 2). Thus, the reaction using P(OEt)3 or PPh3 as ligands gave an identical conversion and the same selectivity as that obtained using larger amounts of IL (compare entries 2 and 3 and entries 4 and 5, Table 2). However, the time for total conversion increases when the amount of IL decreases, indicating that the isomerization is probably under mass transfer control.
In addition, a new family of phosphite ligands (L4 and L5; synthesis is described in Experimental Section) that bear an imidazolium fragment and consist of a versatile ligand class for IL biphasic catalysis was synthesized and applied to the estragole isomerization. The ionophilic phosphine ligands, L1–L3, and the ionophilic phosphite ligands, L4 and L5, were added to the reaction system to prevent the metal contamination in the final oil phase (as previously observed for the system using the [RuHCl(CO)(PPh3)3] precursor) with no pronounced changes in the catalytic results. No meaningful changes were observed for the conversion and selectivity of these systems when compared to those of the parent tag-free analogue (e.g., compare entries 1 and 2, Table 4).
Table 4. Estragole Isomerization in Biphasic Systems Containing PPh3, P(OEt)3, and Ionophilic Ligandsa.
| entry | ligand | conversion (%) | selectivity (%)b |
|---|---|---|---|
| 1 | PPh3 | 99 | 99 |
| 2 | L1 | 99 | 99 |
| 3 | P(OEt)3 | 99 | 98 |
| 4 | L2 | 99 | 96 |
| 5 | L3 | 99 | 95 |
| 6 | L4 | 99 | 98 |
| 7 | L5 | 99 | 98 |
Reaction conditions: estragole 2.0 mmol, estragole/6 ratio of 100, ligand 0.04 mmol, nPr(OH)MI·NTf2 0.1 g, t = 1 h, T = 80 °C, and estragole filtered over aluminum oxide.
Selectivity in trans-anethole.
2.3. Kinetic Modeling Study of Estragole Isomerization
Kinetic studies were carried out to determine a rate law for the systems considered more environmentally friendly, that is, under solventless conditions. Experiments were performed for the homogeneous and biphasic systems containing Ru precursor 1 and estragole. As a first approach, we assumed that the system is composed of a monomolecular reversible first-order reaction according to previous works, such as for the isomerization of butene and pentene (Scheme 3).40
Scheme 3. Monomolecular Model of Estragole Isomerization.

The equilibrium concentrations and thermodynamic properties (ΔG°, ΔH°, ΔS°) of the allylbenzene conversion on its internal isomers at different temperatures have already been reported. For example, at 75 °C the ratio of allylbenzene and cis and trans isomers are 0.157, 3.81, and 96.03%, respectively.41
The concentrations of the substrate ([E]) and products cis-anethole ([C]) and trans-anethole ([T]), with respect to time, are given by the solution of the first-order ordinary differential equations (ODEs), as defined by eqs 1–3
| 1 |
| 2 |
| 3 |
The relative kinetic constants for this model were calculated by the fitting of the ODEs’ numerically integrated solution to the experimental data using the kinetic simulator software Dynafit.42 These constants were evaluated for the model proposed in Scheme 3 at three different temperatures. The agreement between the prediction model and the experimental data could be measured qualitatively by the simultaneous fit of eqs 1–3 to experimental results, and the best agreement was obtained for the reaction performed at 80 °C (Figure 3).
Figure 3.

Simultaneous fit of eqs 1–3 to experimental data for estragole isomerization at 80 °C. Reaction conditions: estragole 2.0 mmol, estragole/1 ratio of 10 000, solventless, and estragole filtered over aluminum oxide. Experimental data: ■, estragole; ▲, trans-anethole; and ○, cis-anethole. Model prediction: blue line, estragole; red line, trans-anethole; and green line, cis-anethole.
This agreement could also be supported quantitatively by the residual sum of squares (S) presented in Table 5. The best S value was obtained for the reaction performed at 80 °C, with a residual sum of squares43 of 457 (entry 3, Table 5). This result supports the assumption that the entire system can be described in terms of the first-order reversible reactions. However, when lower temperatures were used, the difference between the prediction and the experimental data increases (entry 1, Table 5). These results suggest that besides the intrinsic limitation of the monomolecular model, in which virtual kinetic constants are valid only for a catalytic concentration, the model is also not suitable at low temperatures, mainly because the known important steps of the reaction, such as the formation of the catalytic active species, are neglected. Indeed, the formation of the 16-electron catalytic active species [Ru(H)Cl(CO)(PPh3)2] from 1 should be considered (see below).
