

Abstract
Metchnikovellidae are a group of unusual microsporidians that lack some of the defining ultrastructural features characteristic of derived Microsporidia and are thought to be one of their earliest-branching lineages. The basal position of metchnikovellids was never confirmed by molecular phylogeny in published research, and thus far no genomic data for this group were available. In this work, we obtain a partial genome of metchnikovellid Amphiamblys sp. using multiple displacement amplification, next-generation sequencing, and metagenomic binning approaches. The partial genome, which we estimate to be close to 90% complete, displays genome compaction on par with gene-dense microsporidian genomes, but contains an unusual repertoire of unique repeat elements. Phylogenetic analyses of multigene datasets place Amphiamblys sp. as the first branch of the microsporidian lineage following the divergence of a mitochondriate microsporidian Mitosporidium. We find evidence for a mitochondrial remnant presumably functionally equivalent to a mitosome in Amphiamblys sp. and the common enzymatic complement for microsporidian anaerobic metabolism. Comparative genomic analyses identify the conservation of components for clathrin vesicle formation as one of the key features distinguishing the metchnikovellid from its highly derived relatives. The presented data confirm the notion of Metchnikovellidae as a less derived microsporidian group, and provide an additional stepping stone for reconstruction of an evolutionary transition from the early diverging parasitic fungi to derived Microsporidia.
Keywords: Microsporidia, Metchnikovellidae, phylogeny, phylogenomics, comparative genomics, genome evolution
Introduction
Microsporidia is a well-established group of widely distributed and highly specialized obligate intracellular parasites with close phylogenetic ties to fungi. Described members of this phylum, which currently comprises over 1,300 species, are predominantly host-specific parasites of invertebrates, particularly abundant in crustaceans and insects with a fraction of diversity occurring in vertebrates and alveolate protists (Vávra and Lukeš 2013). The broad range of their hosts includes honey bee and silkworm, and a few microsporidian species are recognized as opportunistic parasites of humans, making the group economically and medically relevant. The genome sequencing projects focusing on important pathogenic species of Microsporidia (Katinka et al. 2001; Akiyoshi et al. 2009; Cornman et al. 2009; Corradi et al. 2010; Heinz et al. 2012) revealed the remarkable extent of reductive evolution that shaped their genomes. Complete reliance of microsporidians on their hosts manifests itself in highly streamlined biology with significantly diminished gene complement compared with their fungal relatives (Katinka et al. 2001), accelerated rates of evolution (Thomarat et al. 2004), reduction of cellular organelles (Williams et al. 2002) and loss of core metabolic pathways (Keeling et al. 2010). The exceedingly derived nature of microsporidians had hindered attempts to firmly place them in the phylogenetic tree, and the details of their relationship to fungi have started to resolve only recently with phylogenomic studies uniting microsporidians with a novel fungal lineage Cryptomycota (James et al. 2013). The discovery of an early diverging microsporidian with a mitochondrial genome, Mitosporidium daphniae (Haag et al. 2014), has opened the possibility to explore the evolutionary changes that led to the emergence of highly derived Microsporidia from their cryptomycete relatives in greater detail. To further expand the database for comparative genomics of early diverging Microsporidia another promising candidate for a phylogenomic study is a group of putatively primitive microsporidians of the Metchnikovellidae family.
