

Abstract
Mcl-1 is an anti-apoptotic member of the Bcl-2 family of proteins that when overexpressed is associated with high tumor grade, poor survival, and resistance to chemotherapy. Mcl-1 is amplified in many human cancers, and knockdown of Mcl-1 using RNAi can lead to apoptosis. Thus, Mcl-1 is a promising cancer target. Here, we describe the discovery of picomolar Mcl-1 inhibitors that cause caspase activation, mitochondrial depolarization, and selective growth inhibition. These compounds represent valuable tools to study the role of Mcl-1 in cancer and serve as useful starting points for the discovery of clinically useful Mcl-1 inhibitors.
Keywords: Structure Based Drug Design, apoptosis, cancer, Mcl-1, drug discovery
Introduction
The Bcl-2 family of proteins regulates apoptosis, a process of programmed cell death used to eliminate unwanted cells such as cancer cells[1–3, 4 ]. Abnormal cells can dysregulate this process to avoid cell death[1, 2]. One of the ways that cancer cells can accomplish this is by up regulating the anti-apoptotic members of the Bcl-2 family of proteins[1, 5, 6]. Indeed, amplification of the gene encoding the anti-apoptotic Bcl-2 family protein myeloid cell leukemia-1 (Mcl-1) is one of the most common genetic aberrations in human cancer[7], and Mcl-1 over-expression in human cancers is associated with high tumor grade and poor survival[8–10]. In addition, Mcl-1 activity has also been implicated in the resistance to multiple therapies, including_paclitaxol, and venetoclax which are prescribed for cancer patients[8, 11]. Targeted inhibition of Mcl-1 has been demonstrated to cause tumor cell death in Mcl-1-dependent cancers and to increase the sensitivity of many standard chemotherapeutics in cancers that have developed resistance to the normal apoptotic process[12, 13]. Thus, Mcl-1 represents a promising target for cancer drug discovery. However, Mcl-1 mediates its effects through protein-protein interactions[14] and is considered difficult to target with small molecules. Nevertheless, progress has been made towards this goal. Several groups have reported Mcl-1 inhibitors that have a range of binding affinities and activity in cell based assays including, UM-36 (University of Michigan)[15], EU5346 (Eutropics Pharmaceuticals)[16], Patent EP2886545A1 (Servier/Venalis), Patent WO2016033486 (Amgen), AZ-Mcl1 (Astra Zeneca)[17], Dual Mcl-1/Bcl-xl inhibitors (Takeda)[18], and the AbbVie inhibitor, A-1210477[19, 20]. We have also reported on the discovery of small molecules that bind to Mcl-1 with high affinity that were discovered using fragment-based screening and structure-based design[21, 22]. Here, we describe how these compounds were further optimized to obtain Mcl-1 inhibitors that display picomolar binding affinities and on-target mechanism-based activity in cells. We also show that these compounds have activity in primary cancer patient derived cell lines.
Methods
Chemistry
Synthesis protocols and compound characterization have been reported in the Supplementary Materials The indole amides (2–5) were described in detail in publication No. WO/2015/148854 titled “SUBSTITUTED INDOLE MCL-1 INHIBITORS.”All samples were of ≥95% purity as analyzed by LC−UV/vis-MS. All reagents were purchased from chemical suppliers and used without purification.
Protein expression and purification for Assays and X-ray Structures
Protein preparation was described previously[21]. Briefly, a previously reported [23] construct was sub-cloned into an expression vector (pDEST-HisMBP) expressed in Escherichia coli BL21 CodonPlus (DE3) RIL (Stratagene) and purified through nickel-column and size-exclusion chromatography sequentially.
Protein crystallization, data collection, and structure refinement
Structural studies were performed as previously described[21]. Briefly, Mcl-1 protein (15 mg/mL) was mixed with a 1.2x excess of ligand in solution (25–30% PEG 3350, 0.1 M Bis-TRIS pH 6.5, 0.2 M MgCl2) by hanging drop followed by flash freezing after cryo-protection using 10–20% glycol.
Data were collected at Life Sciences Collaborative Access Team (LS-CAT) 21-ID-G beamline, Advanced Photon Source (APS), Argonne National Laboratory. Indexing, integration and scaling were performed with HKL2000 (HKL Research)[23], phasing by molecular replacement with Phaser (CCP4)[24, 25] using the structure (PDB: 4HW2) as a model, refinement used Phenix[26]. Structural statistics are given in the Supplementary Material. Figures were prepared with PyMOL (Schrödinger, LLC: New York, 2010)[27].