Table 5. Rate Constants Fitted to the Monomolecular Model of Estragole Isomerization at Different Temperatures and the Residual Sum of Squares (S)a.
Reaction conditions: estragole 2.0 mmol and estragole/1 ratio of 10 000.
Estragole 2.0 mmol, estragole/1 ratio of 10 000, L1 2 equiv, BMI·PF6 0.5 g, and estragole filtered over aluminum oxide.
See Table S9 for details.
The kinetic constants determined for the reaction performed at 80 °C are shown in Table 6. The relative constants, k1 and k2, rule the selectivity of the isomerization at the beginning of the reaction. The undesired cis isomer is formed at a rate that depends mainly on k2 and the substrate concentration, as k2 ≫ k3. However, the removal rate of the cis isomer is related to the relative kinetic constant, k–3, as k–3 ≫ k–2. Therefore, as soon as the substrate is consumed, the liquid rate of cis formation decreases due to cis → trans conversion, causing the appearance of a maximum of around 25% of the cis-anethole relative concentration (see green curve, Figure 3). In the equilibrium, the selectivity is controlled mostly by the ratio K = k–3/k3 ≈ 19 (95% trans-anethole), as k–1 and k–2 are approximately zero. Considering that k2 < k1 and K > 1, the trans isomer is the kinetic and thermodynamic product in this reaction.
Table 6. Rate Constants Fitted to the Monomolecular Model of Estragole Isomerization at 80 °Ca.
| entry | constant (min–1) | fitting (×104) | s.d. (×104)b |
|---|---|---|---|
| 1 | k1 | 2856 | 197 |
| 2 | k–1 | ≈0 | |
| 3 | k2 | 2130 | 219 |
| 4 | k–2 | ≈0 | |
| 5 | k3 | 135 | 39 |
| 6 | k–3 | 2548 | 379 |
Reaction conditions: estragole 2.0 mmol and estragole/1 ratio of 10 000.
Standard deviation.
An interesting result about this model is its good agreement for the two-phase system containing IL (entry 4, Table 5 and Figure 4). Obviously, biphasic systems are strongly influenced by mass transfer between the organic and catalytic phases,44 and to neglect this can be problematic. However, for a preliminary fitting of the concentration of the reaction components over time, this approach can be useful. At short times, it is likely that the reaction is slow due to the low concentration of the substrate in the catalytic phase of the IL. This may be a consequence of the slow diffusion and low solubility of the substrate in the IL.
Figure 4.

Simultaneous fit of eqs 1–3 to experimental data for biphasic estragole isomerization at 80 °C. Reaction conditions: estragole 2.0 mmol, estragole/1 ratio of 10 000, L1 2 equiv and BMI·PF6 0.5 g. Experimental data: ■, estragole; ▲, trans-anethole; and ○, cis-anethole. Model prediction: blue line, estragole; red line, trans-anethole; and green line, cis-anethole.
Therefore, this model is adequate for describing the kinetics of the reaction performed in ILs, and the complex behavior of the biphasic systems could be mathematically condensed in the relative kinetic constants. However, as demonstrated, this monomolecular model is not adequate for describing the experimental kinetic data obtained at 40 °C (Table S9).
In the face of the limitations found using the monomolecular model, a simplified model based on the metal hydride mechanism (Scheme 4) was evaluated.
Scheme 4. Metal Hydride Mechanism.

Steps IV, VII, and VIII were omitted, and the sequential monomolecular steps (coordination, insertion/migration, β-elimination, and discoordination) were combined in single steps, characterized by kinetic constants, as follows: k5/–5 (steps II and III); k6/–6 (steps VI and X); and k7/–7 (steps V and IX). Thus, a simplified mechanistic model could be proposed for catalytic estragole isomerization (Scheme 5).
Scheme 5. Mechanistic Model Proposed for Catalytic Estragole Isomerization (Herein, X0 are 16-Electron Ru–H Catalytic Active Species ([Ru(H)Cl(CO)(PPh3)2]).