Metchnikovellidae, a distinct group of Microsporidia with only a few described species, are known exclusively as parasites of gregarines inhabiting the intestinal tract of marine annelids (Vivier 1975). Spores of metchnikovellids lack some of the main distinguishing ultrastructural features of microsporidians—the coiled polar filament and a stack of membranes termed the polaroplast, which form a complex of organelles for host cell invasion in Microsporidia (Xu and Weiss 2005). The polar filament in metchnikovellid spores is short and straight and is recognized as a rudimentary version of the microsporidian invasion apparatus, delivering sporoplasm to the host cell by eversion in a manner similar to typical microsporidia (Sokolova et al. 2014). Unusual for microsporidians, the life cycle of metchnikovellids lacks merogonial stages—spores are produced by sporogony, often endogenously inside cysts or spore sacs (Larsson 2000; Larsson and Køie 2006; Sokolova et al. 2013), the size and shape of which serve as a diagnostic trait for classification. The peculiarities of metchnikovellid spore structure and intracellular development have earned them a status of aberrant microsporidians with unclear relationship to the rest of the group (Sprague 1977). Although metchnikovellids have been known for over a hundred years (Caullery and Félix 1897) and are acknowledged for their potential significance to evolutionary studies (Vossbrinck and Debrunner-Vossbrinck 2005; Corradi and Keeling 2009), the group remains understudied, especially when it comes to molecular data. Like many noncultivable organisms, they present a challenge for direct application of sequencing technology with the paucity of available material.
In this study, we introduce genomic data of Amphiamblys sp., a representative of one of the three metchnikovellid genera. We obtained the partial genome of Amphiamblys sp. using next-generation sequencing and a whole genome amplification approach to tackle the issue of insufficient sample material. We find that, although the prepared sequencing libraries are heavily contaminated by sequences of primarily prokaryotic origin, it is possible to identify sequences of Amphiamblys sp. in the assembly fairly efficiently using metagenomic binning. We estimate the nonrepetitive fraction of the genome to be 90% complete, and perform genome annotation and preliminary comparative analysis. Our data reinforce the notion of Metchnikovellidae as a less derived microsporidian group and support its early divergence from the microsporidian lineage.
Materials and Methods
Biological Material
Amphiamblys
Cells of Ancora sagittata, the gregarine host for metchnikovellid Amphiamblys sp., were collected from the intestinal tract of polychaetes Capitella capitata, inhabiting the sublittoral area near the White Sea Biological Station (WSBS) of Lomonosov Moscow State University, Kandalaksha Gulf of the White Sea (66°33′13″N, 33°06′22″E). The collected gregarine trophozoites were rinsed in filtered seawater and inspected for infection under a light microscope. The infected cells observed in samples gathered in 2006 (WSBS2006) were immediately lysed following an alkaline digestion protocol (Floyd et al. 2002). The cells collected in 2011 (WSBS2011) were fixed with the RNAlater reagent (Life Technologies) for later extraction. DNA extraction for the fixed sample was performed with NucleoSpin Tissue kit (MACHEREY-NAGEL) using the manufacturers protocol.
Amphiacantha
The metchnikovellid-infected cells of gregarines Lecudina cf. elongata and Lecudina cf. longissima were collected in 2006 from the intestinal tract of polychaetes Lumbrineris cf. fragilis, inhabiting the sublittoral area of the Kandalaksha Gulf of the White Sea (66°31′20″N, 33°10′40″E) at depth of 25 m. The gregarine cells were rinsed in filtered seawater and observed under a light microscope. The cells of L. cf. elongata and L. cf. longissima were sorted into separate tubes and lysed following an alkaline digestion protocol (Floyd et al. 2002).
Unidentified Sequence p1_44
The ribosomal RNA of an unidentified organism that displays phylogenetic affinity to the metchnikovellid sequences was discovered in several enrichment cultures of freshwater algae collected in the Leningrad Oblast by S.A. Karpov. The cultures also contained filamentous fungi, amoeboid organisms and other eukaryotes; the host for the unidentified metchnikovellid was not established.
Small Subunit rDNA Sequencing
The small subunit rRNA genes were PCR amplified on a DNA Engine Dyad thermal cycler (Bio-Rad) using an Encyclo PCR kit (Evrogen) or HotTaq polymerase kit (Sileks), and the following pair of primers: 5′-GTATCTGGTTGATCCTGCCAGT-3′, 5′-GGAAACCTTGTTACGACTTTTA-3′. PCR products were electrophoretically separated in 1% agarose gel and purified using Gel Extraction & PCR Cleanup Kit (Cytokine). The gel extracted PCR products were either sequenced directly using the amplification primers on a 3730 DNA Analyzer with BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) or first cloned with an InsTAclone PCR Cloning Kit (Fermentas) and sequenced using the universal plasmid primers.