Competition Binding Assays
A fluorescein isothiocyanate (FITC)-labeled BH3 peptide derived from Bim (FITC-Bim; FITC-AHx- EARIAQELRRIGDEFNETYTR-NH2) or Bak (FITC-Bak; FITC-AHx-GQVGRQLAIIGDDINR-NH2)were purchased (Genscript). FPA measurements used 384-well, black, flat-bottom plates (Greiner Bio-One) and a BioTek Cytation 3. FITC-Bim assay conditions: 20 mM TRIS pH 7.5, 50 mM NaCl, 3 mM DTT, 0.01% CHAPS, FITC-Bim peptide at 1 nM and His6-MBP Mcl-1 at 1.5 nM. Bcl-xl or Bcl-2 assay conditions: 10 nM FITC-Bak peptide incubated with either 15 nM Mcl-1, 4 nM Bcl-xL or 4 nM Bcl-2 in 20 mM TRIS pH 7.5, 50 mM NaCl, 3 mM DTT, and 5% final DMSO. 1% fetal calf serum (FBS) is added in 1% FBS assay. Compounds were diluted in DMSO, (10-point, 3-fold serial dilutions) added to assay plates, and incubated for 0.5 h at room temperature. The TR-FRET assay used the assay buffer described above plus 300 nM FITC BAK, 1 nM Mcl-1-MBP fusion, 1nM MBP-terbium (Cisbio, Bedford,Ma) and 0.05% Pluronic F-68 (Sigma). Mixtures were incubated for 3 hours and signal (Delta F) was measured on the Biotek Cytation 3 equipped with a filter cube containing an Ex 340/30 nM Em 620/10 filter and an Ex 340/30 Em 520 filter.
IC50 values were calculated by fitting anisotropy using XLFit (IDBS) and converted into a binding dissociation constant[28] to give Ki. Two or more repeats were obtained and average Ki values are reported.
JC1 BH3 profiling and intracellular BH3 (iBH3) profiling
Synthetic peptides for MS-1[29] (ac-RPEIWMTQGLRRLGDEINAYYAR-NH2), Bim-BH3 (ac-MRPEIWIAQELRRIGDEFNA-NH2 ), HRK (Ac- WSSAAQLTAARLKALGDELHQ - NH2) and Bad-BH3 (ac-LWAAQRYGRELRRMSDEFEGSFKGL-NH2 ) were purchased (Genscript). JC1 BH3 profiling for figures 4A and 4B was performed as described previously[30]. For figure 4C cytochrome c loss was measured by iBH3 profiling as described earlier [11]. Following cell fixation and cell quenching, cells were stained with of 1:100 dilution of anti-cytochrome c –Alexa647 (clone 6H2.B4; #612310, Biolegend) in a 10X staining buffer (20% FBS, 10% BSA, 1% Saponin, 3 mM Sodium Azide in PBS) to measure cytochrome c loss. Cytochrome c retention was measured on BD LSRII after overnight incubation with antibody and cytochrome c retention was measured using the following equation:
Figure 4.

Mitochondrial Depolarization studies. BH3 profiling with BAD (green) a Bcl-2,Bcl- xL binding peptide, MS-1 (red) a Mcl-1 selective binding peptide, HRK (magenta) a Bcl- xL selective binding peptide, and Bim (blue) a pan anti apoptotic (e.g. Bcl-2,Bcl- xL and, Mcl-1) binding peptide and with compound 4 (orange) and 5 (black) in (A) NCI H929 (B) K562 cells. (C) Comparison of cytochrome c release after dosing with the MS-1 peptide and compound 4 in a panel of Multiple Myeloma (MM) and Acute Myeloid Leukemia (AML) cell lines. (D) IC50 values from a three day cell viability study after dosing compound 4 and 5 in a panel of AML and MM cell lines.
Cell Line Proliferation Assay
Cells were dispensed into 96 well plates at a concentration of 1000 cells per well in RPMI supplemented with 10% FBS and 0.05 mM 2-Mercaptoethanol and incubated overnight at 37 °C in a tissue culture incubator. Compounds were diluted in DMSO [0.5%] and added to the cells. Plates were incubated for 72 hours, and cell viability was measured using the Cell TiterGlo reagent. %Viability was defined as relative luminescence units RLU of each well divided by the RLU of cells on day 0. Dose response curves were generated, and GI50 values were determined using XLFit (IDBS) software.
Caspase Activation Assay
Cells were dispensed in 96 well plates as described in the proliferation assay methods with 5% FBS and a cell concentration of 5000 cells per well. Plates are incubated with compound for 3 hours, 100 μL of Caspase-Glo (Promega) reagent is added, and the mixture incubated at room temperature in the dark for 30 minutes. Luminescence is measured (Biotek Cytation 3) and analyzed using XLFit (IDBS) to generate EC50 values.
co-IP experiment Mcl-1/Bim NCI H929 Cell Line
10,000 NCI H929 cells were treated with compounds 4 and 5 at 4.2 and 6 μM, respectively for 90 minutes. An additional sample was treated with 0.1% DMSO alone as a vehicle control. Cells were pelleted, washed in PBS, and lysed in non denaturing lysis buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 2.5 mM MgCl 0.5% NP40, protease/phosphatase inhibitor), and 800 μg of total cell lysate in a 400 μL volume was incubated with 4 μg of Biotin conjugated anti Mcl-1 clone RC-13 (MA5–13929, Thermo Fisher scientific), 4 ug of Biotin conjugated anti Bcl xLclone 7B2.5 (AB25062, Abcam), or biotin conjugated mouse IgG1 Isotype control (PA-5–33199, Thermo Fisher Scientific), followed by addition of 30 μL of washed Dynabeads® M-280 Streptavidin (11205D, Thermo Fisher Scientific). Beads were washed followed by addition of LI-COR protein loading buffer (LICOR, 928–40004) diluted to in RIPA buffer at 95°C. Bim and Mcl-1 was measured by western blot using rabbit anti Bim Clone Y36(AB32158, Abcam), polyclonal anti Mcl-1 (SC-819, Santa Cruz Biotechnology), and monoclonal anti-rabbit Bcl-xl Clone 54H6 (Cell Signaling Technologies, #2764) followed by IRDye secondary antibodies. Blots were scanned and image analysis performed on a LI-COR Odyssey.