Assuming a first-order dependence of the reaction rate with the catalyst and substrate concentrations, the system of ODEs that describe the kinetic model can be defined by eqs 4–10
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
A good fit was obtained for estragole isomerization at all temperatures used (Figures 5 and S9), as characterized by low values of S (Table S10). This result reinforces the hypothesis that the step of formation of active species should be considered, especially at low temperatures (e.g., compare k4 and k5 for the reaction performed at 40 °C, entries 1 and 3, Table 7). In addition, values k5, k6, and k7 were in agreement with the literature, that is, the relative kinetic constant for estragole coordination (k5) is greater than the cis- and trans-anethole coordination (k6 and k7). Also, as expected, k6 is greater than k7.
Figure 5.

Simultaneous fit of eqs 4–10 to experimental data for estragole isomerization at 40 °C. Reaction conditions: estragole 2.0 mmol and estragole/1 ratio of 10 000, solventless, and estragole filtered over aluminum oxide. Experimental data: ■, estragole; ▲, trans-anethole; and ○, cis-anethole. Model prediction: blue line, estragole; red line, trans-anethole; and green line, cis-anethole.
Table 7. Rate Constants Fitted to the Simplified Mechanistic Model of Estragole Isomerization at 40 °Ca.
| entry | constant | fitting | s.d.b |
|---|---|---|---|
| 1 | k4 (min–1) | 0.139 | 0.005 |
| 2 | k–4 (L mol–1 min–1) | ≈0 | |
| 3 | k5 (L mol–1 min–1) | 39.87 | 1.63 |
| 4 | k–5 (min–1) | 23.01 | 4.79 |
| 5 | k6 (L mol–1 min–1) | 18.01 | 0.53 |
| 6 | k–6 (min–1) | 129.3 | 10.4 |
| 7 | k7 (L mol–1 min–1) | 1.537 | 0.124 |
| 8 | k–7 (min–1) | 231.0 | 17.4 |
Reaction conditions: estragole 2.0 mmol and estragole/1 ratio of 10 000, solventless, and estragole filtered over aluminum oxide.
Standard deviation.
The proposed simplified mechanistic model was also evaluated for considering the steady-state approximation for the concentration of reaction intermediates and the mass balance of metal species using the mathematical Software Maple 15.00 (Figure S10). A good agreement between the experimental data and the reaction rate based on the Bodenstein approximation was also achieved. Thus, the reaction rate could be described as presented in eq 11, where [XT] is the metal complex concentration used.
| 11 |
3. Conclusions
The isomerization of estragole to trans-anethole can be easily accomplished by Ru-based catalysts under homogeneous, IL biphasic, and “solventless” conditions with selectivities of >99% at complete estragole conversions. TOFs of 125–500 min–1 can be achieved using simple Ru(II) or Ru(IV) catalyst precursors, such as [RuHCl(CO)(PPh3)3] or [RuCl(μ-Cl)(η3:η3-C10H16)]2 in toluene, ethanol, ILs, or under solventless conditions. Although this reaction can be transposed to IL biphasic conditions using ionic-tagged P-ligands, the system cannot be reused in view of the high sensitivity of the catalytic active species. The removal of the peroxides present in the substrate is essential to not deactivate the catalytic active species. The selectivity of this reaction is more sensitive to the solvent/support used than the ligands associated with the metal catalyst. The monomolecular kinetic model (monomolecular reversible first-order reaction) is adequate for fitting the homogeneous reaction at 80 °C and under biphasic conditions. However, the kinetics of the reaction is better described if a mechanistic model containing elemental steps (such as migratory-insertion and β-elimination) is used, indicating that the step of formation of active species should be considered, especially at 40 °C, along with the generation of unsaturated 16-electron Ru–H catalytic active species.
Considering Bondenstein approximation, the reaction
follows the
rate law 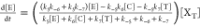 . Therefore, the use of well-defined 16-electron
Ru–H catalyst precursors is key for obtaining a more easily
recyclable catalytic system for the isomerization of alkylbenzenes.
. Therefore, the use of well-defined 16-electron
Ru–H catalyst precursors is key for obtaining a more easily
recyclable catalytic system for the isomerization of alkylbenzenes.