Total DNA Amplification and Sequencing
The DNA sample extracted from the infected cells of Ancora sagittata WSBS2006 was amplified with the REPLI-g Midi kit (QIAGEN) following the manufacturers protocol. Two amplification reactions were performed with an estimated 1 ng of starting material, which yielded 10 and 23 μg of DNA. DNA quantity assessments were performed with Qubit fluorometric quantification (Life Technologies). The amplified DNA samples were used to prepare two sequencing libraries for the Illumina platform. Paired-end libraries with estimated insert lengths of 333 bp (41 bp SD) and 424 bp (63 bp SD) were constructed using TruSeq library preparation protocol (Illumina) and sequenced on a HiSeq 2000 instrument. A total of 38M and 44M 100-bp paired end reads were obtained for the two libraries, respectively. The reads were adapter trimmed with Trimmomatic v0.30 in Paired End mode (Bolger et al. 2014), requiring a minimal length of 55 bp for each read to keep the pair.
Contig Assembly and Read Filtering
Paired-end reads from both libraries were assembled by SPAdes 3.0.0 in single-cell mode, designed to handle data resulting from multiple displacement amplification (Bankevich et al. 2012). Two rounds of assembling were performed, each employing iterative assembly with three k-mer values (21, 33, 55), read error correction and contig mismatch correction. In the first round, we used all reads from the two libraries to obtain a 62-Mb initial assembly, containing a mix of contigs originating from eukaryotic and prokaryotic sources. In the second round, we discarded reads that mapped on contigs identified as prokaryotic, reducing the size of assembly to 12 Mb. To identify sequences originating from prokaryotic sources, we extracted a set of MetaGene-predicted (Noguchi et al. 2006) open reading frames from each contig of the initial assembly and performed a similarity search with BLASTp (BLAST 2.2.27+) (Altschul et al. 1997) against the proteomes of 61 eukaryotic organisms (39 holomycots, 8 holozoans, 14 from other eukaryotic groups) and the prokaryotic RefSeq database (Pruitt et al. 2007), ruling the likely taxonomic domain of origin for each contig from hit bitscore values of its respective ORFs. A contig was classified as prokaryotic if more than half of its ORFs with significant hits (bitscore threshold = 50) had the highest bitscore value for hits in the prokaryotic RefSeq database. Likewise, contigs with more than half of ORFs having highest bitscore values in databases of eukaryotic organisms were classified as eukaryotic. Contigs with a cumulative length of predicted ORFs longer than 3,000 nucleotides were inspected for chimeric assembly artifacts. To filter out contaminating reads the read pairs were mapped to contigs with Bowtie 2.2.1 (Langmead and Salzberg 2012). Pairs that failed to align to contigs classified as prokaryotic with both of their reads were selected for the second round of assembling.
Amphiamblys sp. Genome Identification
Identification of candidate Amphiamblys sp. contigs in the initial assembly was assisted by contig clustering method utilizing relative synonymous codon usage (RSCU) values. The RSCU-based contig clustering was implemented using the following procedure: (1) concatenate all MetaGene-predicted (Noguchi et al. 2006) ORFs for each contig; (2) calculate RSCU distance matrix for the concatenates with GCUA 1.0 (McInerney 1998); and (3) construct a dendrogram for the obtained distance matrix using Neighbor-Joining method of the PHYLIP 3.695 package (Felsenstein 2005). Contigs with ORF concatenates shorter than 3,000 nucleotides were excluded from the RSCU analysis. The resulting dendrogram accounted for 2,885 contigs with cumulative length of 27 Mb, and was visualized with TopiaryExplorer (Pirrung et al. 2011). A set of 40 contigs with cumulative length of 1.3 Mb derived from the cluster of eukaryotic contigs were examined to ensure microsporidian affinity using BLASTp searches against the NCBI database and phylogenetic inference with a sample of predicted ORFs. These contigs were selected for training a composition-based classifier for metagenomic sequences, ClaMS (Pati et al. 2011), after manual inspection for cases of chimeric assembly artifacts. Scaffolds of the 12-Mb assembly were queried against the reference set by ClaMS with a de Bruijn chain (DBC) genomic signature (Heath and Pati 2007) and a k-mer of length 2. Pearson distance cutoff value of 0.1 was selected for binning sequences. Synonymous codon usage frequencies in the ORFs of reference contigs were calculated using Sequence Manipulation Suite (Stothard 2000), and genome assembly statistics were calculated by QUAST 2.3 (Gurevich et al. 2013).