Generation of Engineered Cell Lines
As described previously[31] human BCL-XL, BCL-W, BCL-2 and BFL-1 cDNAs and a BCR-ABL (p185) oncofusion plasmid were stably expressed in Mcl-1 conditional Arf-deficient bone marrow by retroviral transduction and puromycin selection (Sigma Aldrich, MO, 2 μg per ml). Endogenous murine Mcl-1 was deleted by transduction with Cre recombinase.
Cell Death Experiments in Engineered Cell lines
BCR-ABL p185+ B-ALL cell lines re-programmed with human anti-apoptotic BCL-2 family members were seeded in 96-well round bottom plates (6×104 cells/well). Mcl-1 inhibitors were solubilized in DMSO or DMSO vehicle controls were added at the indicated concentrations, and cells were in complete RPMI media with 1% fetal calf serum. After 24 hours, the plates were centrifuged and cell viability was determined by staining with Annexin-V-APC and propidium iodide (BD Biosciences) and measured by flow cytometry using the high throughput sampler (HTS) on a FACSCanto II (BD Biosciences, CA).
Proliferation/Immunoprecipitation in MM Patient Samples
Bone marrow aspirates were collected following a Emory University Institutional Review Board–approved protocol from consenting myeloma patients and prepared as described earlier[32]. Briefly, aspirates were diluted to 20 mL with PBS, and underlaid with lymphocyte separation medium (Mediatech). The buffy coat was washed, resuspended in culture medium, and stained with anti–CD38-phycoerythrin, anti–CD45-allophycocyanin-Cy7, and anti-CD138-fluorescein isothiocyanate antibodies (BD Biosciences) for FACS analysis. Plasma cells were prepared for purification using MACS Cell Separation MS Columns and CD138 magnetic microbeads per the manufacturer’s protocol (Miltenyi Biotec). Once isolated, CD138-positive cells were checked for purity via flow cytometry. Cells (0.25 × 106 cells/mL) were treated with the indicated concentrations of compound 5 for 24 hours and apoptosis determined by staining with anti–CD38-phycoerythrin and anti–CD45-allophycocyanin-Cy7 antibodies and Annexin-V-fluorescein isothiocyanate; three million cells were used for co-immuoprecipitation analysis.
Immunoprecipitation experiments on myeloma cells were performed using the Exacta- Cruz™ C Kit (Santa Cruz Biotechnology, Inc.) following the manufacturer’s instructions as previously described[32]. The following primary antibodies were used: mouse anti-Mcl-1 mAb (BD Biosciences, San Jose, CA); hamster anti-Bcl-2 mAb (BD Biosciences, San Jose, CA); mouse anti-Bcl-xL mAb (7B2.5[32]). The resulting complexes were analyzed by Western blotting using a rabbit anti-Bim pAb (EMD Millipore, Temecula, CA). The secondary antibody was provided in the Exacta- Cruz™ C Kit (Santa Cruz Biotechnology, Inc.).
Proliferation of AML Patient Samples
Bone marrow aspirates were collected following a Vanderbilt University Institutional Review Board–approved protocol from consenting AML patients. Bone marrow aspirates in acid citrate dextrose tubes were added to ACK lysis buffer at room temperature for 5 minutes. Samples were then diluted with 20–40mL of PBS, and centrifuged at 200G for 10 minutes. Supernatant was discarded, and cell pellet was re-suspended in 5mL of PBS. Cells were dispensed into 96 well plates at a concentration of 6000 cells per well in RPMI supplemented with 10% FBS. Compound was diluted in DMSO and added to the cells at the given concentrations with a final DMSO concentration of 0.03%. Compound-treated plates were incubated at 37 °C for 24 hours. Cell viability was measured using the Cell TiterGlo reagent (Promega), and the relative luminescence units (RLU) were measured. Viability was defined as RLU of each well divided by the RLU of cells treated with DMSO vehicle. Dose response curves were generated using Prism (GraphPad) software.
Results and Discussion
Discovery of Mcl-1 inhibitors
The progression of our previously reported lead compound to our picomolar Mcl-1 inhibitors (4, 5) that possess mechanism-based activities in cells is summarized in Figure 1. Compound 1 was discovered by merging together two hits obtained in a fragment-based screen followed by further potency optimization using structure-based design[21]. This compound displaced a fluorescently labeled peptide derived from the pro-apoptotic protein Bak with a Ki of 55 nM. However, this was not potent enough to elicit cellular activity. The necessity of picomolar binding affinity to exhibit robust activity in cells is consistent with previously reported inhibitors for other Bcl-2 family members[20, 33]. Another problem with compound 1 is it showed reduced binding to Mcl-1 in the presence of serum, suggesting that it binds tightly to serum proteins. Anionic lipophilic compounds like 1 generally bind tightly to albumin[34, 35]. Compounds that bind to plasma proteins have less free fraction available to interact with cells in the in vitro cell assay and we and others have found that adding FBS to the biochemical binding assay is a useful way to gauge progress on reducing plasma binding.
Figure 1.

Chemical structures illustrating compound progression and chemical changes from an initial merged fragment lead 1, to the cell active compounds 4 and 5.