4. Experimental Section
4.1. General
All manipulations of complexes were conducted under Ar using dry boxes or standard Schlenk techniques. Chemicals were treated as follows: toluene distilled from Na/benzophenone, [RuH2(CO)(PPh3)3], [RhCl(PPh3)3], and [PdCl2(NCPh)2] from Aldrich, acetone-d6 and CDCl3 (Cambridge Isotope) and other solvents were used as received. [RuHCl(CO)(PPh3)3],45 ILs (BMI·PF6, BMI·BF4, and BMI·NTf2),46L1,47L2,47 and L3(48) were synthesized according to literature procedures. Gas chromatography (GC) analyses were performed using a Shimadzu GC-2010 instrument equipped with a capillary column RTx-50 (30 m; 0.25 mm i.d.) using 4-methoxy-1-propylbenzene as an external standard. GC–MS data were recorded with a Shimadzu QP2010. ESI-MS experiments were performed using a Micromass Q-Tof micro. NMR spectra were recorded on a Varian instrument at 400 MHz (1H), 100 MHz (13C), or 162 MHz (31P) using SiMe4 or 85% H3PO4 as standards.
4.2. Typical Isomerization Procedure in the Homogeneous System
In a glovebox, estragole (2.0 mmol, 298 mg), the isomerization catalyst (0.020 mmol), and the solvent (0.5 g) were charged in a Schlenk flask. The system was heated to 80 °C and kept under stirring for predetermined times. Samples were taken periodically for GC analysis. The reaction products were identified by nuclear magnetic resonance of hydrogen (1H NMR) (Figure S3) and quantified by GC (Figure S4).
4.3. Typical Isomerization Procedure in the Biphasic System
In a glovebox, estragole (2.0 mmol, 298 mg), the catalyst precursor (0.020 mmol), the solvent (0.5 g), and IL (0.5 g) were added in a Schlenk flask. The system was heated to 80 °C and kept under stirring for predetermined times. The reaction products were quantified by GC by removing the top phase (organic phase) diluted in diethyl ether.
4.4. Hydrogenation of Estragole for 1-Methoxy-4-propylbenzene
In a one-type reactor (Fisher Porter stainless) were added 15 g of estragole and 0.5 g of Pd/C 10% (0.464 mol % Pd). Total conversion was observed for the p-propilanisol (1-methoxy-4-propylbenzene) after 3 h at a temperature of 85 °C and 26 bar hydrogen pressure. The product was characterized by GC–MS and stored to be used as an external standard (ES) in the chromatographic analysis.
4.5. Determination of Estragole and Anethole Solubilities in ILs
To a glass reactor with an external heating jacket (Figure S11), 0.700 g of IL and 1.500 g of estragole or anethole (trans/cis: 96/4) were added. The system temperature was raised by circulating warm water in the external jacket and was monitored with a thermometer inserted in the biphasic mixture. After the mixture was allowed to stand for 10 min at a temperature of analysis under vigorous stirring (600 rpm) using a magnetic bar, the agitation was reduced and maintained at approximately 60 rpm for 5 min to allow phase separation. With the aid of a glass Pasteur pipette, previously heated with a heat gun, samples were removed from 200 mg of the ionic phase. About 100 mg of 1-methoxy-4-propylbenzene was added as an external standard, and three extractions with diethyl ether were carried out. The extracted fractions were pooled and analyzed by GC. The solubility of estragole or anethole in IL was calculated by eq 12. In cases in which the preheating of the Pasteur pipette was not sufficient to prevent phase separation, due to cooling of the mixture, the pipette containing the sample was heated with the heat gun until the mixture became homogenous again avoiding preferential loss of a phase on the walls of the pipette. Solubility measurements were made in duplicate and are presented in a mean ± standard deviation format.
| 12 |
4.6. Determination of Metal Content in Samples
The decomposition of the samples was assisted by microwaves (MAD) using Multiwave 3000 oven (Anton Paar, Austria) and quartz bottles. The sample was weighed with the aid of a pipette into a small glass container, which was placed inside the quartz vial. The samples were decomposed in triplicate, using 5 mL of HNO3 and 1 mL of HCl. The determination of Ru was performed using ICP-OES equipment (Perkin Elmer Optima 4300 DV) with a primary argon flow of 15 L/min, an argon auxiliary of 0.2 L/min, and an argon nebulizer of 0.7 L/min. The wavelengths used were 240.272, 349.894, and 279.535 nm.