Survey of repetitive elements in the assembly was performed with RepeatModeler 1.0.7 (Benson 1999; Bao and Eddy 2002; Price et al. 2005; Smit 2008–2015) for nucleotide sequences and BLASTp (BLAST 2.2.27+) (Altschul et al. 1997) for translated sequences of predicted genes. Repeat families discovered by RepeatModeler were aligned to sequences in the assembly with RepeatMasker 4.0.3 (Smit 2013–2015), and the distribution of repetitive sequences was inspected for each family. Repeats appearing consistently on scaffolds attributed to the genome of Amphiamblys sp. on the basis of genomic signature similarity were used as an indicator of Amphiamblys sp. genomic sequence for scaffolds outside the 0.1 Pearson distance binning threshold. Amphiamblys-specific repetitive elements found at the amino acid level were queried against the assembly by tBLASTn with an e-value cutoff of 1E-10.
Genome Annotation
Ab initio gene models were predicted independently using three gene prediction programs: GeneMarkS 4.6b (Besemer et al. 2001), GeneMark-ES 2.3c (Ter-Hovhannisyan et al. 2008) and Augustus 3.0.1 (Stanke and Waack 2003) trained on a gene set inferred with CEGMA 2.5 (Parra et al. 2007). Predictions were inspected using GenomeView v2450 (Abeel et al. 2012) with the assistance of BLASTx (BLAST 2.2.27+) (Altschul et al. 1997) search against the proteomes of several eukaryotic organisms. The introns introduced by CEGMA, Augustus or GeneMark-ES found no support in the search results or were refuted as erroneous. Consequently, GeneMarkS was selected for exclusive annotation of Amphiamblys sp. genomic sequences. Genes for transfer RNAs were predicted by tRNAscan-SE 1.3.1 (Lowe and Eddy 1997) and ARAGORN 1.2.36 (Laslett and Canback 2004). Repetitive sequences were discovered by RepeatModeler 1.0.7 (Benson 1999; Bao and Eddy 2002; Price et al. 2005; Smit 2008–2015) and MITE-Hunter v11-2011 (Han and Wessler 2010) programs. The analysis of intragenic repeats was assisted by the RADAR (Heger and Holm 2000) service of the EBI website (Goujon et al. 2010). Intergenic regions were examined for regulatory motifs using the MEME 4.9.1 program (Bailey et al. 2006). The sequence logos were generated using WebLogo 3.4 (Crooks et al. 2004). Analysis of microsporidian genome assembly and annotation statistics was performed by the Genome Annotation Generator 1.0 (Hall et al. 2014).
Estimation of genome completeness was performed for the predicted genes by BUSCO 1.1b (Simão et al. 2015) in the gene set assessment mode (-m OGS). The estimates with the 429 core eukaryotic orthologs give values in the range of 50–60% for completely sequenced microsporidian genomes and the partial genome of Amphiamblys sp. (supplementary table S1, Supplementary Material online). To adjust for the highly reduced gene complement of microsporidian genomes, we narrowed down the initial set of orthologs to 147 orthologs that were required to be present as either complete or fragmented and single-copy or duplicated in each of the following genomes: Edhazardia aedis USNM 41457, Encephalitozoon cuniculi GB-M1, Nematocida parisii ERTm1, Nosema ceranae BRL01, Vavraia culicis subsp. floridensis, Vittaforma corneae ATCC 50505, and Mitosporidium daphniae. The completeness estimate for Amphiamblys sp. 5.6 Mb genome using the microsporidia-specific set of universal orthologs evaluates roughly to 90%: 85 complete single-copy, 25 fragmented, 22 duplicated, and 15 missing.