To further improve the affinity of 1 to Mcl-1, we screened substituted aromatic heterocyclic groups at the 7-position of the indole and found that various methyl pyridines, 3,5 di-methyl oxazoles as well as the 1,3,5-tri-methyl-4-pyrazolo group improved affinity while maintaining desirable pharmaceutical properties. A similar approach was also employed by the AbbVie group[19]. However, the resulting indole acid was not able to show convincing activity in the Mcl-1 sensitive cell line (NCI H929).
The X-ray co-crystal structure of 1 complexed to Mcl-1 (Figure 2A) revealed the key charge-charge interactions between the carboxylate of 1 and guanidinium of R263, which was crucial for binding. This analysis led us to consider the expansion of an inhibitor beyond the P2 pocket first using a sulphonamide[36] and then an amide linkage in which the carbonyl moiety could maintain the favorable hydrogen bond with R263.
Figure 2.

Structural data on lead compounds used to drive compound design. Mcl-1 residues R263 and N260 are labeled. (A) A superposition of the X-ray structures of lead acid 1 (white stick) and one pose for the potent amide 2 (cyan). (B) Superposition of the two conformations of compound 2 observed in the X-ray structure. Expansion of circled region shown on right hand side. (C) X-ray structures of lead molecules 2 and 5 and a superposition of their structures are shown. (D) A superposition of the X-ray structure of compound 5 and the corresponding phenyl acid pose of compound 2.
Through extensive SAR studies, compound 2 containing the 3-benzoic acid amide was discovered. Initially, the amide series of our compounds suffered from a considerable loss in affinity when the anionic carboxylate was replaced with the neutral amide group. However, introduction of a suitable aromatic acid through an amide linkage fully recovered the affinity loss by creating new binding interactions. It is also noteworthy that compound 2 showed almost 20-fold higher potency than the parent indole acid 1 in the presence of 1% fetal calf serum (FBS) despite their comparable binding affinities without serum (Table 1). These results suggested that the 2-indole amide inhibitor series offered a new opportunity with greater expandability and could have significantly lower non-specific serum protein binding than the 2-indole acids.
Table 1.
Compound characterization in biochemical and cell-based assays.
| 1 | 2 | 3 | 4 | 5 | ||
|---|---|---|---|---|---|---|
| Ki (nM) | Mcl-1 | 55 | 23 | 2.1 | <1.0 (0.7+/− 0.1)3 | <1.0(0.5+/− 0.09)3 |
| Mcl-1 + 1%FBS | 1900 | 108 | 7.9 | 2.3 | 2.8 | |
| Bcl-xL | 2190 | 4600 | 8900 | >10000 | >10000 | |
| Bcl-2 | 928 | 1540 | 1900 | 576 | 950 | |
| GI50 (μM)1 | H929 | 6.4 | 1.4 +/− 0.3 | 2.0 +/− 0.3 | ||
| K562 | >20 | 11.5 +/− 1.3 | 11.5 +/− 1.5 | |||
| Caspase 3/7GI50 (μM)2 | H929 | 3.7 | 0.75 | 0.76 | ||
| K562 | >50 | >50 | >50 | |||
| Aq. Sol. (μg/mL) | 28 | 58 | 47 | 4.2 | ||
| PAMPA (e−6 cm/sec) | <3.3 | 307 | 21 | 13 | ||
| Rat Microsomal Stab. (min) | >30 | >30 | >30 | >30 | ||
Cells supplemented with 10 % FBS.
Cells supplemented with 5% FBS.
Measurement in TR-Fret Assay.
To understand the binding interactions for further optimization, we obtained an X-ray structure of 2 complexed with Mcl-1. When the crystal structure of 2 is superimposed with 1 (Figure 2A), we find that the indole cores of both molecules superimpose and that the carbonyl of compound 2 maintains polar interactions with R263. The crystal structure contains 4 copies of the Mcl-1 protein in an asymmetric unit, and two equally populated 3-benzoic acid conformations of 2 (Figure 2B). In both conformations, the phenyl group sits over the guanidinium moiety of R263 in a large hydrophobic shelf formed by the loop and the top of helix 4 and 5 of the protein to form a cation-π stacking which likely enhances the stability of the complex[37]. However, the carboxylate moiety in each conformation utilizes different polar contacts in the perimeter of the shelf. In the first pose (cyan) (Figure 2A, B), the carboxylate is positioned above the guanidinium of R263 to engage in a favorable charge-charge interaction. In contrast, the group is flipped to form a H-bond with the side chain of N260 in the second pose of compound 2 (olive Figure 2B). In both cases, these newly introduced binding interactions involving the 3-benzoic acid group are important for binding and overcoming the large loss in affinity to Mcl-1 when the carboxylate that was directly attached to the indole core of 1 was removed.
Inspection of the Mcl-1 bound conformation of 2 (Figure 2C) also indicated a near planar arrangement of the amide and indole. Furthermore, the NH’s of both groups point in the same direction, which is ideal for tethering. Based on the X-ray structure, these two atoms were bridged to form a tricyclic indole lactam 3 to lock the binding conformation. This modification resulted in multiple benefits for the series by optimizing both potency and the pharmaceutical properties of the compounds[38, 39]. As shown in Table 1, the indole lactam 3 exhibited >10 fold enhanced binding affinity when compared to the open chain analog 2. Also, by removing two rotatable bonds and two H-bond donors, this significantly improved passive permeability as assessed by a parallel artificial membrane permeability assay (PAMPA). This change also reduced the interference from serum in our binding assay to a single digit nanomolar Ki and improved the compound’s aqueous solubility. Finally, it is possible that our tethering strategy enhances oral bioavailability[40] by effectively eliminating a secondary peptide bond that would be vulnerable to proteolysis in the GI tract.