4.7. Synthesis of Ionophilic Ligand L4
In a typical synthesis, a round-bottom Schlenk flask was charged with dry 1-(3-hydroxypropyl)-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide (2.80 g, 4.13 mmol), triethylamine (0.420 g, 4.13 mmol), and dry CH2Cl2 (20 mL) and cooled to 0 °C. Then, freshly distilled PCl3 (0.190 g; 1.38 mmol) in dry CH2Cl2 (50 mL) was added dropwise; the ice bath was removed, and the reaction was stirred for 30 min at room temperature. The solvent was removed under vacuum, and the product was extracted with dry acetone (3 × 5 mL) to remove the insoluble HNEt3Cl. Yield: 1.71 g, 93%.
1H NMR (400 MHz, acetone-d6): δ ppm 7.60 (d, J = 2.8 Hz,
H3 and H4), 4.40 (t, J = 5.2 Hz, H6), 3.92 (s, H1),
3.98 (q, J = 5.2 Hz, H8), 2.78 (s, H5), 2.22 (q, J = 5.2 Hz, H7). 13C NMR (100 MHz, acetone-d6) δ ppm 137.8 (C2); 124.6 (C3 or C4);
123.8 (C3 or C4); 120.8 (q, JCF = 320.0
Hz, NTf2); 62.0 (C6); 50.4 (C8); 46.0 (C1); 35.8 (C5);
9.4 (C7). 31P NMR (162 MHz, acetone-d6): δ ppm 139.5 (POR3, 92%), 8.0 (O=POR3, 8%). ESI(+)–MS: m/z calcd. [C24H42N6O3P]3+ 164.4352, expt. 164.4275.
4.8. Synthesis of Ionophilic Ligand L5
In a typical synthesis, a round-bottom Schlenk flask was charged with dry 1-(3-hydroxyhexyl)-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide (1.97 g, 4.13 mmol), triethylamine (0.420 g, 4.13 mmol), and dry CH2Cl2 (20 mL) and cooled to 0 °C. Then, freshly distilled PCl3 (0.190 g; 1.38 mmol) in dry CH2Cl2 (50 mL) was added dropwise; the ice bath was removed, and the reaction was stirred for 30 min at room temperature. The solvent was removed under vacuum, and the product was extracted with dry acetone (3 × 5 mL) to remove the insoluble HNEt3Cl. Yield: 1.81 g, 90%.
1H NMR (400 MHz, acetone-d6): δ ppm 7.60 (d, J = 2.4 Hz,
H3 and H4), 4.27 (t, J = 5.2 Hz, H6), 3.94 (s, H1),
3.76 (q, J = 5.2 Hz, H11), 2.76 (s, H5), 1.89 (q, J = 5.2 Hz, H7), 1.59 (q, J = 5.2 Hz, H10),
1.43 (m, J = 5.2 Hz, H8 e H9). 13C NMR
(100 MHz, acetone-d6): δ ppm 137.3
(C2); 124.7 (C3 or C4); 123.3 (C3 or C4); 120.8 (q, JCF = 320.0 Hz, NTf2); 66.0 (C6); 50.3 (C11);
48.9 (C1); 36.5 (C5); 30.5 (C7); 26.1 (C10); 25.4 (C8) and 9.2 (C9). 31P NMR (162 MHz, acetone-d6):
δ ppm 139.2 (POR3, 88%), 7.8
(O=POR3, 12%). ESI(+)–MS: m/z calcd. [C33H60N6O3P]3+ 206.4821, expt. 206.4796.
Acknowledgments
Thanks are due to the following agencies for financial support: CNPq, CAPES, INCT-Catal., and FAPERGS.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00078.
Detailed description of the material (PDF)
Author Present Address
† School of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, U.K. (J.D.).