Phylogenetic Analyses
Tree reconstructions for the alignment of small subunit rRNA gene sequences and the concatenated alignments of small and large subunit rRNA genes were performed by MrBayes 3.2.2 (Ronquist et al. 2012) using a GTR substitution model with eight categories of Gamma-distributed among site rate variation, calculation of proportion of invariable sites, and a covarion model. Four independent runs of 4 Metropolis-coupled Markov chains were sampled across 10 million generations and summarized with a 50% burn-in for both tree inferences.
The multigene phylogenetic analyses were conducted with a set of 303 genes that were selected by establishing orthology relations between genes from 38 representatives of the Holomycota lineage and 11 eukaryotic species comprising the outgroup. The proteomes were obtained from three resources: NCBI’s GenBank database (http://www.ncbi.nlm.nih.gov/genbank; last accessed September 27, 2016), JGI Genome Portal (http://genome.jgi.doe.gov/; last accessed September 27, 2016), and Broad Institute genome annotation projects (http://www.broadinstitute.org/scientific-community/data; last accessed September 27, 2016). The alignments were prepared with MAFFT v7.130b (Katoh and Standley 2013) and inspected manually using BioEdit 7.2.5 alignment editor (Hall 1999). A custom mask was designed for each alignment to specifically account for the divergent microsporidian sequences. The alignments were trimmed according to the mask and concatenated with SCaFoS 1.2.5 (Roure et al. 2007). The ML analysis for the concatenated dataset was performed with RAxML 8.0.0 (Stamatakis 2014) utilizing a rapid search procedure (-f a) under a GTR model with Gamma-distributed rate variation across sites; node support was evaluated with 100 bootstrap replicates. The Bayesian inference was performed with PhyloBayes MPI 1.5a (Lartillot et al. 2013) using the CAT-Poisson model and 4 discrete Gamma rate categories. Four Phylobayes chains were run for 10,000 cycles each and summarized with a 25% burn-in.
Individual gene trees (retrotransposon sequences, CMGC kinases, and malate/lactate dehydrogenases) were reconstructed using RAxML 8.0.0 (Stamatakis 2014) following the choice of best-fit model by ProtTest 3.2 (Darriba et al. 2011). The alignments were prepared using MAFFT v7.130b (Katoh and Standley 2013) and BioEdit 7.2.5 (Hall 1999) programs, and tree inferences were performed using a rapid search procedure (-f a) with node support evaluation in 1,000 bootstrap replicates. All trees were visualized using MEGA 5.2.2 (Tamura et al. 2011).
Functional Annotation
The initial assignment of Gene Ontology (GO) terms for predicted proteins was performed using the UniProtKB/Swiss-Prot database (UniProt Consortium 2015) and the third module of the Sma3s program (Muñoz-Mérida et al. 2014), modified to accept only reciprocal BLAST hits for annotation. The initial annotations were merged with GO assignments provided by the InterPro database (Mitchell et al. 2015) search using the InterProScan application (Jones et al. 2014) and the functionality of the Blast2GO 3.0 program (Conesa et al. 2005). To reduce the GO annotations to the most relevant terms a GO-Slim option of the Blast2GO program was used with a Yeast GO-Slim mapping. For comparative analysis of gene counts in each GO term the proteomes were annotated individually using an identical procedure. KEGG (Kanehisa and Goto 2000) orthology assignments and pathway mappings for proteomes were performed with the KEGG Automatic Annotation Server (Moriya et al. 2007) using best bi-directional hit method and the default bit score cutoff of 60. Pfam 27.0 database (Finn et al. 2014) searches were carried out by the HMMER 3.1b1 software (Eddy 2011) with the assistance of the publicly available pfam_scan.pl script.