The structure of 2 (Figure 2B) also suggested that the benzoic acid head group only accesses a small portion of the hydrophobic shelf. This suggests that a larger fused bicyclic aromatic ring may be better suited to bind to this site. To test this hypothesis, compounds 4 and 5 were synthesized using an indole. In both compounds, the structural motif of the 3-benzoate of 2 was preserved to capture all of the favorable interactions while the fused methyl-pyrrole portion was introduced with different trajectories. When tested, both compounds had significantly higher potency than 3 by exhibiting picomolar binding affinities to Mcl-1, which was below the level able to be accurately determined in our Fluorescence Polarization Anisotropy (FPA), assay conditions. They also maintained low nanomolar Ki’s in the presence of 1% FBS.
To understand the gains in potency, we obtained a structure of compound 5 in complex with Mcl-1 and found only one conformation for the indole acid of 5. As shown in Figure 2D, the meta-benzoate portion of indole adopts the same binding pose as the first conformation of 2 (cyan) to preserve all of the desirable binding attributes. The methyl-pyrrazole moiety is extended toward N260 to cover a wider area of the hydrophobic binding interface over the propylene chain of R263 which likely contributes to its improved binding affinity. The contacts of the indole acid headpiece, as shown in the high resolution X-ray structure, contributes significantly to affinity which is consistent with the SAR of related analogs. For example, analogs without the acid on the indole or removal of the indole acid headpiece leads to a ten to hundred fold decrease in affinity (data not shown).
Demonstration of mechanism-based activity
Programmed cell death can occur due to non-specific cytotoxicity. Thus, it was imperative to prove that our compounds exhibit the mechanism-based activity expected for a Mcl-1 inhibitor [5, 20]. One piece of evidence to support this claim is if the compounds exhibit more potent cellular activity in cell lines that are Mcl-1 dependent compared to those that are not. To identify cell lines that were very sensitive to Mcl-1 inhibition, we searched the hematologic cancer cell lines profiled in the CCLE (Broad Institute)[41] that had a profile of protein expression that we had found earlier to predict Mcl-1 selective activity[42]. One of the most sensitive cell lines predicted from this analysis was NCI H929 a multiple myeloma (MM) cell line shown in previous studies to be Mcl-1 sensitive[20]. We also predicted that chronic myelogenous leukemia (CML) cell line K562 would be resistant to Mcl-1 inhibitors and could serve as an inactive control for off target toxicity.
We used the NCI H929 cell line to test if our compounds acted like a BH3 mimetic and displaced Bim from Mcl-1 [13, 43]. As shown in Figure 3 dosing cells with compounds 4 or 5 causes a near complete displacement of Bim from Mcl-1when compared to the DMSO vehicle in a co-IP experiment. A similar dose dependent displacement of Noxa from Mcl-1 is shown in the Supplementary Materials Figure S1. These data shows that the mechanistically expected displacement of Bim or Noxa from Mcl-1 is observed after compound dosing. Unexpectedly, we also observed that compound dosing with our Mcl-1 inhibitor causes an increase in total Mcl-1 protein. As shown in the Supplementary material Figure S1, the increase in Mcl-1 levels is as much as 1.5 fold over vehicle after dosing with 4. This was also observed in an earlier study and it was hypothesized to be due to compound displacement of Noxa, a BH3 protein involved in Mcl-1 turnover [20]. The compound selectivity for Mcl-1 over another family member, Bcl-xl, is shown in Figure 3B. No decrease is seen in the Bcl-xl/Bim band, in fact, an increase in Bim bound to Bcl-xl is observed, presumably due to displacement of Bim from Mcl-1.
Figure 3.

Co-IP of Mcl-1 and Bim (A), or Bcl-xl and Bim (B) treated with three times the GI50 from the proliferation assay in H929 cells, compound 4 (4.2 μM) and 5 (6.0 μM). The vehicle control, V, was treated with 0.1% DMSO.