Author Contributions
The manuscript was written with contributions from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Hassam M.; Taher A.; Arnott G. E.; Green I. R.; van Otterlo W. A. Isomerization of allylbenzenes. Chem. Rev. 2015, 115, 5462–569. 10.1021/acs.chemrev.5b00052. [DOI] [PubMed] [Google Scholar]
- Tuan D. Q.; Ilangantileket S. G. Liquid CO2 extraction of essential oil from Star anise fruits (Illicium verum H.). J. Food Eng. 1997, 31, 47–57. 10.1016/S0260-8774(96)00030-1. [DOI] [Google Scholar]
- Soares P. M. G.; Lima R. F.; de Freitas Pires A.; Souza E. P.; Assreuy A. M. S.; Criddle D. N. Effects of anethole and structural analogues on the contractility of rat isolated aorta: Involvement of voltage-dependent Ca2+-channels. Life Sci. 2007, 81, 1085–1093. 10.1016/j.lfs.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Reineccius G.Flavor Chemistry and Technology, 2nd ed.; CRC Press: Boca Raton: 2006; pp 201–220. [Google Scholar]
- Kouznetsov V. V.; Merchan Arenas D. R. First green protocols for the large-scale preparation of γ-diisoeugenol and related dihydro(1H)indenes via formal [3 + 2] cycloaddition reactions. Tetrahedron Lett. 2009, 50, 1546–1549. 10.1016/j.tetlet.2009.01.038. [DOI] [Google Scholar]
- Glenert U. Effects of chronic anethole trithione and amitriptyline treatment on rat parotid gland signalling. Eur. J. Pharmacol. 1992, 226, 43–52. 10.1016/0922-4106(92)90081-6. [DOI] [PubMed] [Google Scholar]
- Khan I. A.; Abourashed E. A.. Leung’s Encyclopedia of Common Natural Ingredients Used in Food, Drugs and Cosmetics, 3rd ed.; John Wiley & Sons: Hoboken, 2010; pp 1–810. [Google Scholar]
- Taylor J. M.; Jenner P. M.; Jones W. I. A Comparison of the Toxicity of some Allyl, Propenyl, and Propyl Compounds in the Rat. Toxicol. Appl. Pharmacol. 1964, 6, 378–387. 10.1016/S0041-008X(64)80002-8. [DOI] [PubMed] [Google Scholar]
- Boissier J. R.; Simon P.; Bourhis B. L. Experimental psychotropic effect of isomeric cis- and trans-anetholes. Therapie 1967, 22, 309–323. [PubMed] [Google Scholar]
- Hansel R.; Holzl J.. Lehrbuch der Pharmazeutishen Biologie; Springer-Verlag: Berlin, 1996; pp 282–352. [Google Scholar]
- Pines H.; Stalick W. M.. Base-Catalyzed Reactions of Hydrocarbons and Related Compounds; Academic Press Inc.: New York, 1977. [Google Scholar]
- Wagner A. P. Anethole Production, Properties and Uses. Manuf. Chem. 1952, 23, 56–59. [Google Scholar]
- Erdogan G.; Grotjahn D. B. Mild and Selective Deuteration and Isomerization of Alkenes by a Bifunctional Catalyst and Deuterium Oxide. J. Am. Chem. Soc. 2009, 131, 10354–10355. 10.1021/ja903519a. [DOI] [PubMed] [Google Scholar]
- Scarso A.; Colladon M.; Sgarbossa P.; Santo C.; Michelin R. A.; Strukul G. Highly Active and Selective Platinum(II)-Catalyzed Isomerization of Allylbenzenes: Efficient Access to (E)-Anethole and Other Fragrances via Unusual Agostic Intermediates. Organometallics 2010, 29, 1487–1497. 10.1021/om100033a. [DOI] [Google Scholar]
- Lastra-Barreira B.; Crochet P. Ruthenium-catalyzed estragole isomerization: high trans-selective formation of anethole. Green Chem. 2010, 12, 1311–1314. 10.1039/c003901b. [DOI] [Google Scholar]
- Lastra-Barreira B.; Francos J.; Crochet P.; Cadierno V. Ruthenium(iv) catalysts for the selective estragole to trans-anethole isomerization in environmentally friendly media. Green Chem. 2011, 13, 307–313. 10.1039/C0GC00417K. [DOI] [Google Scholar]
- Jinesh C. M.; Antonyraj C. A.; Kannan S. Isomerization of eugenol and alkenyl aromatics of perfumery interest over Ni-containing layered double hydroxides as solid base catalysts. Catal. Today 2009, 141, 176–181. 10.1016/j.cattod.2008.03.023. [DOI] [Google Scholar]