Gain and Loss Analysis
To construct protein families for the gain/loss analysis, we selected 20 annotated opisthokont genomes including the metchnikovellid partial genome, and performed protein orthology clustering using the OrthoMCL v2.0.9 pipeline (van Dongen 2000; Li et al. 2003). BLAST (Altschul et al. 1997) similarity search for OrthoMCL was carried out with an e-value cutoff of 1E-05, and Markov clustering was performed with an inflation parameter of 1.5. The inflation parameter was selected using the F-measure (Paccanaro et al. 2006) on the basis of the ortholog dataset used for phylogenetic analyses (best F-measure = 0.932 with an inflation parameter of 1.5). The clustering returned 9,013 orthogoups that were shared by at least 2 genomes and 48,901 genome-specific orthogoups or single gene groups.
Evolution of protein families in the cryptomycote/microsporidian lineage was modelled using the OrthoMCL-generated orthology groups and the Dollo parsimony method of the Count software (Csűrös 2010). The tree topology for the analysis is based on the Bayesian inference result for the multigene dataset with branch lengths calculated by RAxML 8.0.0 (Stamatakis 2014) using the reduced version of the concatenated multigene alignment and the previously optimized GTR model. For GO enrichment analysis of the ancestral proteomes, we selected all orthogoups appearing in the microsporidian lineage (10,366 orthogroups) and performed GO assignments by selecting a representative sequence for each orthogroup and reproducing the functional annotation procedure. The sets of annotated orthogroups inferred to be present in the microsporidian ancestor before, at the time of, and after the divergence of metchnikovellids were compared by the Blast2GO 3.0 program (Conesa et al. 2005) using two-sided Fisher’s Exact test with a P value filter of 0.05; the resulting tables were reduced to the most specific GO terms.
Results and Discussion
Metchnikovellid Ribosomal RNA Sequencing and Analysis
Starting off the study several samples of metchnikovellid-infected gregarine cells, extracted from the intestinal tract of White Sea polychaetes, were examined with light-microscopy and characterized using PCR-amplified small subunit rDNA sequences. We obtained metchnikovellid rDNA sequences from two samples of infected gregarines Ancora sagittata, collected from the polychaetes Capitella capitata. Two sequences (one per sample) display 80% identity and were both attributed to genus Amphiamblys based on host specificity (Vivier 1975; Rotari et al. 2015). Two more metchnikovellid sequences were obtained from gregarines Lecudina cf. elongata and Lecudina cf. longissima, parasitizing Lumbrineris cf. fragilis, and were attributed to genus Amphiacantha. The small subunit rDNA sequences of Amphiacantha spp. have 73% identity and share from 47% to 55% identity with the sequences of Amphiamblys spp. Consistent with the expected phylogenetic position of Metchnikovellidae, the obtained rDNA sequences form a strongly supported monophyletic group that branches near the base of the microsporidian tree preceded by the divergence of Mitosporidium daphniae (fig. 1 and supplementary fig. S1, Supplementary Material online). Metchnikovellid rDNA sequences are highly derived and have the distinctive features of microsporidian ribosomes, conspicuous for reduction of several eukaryotic core structure helices (Wuyts et al. 2001). The sequence linking the conserved 5.8S and 23S rRNA gene regions in Amphiamblys sp. is short compared with the majority of its eukaryotic counterparts and only around 60 bases longer than the corresponding region in other microsporidians, suggesting that metchnikovellids share the characteristic 5.8S/23S gene fusion seen in Microsporidia (Vossbrinck and Woese 1986).