We next used BH3 profiling experiments to monitor mitochondrial depolarization and cytochrome c release caused by BH3 peptides or our small molecule BH3 mimetic[30]. As shown in Figure 4A, the NCI H929 cell line is not affected when exposed to HRK a Bcl-xl selective peptide, or Bad a Bcl-xl and Bcl-2 peptide. However, mitochondrial depolarization was observed after addition of the Mcl-1 specific peptide MS-1 and to Bim, a pan pro-apoptotic peptide. When compound 4 and 5 were dosed in the NCI H929 cell line mitochondrial depolarization nearly equipotent to the MS-1 peptide, which is one of the best Mcl-1 specific probes, reported in the literature[29, 44]. In Figure 4B we show that the K562 heme cell line is insensitive to MS-1 and most other BH3 peptides with only moderate activity to the pan Bcl-2 family inhibitor Bim. The K562 cell line is also much less sensitive to compound 4 and 5. However, some activity is observed at high concentrations which may indicate some additional non BH3 mimetic activity. When compound 4 activity was compared to the Mcl-1 specific MS-1 peptide in a larger panel of MM and AML cancer cell lines we see parallel activity of compound 4 and MS-1 to cause Cytochrome C release due to mitochondrial depolarization. The cell line activity of our compounds observed in the BH3 profiling experiments is also reflected in proliferation assays (Table 1) on NCI H929/K562 cells and in a larger panel of AML and MM cancer cell lines shown in Figure 4D. In the NCI H929/K562 cells, we found that dosing cells with compound 4 or 5 resulted in low micromolar GI50 in the MS-1 sensitive cell line (NCI H929) and a 5- to 8-fold lower (i.e. GI50( K562)/GI50( H929) ) GI50 activity for the MS-1 insensitive line (K562). Looking at the larger set of cell lines, the overall correlation between cytochrome c release (Figure 4C) and activity of compounds in a proliferation assays (4D) is good but not perfect (e.g. MV411) which likely reflect additional cell line specific factors or some additional non BH3 mimetic activity that influence how primed cell lines are for death.
We also tested compounds in a caspase activation assay ( supplemented with only 5% FBS to reduce plasma interference ) and found rapid activation at low concentrations only in the NCI H929 sensitive cell line. The difference observed in the proliferation and caspase assays between a BH3 MS-1 sensitive and insensitive cell line suggests that most of the observed cell killing of the compound in the proliferation assay is indeed due to on-target Mcl-1-mediated activity.
As another test for specific on-target activity we tested compounds 4 and 5 in a panel of re-engineered cell lines. These cell lines were re-engineered BCR-ABL+ B-ALL cells modified to only contain specific exogenous human Bcl-2 family proteins[31]. As previously reported, these cell lines are able to unambiguously demonstrate on-target activity for Bcl-2 family inhibitors. When these cell lines were dosed with compounds 4 (Figure 5A) or 5 (Figure 5B), a clear separation was observed for the activity of the compounds against cell lines with different Bcl-2 family proteins, with Mcl-1 having the lowest observed EC50 followed by its closest related family member BFL-1. Both compounds show two- to three-fold less activity against the Bcl-2 and Bcl-xl engineered lines, respectively. The compounds do exhibit some off target toxicity at high concentrations based on the killing of the DKO (double Bax, Bak knockout), TKO (triple Bax, Bak, Mcl-1 knockout) cell lines at high micromolar concentrations. As seen in Figure 5, the compounds have reasonable selectivity for tool compounds but they are not close to the specificity achieved by venetoclax, an approved BH3 mimetic, that targets Bcl-2
Figure 5.
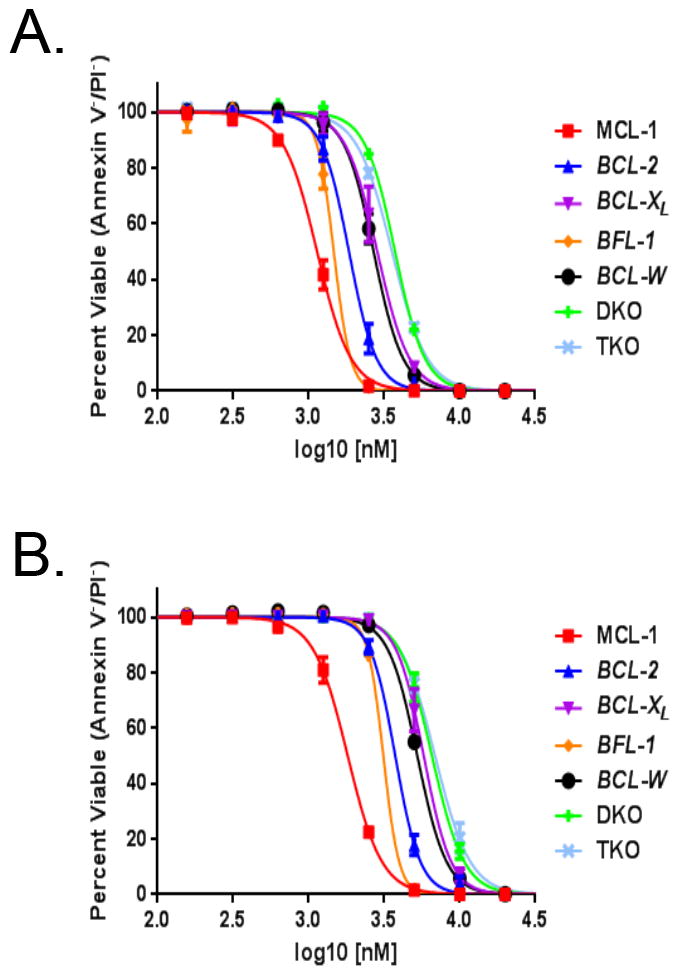
Viability studies of specific human Bcl-2 family engineered cell lines after dosing with Mcl-1 inhibitors. (A,B) a panel of re-engineered BCR-ABL+ B-ALL Cells modified to have the indicated anti-apoptotic exogenous Human Bcl-2 family member or DKO (double Bax,Bak knockout), TKO (triple Bax,Bak,Mcl-1 knockout) dosed with (A) compound 4 (B) compound 5.