- Sharma S. K.; Parikh P. A.; Jasra R. V. Ruthenium containing hydrotalcite as a solid base catalyst for >CC< double bond isomerization in perfumery chemicals. J. Mol. Catal. A: Chem. 2010, 317, 27–33. 10.1016/j.molcata.2009.10.017. [DOI] [Google Scholar]
- Poliakoff M.; Fitzpatrick J. M.; Farren T. R.; Anastas P. T. Green chemistry: science and politics of change. Science 2002, 297, 807–10. 10.1126/science.297.5582.807. [DOI] [PubMed] [Google Scholar]
- Anastas P. T.Handbook of Green Chemsitry; John Wiley and Sons, 2001–2015; Vol. 1–9. [Google Scholar]
- Lancaster M.Green Chemistry: An Introductory Text; RSC Editions: Cambridge, 2002; pp 84–129. [Google Scholar]
- Dupont J.; de Souza R. F.; Suarez P. A. Z. Ionic Liquid (Molten Salt) Phase Organometallic Catalysis. Chem. Rev. 2002, 102, 3667–3692. 10.1021/cr010338r. [DOI] [PubMed] [Google Scholar]
- Sheldon R. Catalytic reactions in ionic liquids. Chem. Commun. 2001, 23, 2399–2407. 10.1039/b107270f. [DOI] [PubMed] [Google Scholar]
- Scholten J. D.; Neto B. A. D.; Suarez P. A. Z.; Dupont J.. Ionic Liquids as Versatile Media for Chemical Reactions. In Environmentally Friendly Syntheses Using Ionic Liquids, 1st ed.; CRC Press: Boca Raton, 2015; pp 109–138. [Google Scholar]
- Dupont J.Ionic Liquids: Structure, Properties and Major Applications in Extraction/Reaction Technology. In Green Separation Processes. Fundamentals and Applications, 1st ed.; Wiley-VCH: Weinheim, 2005; pp 229–249. [Google Scholar]
- Neto B. A. D.; Spencer J. The impressive chemistry, applications and features of ionic liquids: properties, catalysis & catalysts and trends. J. Braz. Chem. Soc. 2012, 23, 987–1007. 10.1590/S0103-50532012000600002. [DOI] [Google Scholar]
- Limberger J.; Leal B. C.; Monteiro A. L.; Dupont J. Charge-tagged ligands: useful tools for immobilising complexes and detecting reaction species during catalysis. Chem. Sci. 2015, 6, 77–94. 10.1039/C4SC02151G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronger R. P.; Silva S. M.; Kamer P. C.; van Leeuwen P. W. A novel dicationic phenoxaphosphino-modified xantphos-type ligand--a unique ligand specifically designed for a high activity, selectivity and recyclability. Chem. Commun. 2002, 24, 3044–5. 10.1039/B209486J. [DOI] [PubMed] [Google Scholar]
- Riisager A.; Fehrmann R.; Flicker S.; van Hal R.; Haumann M.; Wasserscheid P. Very stable and highly regioselective supported ionic-liquid-phase (SILP) catalysis: continuous-flow fixed-bed hydroformylation of propene. Angew. Chem., Int. Ed. 2005, 44, 815–9. 10.1002/anie.200461534. [DOI] [PubMed] [Google Scholar]
- Favre F.; Olivier-Bourbigou H.; Commereuc D.; Saussine L. Hydroformylation of 1-hexene with rhodium in non-aqueous ionic liquids: how to design the solvent and the ligand to the reaction. Chem. Commun. 2001, 1360–1361. 10.1039/b104155j. [DOI] [Google Scholar]
- Oliveira F. F. D.; dos Santos M. R.; Lalli P. M.; Schmidt E. M.; Bakuzis P.; Lapis A. A. M.; Monteiro A. L.; Eberlin M. N.; Neto B. A. D. Charge-Tagged Acetate Ligands As Mass Spectrometry Probes for Metal Complexes Investigations: Applications in Suzuki and Heck Phosphine-Free Reactions. J. Org. Chem. 2011, 76, 10140–10147. 10.1021/jo201990n. [DOI] [PubMed] [Google Scholar]
- Consorti C. S.; Aydos G. L. P.; Dupont J. Tandem isomerisation-metathesis catalytic processes of linear olefins in ionic liquid biphasic system. Chem. Commun. 2010, 46, 9058–9060. 10.1039/c0cc03891a. [DOI] [PubMed] [Google Scholar]
- Luza L.; Rambor C. P.; Gual A.; Bernardi F.; Domingos J. B.; Grehl T.; Brüner P.; Dupont J. Catalytically Active Membranelike Devices: Ionic Liquid Hybrid Organosilicas Decorated with Palladium Nanoparticles. ACS Catal. 2016, 6, 6478–6486. 10.1021/acscatal.6b01813. [DOI] [Google Scholar]