Fig. 1.—
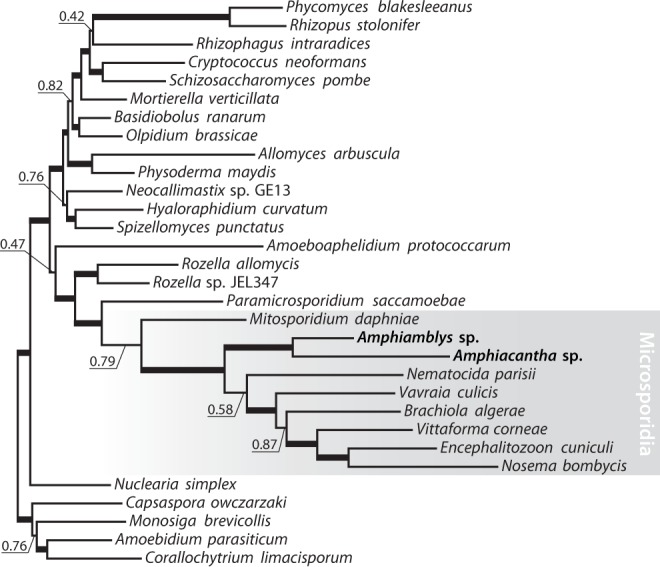
Phylogeny of the fungal lineage based on rDNA sequences. The tree was reconstructed from concatenated alignments of small and large subunits of ribosomal RNA genes using the Bayesian inference approach. Nodes with posterior probability ≥0.95 are marked with thick lines and their support values are omitted. The branches of the microsporidian subtree following the divergence of Mitosporidium are shortened by a factor of 10 relative to the rest of the tree. Amphiacantha sp. is represented only by the small subunit rRNA gene sequence in the concatenated dataset.
Assembly and Identification of Amphiamblys sp. Genomic Sequences
To investigate genomic data, the DNA sample of Ancora sagittata WSBS2006 containing metchnikovellid identified as Amphiamblys sp. was prepared for sequencing by performing total DNA amplification. The amplified sample was sequenced on the Illumina platform and assembled into 67,251 contigs with cumulative length of 62 megabase pairs (Mb). This initial assembly exceeds the range of microsporidian genome sizes and in advance was suspect of being contaminated by sequences of nonmicrosporidian origin. A dominant fraction of the assembly are sequences from prokaryotic sources, which contribute >70% of all reads in the sequenced libraries, by our estimates. By filtering out prokaryotic contamination from the read data the size of the assembly was reduced to 12 Mb in 13,085 contigs.
To discriminate sequences of Amphiamblys sp. from the rest of the assembly, we applied a two-step metagenomic binning procedure limited to the organism of interest (supplementary fig. S2, Supplementary Material online). First, we identified a set of candidate contigs in the initial assembly using relative synonymous codon usage (RSCU) distances (McInerney 1998) for contig clustering. Second, we utilized this set of contigs to train a composition-based classifier for metagenomic sequences, ClaMS (Pati et al. 2011), and interrogated all sequences in the 12 Mb assembly on their conformity to the genomic signature of the training set. The rationale behind using RSCU for clustering stems from the observation that genomes from different species exhibit their own characteristic patterns of synonymous codon usage (Grantham et al. 1980), in some cases granting enough specificity to accurately classify coding sequences (Sandberg et al. 2003). The RSCU-based clustering of sufficiently long contigs in the initial assembly produced a prominent cluster comprising 258 contigs with a cumulative length of 2.8 Mb, the majority of which were recognized as eukaryotic after a similarity search (supplementary fig. S3A, Supplementary Material online). This cluster is characterized by a distinctive pattern of synonymous codon usage (supplementary fig. S4, Supplementary Material online) that exists in an otherwise well-balanced nucleotide composition environment, with the G + C content of contigs not deviating strongly from the average 52% for the whole cluster (supplementary fig. S3B, Supplementary Material online). Preliminary phylogenetic analysis with a few conserved genes found among the ORFs in the cluster confirmed that their source was indeed a novel microsporidian genome, and a set of 40 contigs totaling 1.3 Mb were selected to function as a reference for constructing a genomic signature of Amphiamblys sp.