We next expanded our testing to include MM and acute myeloid leukemia (AML) primary patient samples. In Figure 6a we show a co-IP experiment on a patient sample that indicates that the majority of Bim in these cells is sequestered by Mcl-1. The high level of sequestration of Bim by Mcl-1 in this patient sample is consistent with these cell lines being Mcl-1 dependent[32]. When this sample was dosed with compound 4 we observed Annexin V and propidium ioidide positive cells at low micromolar concentrations of compound (Figure 6B). In Figure 6C, we show an AML patient sample dosed using compounds 4 and 5. The AML patient sample also had reduced viability at low micromolar concentrations of our compounds. These two studies show that these primary patient samples are also sensitive to our compounds. However, further characterization of the Mcl-1 dependency of the patient samples is needed to insure that the sensitivity is primarily due to an Mcl-1 based mechanism.
Figure 6.

Compound activity in freshly isolated patient samples. (A,B) Multiple Myeloma patient sample. (A) CD138+ cells were isolated from the bone marrow of a myeloma patient and treated with compound 5 for 24 h. Protein lysates were subjected to co-immunoprecipitation with monoclonal mouse anti-Mcl-1, anti-Bcl-xL, and monoclonal hamster anti-Bcl-2 antibodies. Resulting protein complexes were subjected to Western blot analysis using rabbit antibodies against Mcl-1, Bcl-xL, Bcl-2, and Bim. (B) Ficoll isolated buffy coat cells were treated with the indicated concentrations of compound 4 for 24 h. Apoptosis was determined by staining with antibodies against CD38, CD45, and Annexin-V-FITC. The percent annexin/PI positive cells represent the specific cell death after subtracting the spontaneous cell death. (C) Dose dependent viability decrease in Acute Myeloid Leukemia (AML) patient sample with 4 (triangle) and 5 (open circle).
Discussion
We have described the discovery of picomolar Mcl-1 inhibitors. The activity of these compounds, in multiple experiments is consistent with apoptosis mediated by an Mcl-1 BH3 mimetic. The compounds caused mitochondrial depolarization at single digit nM concentrations and caspase activation in sensitive cancer cell lines. When tested in engineered cell lines containing specific Bcl-2 family members, we found lower IC50 and higher specificity for compounds 4 and 5 than was reported for earlier literature compounds [31]. We also showed that patient samples from MM and AML had a response to compound treatment at low micromolar concentrations. Much more potent Mcl-1 inhibitors could thus be expected to be useful therapeutic agents in these cancers. These results suggest that the Mcl-1 inhibitors described here will be useful as tool molecules to study the role of Mcl-1 in cancer and serve as useful starting points for the discovery of Mcl-1 inhibitors as therapeutic agents.
Supplementary Material
Acknowledgments
The authors thank co-workers at the High-Throughput Screening Core facility of Vanderbilt University, TN, for compound management and Nicolas Pelz for useful discussions. This research was supported by the U.S. National Institutes of Health, NIH Director’s Pioneer Award DP1OD006933/DP1CA174419 to S.W.F., The NCI Experimental Therapeutics (NExT) Program BOA29XS129TO22 under the Leidos Biomed Prime Contract No. HHSN261200800001E, and a career development award to S.W.F. from a NCI SPORE grant in breast cancer (Grant P50CA098131) to C. L. Arteaga. The Biomolecular NMR Facility at Vanderbilt University is supported in part by a NIH SIG Grant 1S-10RR025677-01 and Vanderbilt University matching funds. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
ABBREVIATIONS
- Mcl-1
myeloid cell leukemia 1
- Bcl-2
B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma extra large
- BH3
Bcl-2 homology domain 3
- Bax
Bcl-2-associated X protein
- Bak
Bcl-2 homologous antagonist killer
- BIM BCL2L11
Bcl-2 like protein 11
- Noxa
PMAIP1 gene Phorbol-12-myristate-13-acetate-induced protein 1
- Bad
Bcl-2-associated death promoter
- FITC
fluorescein isothiocyanate
- FPA
fluorescence polarization anisotropy
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interest.
PDB ID codes: Comp. 2: 5IEZ; Comp. 5: 5IF4.