- Weilhard A.; Abarca G.; Viscardi J.; Prechtl Martin H. G.; Scholten J. D.; Bernardi F.; Baptista D. L.; Dupont J. Challenging Thermodynamics: Hydrogenation of Benzene to 1,3-Cyclohexadiene by Ru@Pt Nanoparticles. ChemCatChem 2017, 9, 204–211. 10.1002/cctc.201601196. [DOI] [Google Scholar]
- Geoffroy G. L.; Bradley M. G. Photochemistry of transition metal hydride complexes. 2. Chlorohydro(carbonyl)tris(triphenylphosphine)ruthenium, dihydro(carbonyl)tris(triphenylphosphine)ruthenium, and chlorohydro(dicarbonyl)bis(triphenylphosphine)ruthenium. Inorg. Chem. 1977, 16, 744–748. 10.1021/ic50170a004. [DOI] [Google Scholar]
- Sharma S. K.; Srivastava V. K.; Pandya P. H.; Jasra R. V. Solvent-free isomerization of methyl chavicol to trans-anethole using transition metal complexes as catalysts. Catal. Commun. 2005, 6, 205–209. 10.1016/j.catcom.2004.12.011. [DOI] [Google Scholar]
- Kwini M. N.; Botha J. M. Influence of feed components on the activity and stability of cobalt molybdenum alumina metathesis catalyst. Appl. Catal., A 2005, 280, 199–208. 10.1016/j.apcata.2004.10.043. [DOI] [Google Scholar]
- Prudent Practices in the Laboratory: Handling and Disposal of Chemicals; National Academy Press: Washington, DC, 1995. [Google Scholar]
- Permin A. B.; Petrosyan V. S. The study of alkene isomerization catalyzed by the system: Rhodium dimeric complex,Äîtertiary phosphine,Äîtin dichloride. Russ. Chem. Bull. 1997, 46, 1104–1106. 10.1007/BF02496206. [DOI] [Google Scholar]
- Ewing D. F.; Hudson B.; Webster D. E.; Wells P. B. Homogeneous catalysis of olefin isomerisation. Part II. (a) Isomerisation of pent-1-ene catalysed by hydridochlorotris(triphenylphosphine)-ruthenium(II) in benzene; and (b) a novel procedure for evaluating velocity constants for a network of three first-order reversible reactions. J. Chem. Soc., Dalton Trans. 1972, 12, 1287–1293. 10.1039/dt9720001287. [DOI] [Google Scholar]
- Taskinen E.; Lindholm N. Relative thermodynamic stabilities of the isomeric propenylbenzenes. J. Phys. Org. Chem. 1994, 7, 256–258. 10.1002/poc.610070507. [DOI] [Google Scholar]
- Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 1996, 237, 260–73. 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- Cain J. W.Mathematics of Fitting Scientific Data. In Molecular Life Sciences: An Encyclopedic Reference; Bell E., Ed.; Springer: New York, NY, 2021; pp 1–7. [Google Scholar]
- Wasserscheid P. Continuous reactions using ionic liquids as catalytic phase. J. Ind. Eng. Chem. 2007, 13, 325–338. [Google Scholar]
- Ahmad N.; Levison J. J.; Robinson S. D.; Uttley M. F.; Wonchoba E. R.; Parshall G. W. Complexes of Ruthenium, Osmium, Rhodium, and Iridium Containing Hydride Carbonyl, or Nitrosyl Ligands. Inorg. Synth. 2007, 15, 45–64. 10.1002/9780470132463.ch13. [DOI] [Google Scholar]
- Cassol C. C.; Ebeling G.; Ferrera B.; Dupont J. A Simple and Practical Method for the Preparation and Purity Determination of Halide-Free Imidazolium Ionic Liquids. Adv. Synth. Catal. 2006, 348, 243–248. 10.1002/adsc.200505295. [DOI] [Google Scholar]
- Consorti C. S.; Aydos G. L. P.; Ebeling G.; Dupont J. Ionophilic Phosphines: Versatile Ligands for Ionic Liquid Biphasic Catalysis. Org. Lett. 2008, 10, 237–240. 10.1021/ol702664a. [DOI] [PubMed] [Google Scholar]
- Leal B. C.; Consorti C. S.; Machado G.; Dupont J. Palladium metal nanoparticles stabilized by ionophilic ligands in ionic liquids: synthesis and application in hydrogenation reactions. Catal. Sci. Technol. 2015, 5, 903–909. 10.1039/C4CY01116C. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.