References
- 1.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 3.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA: a cancer journal for clinicians. 2005;55:178–94. doi: 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- 4.Reed JC. Bcl-2 and the regulation of programmed cell death. The Journal of cell biology. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell death and differentiation. 2009;16:360–7. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 6.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature reviews Molecular cell biology. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 7.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC, Dixit VM. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–4. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 9.Wei SH, Dong K, Lin F, Wang X, Li B, Shen JJ, Zhang Q, Wang R, Zhang HZ. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer chemotherapy and pharmacology. 2008;62:1055–64. doi: 10.1007/s00280-008-0697-7. [DOI] [PubMed] [Google Scholar]
- 10.Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell death & disease. 2010;1:e40. doi: 10.1038/cddis.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, Dohner H, Gaidzik VI, Galinsky I, Golfman LS, Haferlach T, Harutyunyan KG, Hu J, Leverson JD, Marcucci G, Muschen M, Newman R, Park E, Ruvolo PP, Ruvolo V, Ryan J, Schindela S, Zweidler-McKay P, Stone RM, Kantarjian H, Andreeff M, Konopleva M, Letai AG. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer discovery. 2014;4:362–75. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell death and differentiation. 2015;22:1071–80. doi: 10.1038/cdd.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS letters. 2010;584:2981–9. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 14.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nature reviews Drug discovery. 2004;3:301–17. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 15.Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, Stuckey JA, Showalter HD, Nikolovska-Coleska Z. 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. Journal of medicinal chemistry. 2014;57:4111–33. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richard DJ, Lena R, Bannister T, Blake N, Pierceall WE, Carlson NE, Keller CE, Koenig M, He Y, Minond D, Mishra J, Cameron M, Spicer T, Hodder P, Cardone MH. Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorganic & medicinal chemistry. 2013;21:6642–9. doi: 10.1016/j.bmc.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MAB Evaluation of Mcl-1 Inhibitors in Preclinical Models of Multiple Myeloma. ASH American Society of Hematology Annual Meeting; San Diego, Ca. 2014. [Google Scholar]
- 18.Tanaka Y, Aikawa K, Nishida G, Homma M, Sogabe S, Igaki S, Hayano Y, Sameshima T, Miyahisa I, Kawamoto T, Tawada M, Imai Y, Inazuka M, Cho N, Imaeda Y, Ishikawa T. Discovery of potent Mcl-1/Bcl-xL dual inhibitors by using a hybridization strategy based on structural analysis of target proteins. Journal of medicinal chemistry. 2013;56:9635–45. doi: 10.1021/jm401170c. [DOI] [PubMed] [Google Scholar]
- 19.Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, Erickson S, Fidanze S, Fry E, Hasvold L, Jenkins GJ, Jin S, Judge RA, Kovar PJ, Madar D, Nimmer P, Park C, Petros AM, Rosenberg SH, Smith ML, Song X, Sun C, Tao ZF, Wang X, Xiao Y, Zhang H, Tse C, Leverson JD, Elmore SW, Souers AJ. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. Journal of medicinal chemistry. 2015;58:2180–94. doi: 10.1021/jm501258m. [DOI] [PubMed] [Google Scholar]
- 20.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, Sheppard GS, Wang L, Gierke S, Kategaya L, Anderson DJ, Wong C, Eastham-Anderson J, Ludlam MJ, Sampath D, Fairbrother WJ, Wertz I, Rosenberg SH, Tse C, Elmore SW, Souers AJ. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell death & disease. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. Journal of medicinal chemistry. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burke JP, Bian Z, Shaw S, Zhao B, Goodwin CM, Belmar J, Browning CF, Vigil D, Friberg A, Camper DV, Rossanese OW, Lee T, Olejniczak ET, Fesik SW. Discovery of tricyclic indoles that potently inhibit Mcl-1 using fragment-based methods and structure-based design. Journal of medicinal chemistry. 2015;58:3794–805. doi: 10.1021/jm501984f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Academic Press; New York: 19977. [DOI] [PubMed] [Google Scholar]
- 24.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta crystallographica Section D, Biological crystallography. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods in molecular biology. 2008;426:419–35. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 27.LLC, S. The Pymol Graphics System in
- 28.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Analytical biochemistry. 2004;332:261–73. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 29.Foight GW, Ryan JA, Gulla SV, Letai A, Keating AE. Designed BH3 peptides with high affinity and specificity for targeting Mcl-1 in cells. ACS chemical biology. 2014;9:1962–8. doi: 10.1021/cb500340w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Koss B, Ryan J, Budhraja A, Szarama K, Yang X, Bathina M, Cardone MH, Nikolovska-Coleska Z, Letai A, Opferman JT. Defining specificity and on-target activity of BH3-mimetics using engineered B-ALL cell lines. Oncotarget. 2016 doi: 10.18632/oncotarget.7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, Kaufman JL, Lee KP, Lonial S, Boise LH. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood. 2011;118:1329–39. doi: 10.1182/blood-2011-01-327197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 34.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. Journal of molecular biology. 2005;353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Wright M, Hop CE. Rational use of plasma protein and tissue binding data in drug design. Journal of medicinal chemistry. 2014;57:8238–48. doi: 10.1021/jm5007935. [DOI] [PubMed] [Google Scholar]
- 36.Pelz NF, Bian Z, Zhao B, Shaw S, Tarr JC, Belmar J, Gregg C, Camper DV, Goodwin CM, Arnold AL, Sensintaffar JL, Friberg A, Rossanese OW, Lee T, Olejniczak ET, Fesik SW. Discovery of 2-Indole-acylsulfonamide Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods. Journal of medicinal chemistry. 2016;59:2054–66. doi: 10.1021/acs.jmedchem.5b01660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gromiha MM, Santhosh C, Ahmad S. Structural analysis of cation-pi interactions in DNA binding proteins. International journal of biological macromolecules. 2004;34:203–11. doi: 10.1016/j.ijbiomac.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Di L, Kerns EH. Profiling drug-like properties in discovery research. Current opinion in chemical biology. 2003;7:402–8. doi: 10.1016/s1367-5931(03)00055-3. [DOI] [PubMed] [Google Scholar]
- 39.Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug discovery today Technologies. 2004;1:337–41. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. Journal of medicinal chemistry. 2002;45:2615–23. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 41.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodwin CM, Rossanese OW, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell death and differentiation. 2015 doi: 10.1038/cdd.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC, Fairlie WD, Hinds MG, Colman PM. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6217–22. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nature chemical biology. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.