Following the metagenomic binning, approximately a quarter of the 12 Mb assembly (3.4 Mb) fell within 0.1 Pearson distance from the reference set, indicating strong correlation with the constructed genomic signature (Heath and Pati 2007). This part of the assembly includes the majority of long scaffolds produced by the assembler (fig. 2) and is predicted to encode approximately 2,400 proteins. Another 2,000 genes were predicted using the same GeneMark model for sequences outside the 0.1 Pearson distance threshold. The sequences outside the threshold are on an average much shorter than the ones within the threshold and are intrinsically more difficult to reliably classify using the statistical properties of their composition.
Fig. 2.—
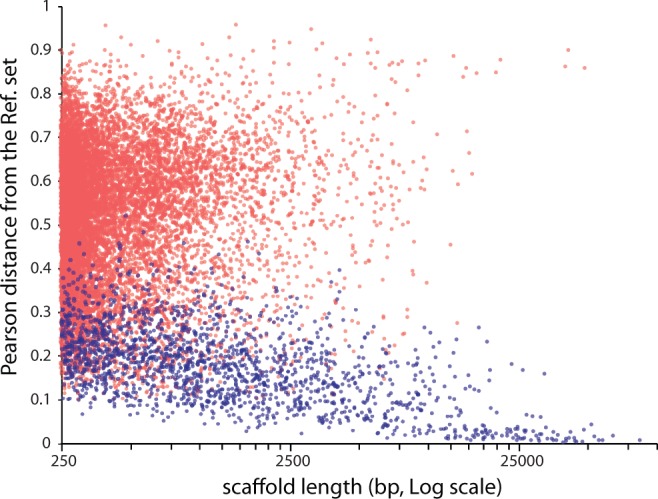
Scatter plot of Pearson distance against sequence length for scaffolds in the 12-Mb assembly. Pearson distance from the reference set of Amphiamblys sp. contigs was calculated using de Bruijn chain genomic signature (Heath and Pati 2007). Blue dots correspond to sequences attributed to the genome of Amphiamblys sp. using genomic signature similarity and repeat finding approaches; red dots represent the rest of sequences in the assembly.
To complement the diminished discriminatory power of the classification method for shorter sequences, we employed an ancillary criterion for identification of Amphiamblys sp. genomic sequences by admitting to its genome any sequence containing repetitive elements discovered in the 3.4-Mb assembly. The repeats identified as Amphiamblys-specific were found in scaffolds well beyond 0.1 Pearson distance from the reference set (fig. 2). A few types of repeats represented by specific ORFs are organized as both interspersed and tandem versions. The latter are a common occurrence in longer scaffolds outside the 0.1 distance threshold, and are presumably responsible for dissociating these sequences from the genomic signature of the less idiosyncratic sequences in the reference set. Incorporating all scaffolds carrying these repeats into the set of sequences identified with Amphiamblys sp. puts the size of its partial genome at 5.6 Mb in 1,743 scaffolds and the total count of gene predictions at 3,647 ( table 1 and supplementary fig. S2, Supplementary Material online).
Table 1.
Microsporidian Genome Assembly and Annotation Statistics
| Amphiamblys WSBS2006 (all | nr)a | E. cuniculi GB-M1 | V. corneae ATCC 50505 | N. parisii ERTm1 | M. daphniae | V. culicis subsp. floridensis | T. hominis | N. apis BRL 01 | A. algerae PRA339 | E. aedis USNM 41457 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assembly size (Mb) | 5.6 | 2.5 | 3.2 | 4.1 | 5.6 | 6.1 | 8.5 | 8.6 | 12.2 | 51.3 | |
| GC (%) | 50.4 | 47.3 | 36.5 | 34.4 | 43.0 | 39.7 | 34.1 | 18.8 | 23.6 | 22.8 | |
| Protein-coding genes | 3,647 | 2,529 | 1,996 | 2,239 | 2,661 | 3,330 | 2,773 | 3,212 | 2,764 | 3,598 | 4,190 |
| Gene density (genes/kb) | 0.66 | 0.46 | 0.81 | 0.71 | 0.67 | 0.59 | 0.46 | 0.38 | 0.32 | 0.30 | 0.08 |
| Protein-coding (%) | 63.8 | 46.0 | 86.3 | 66.9 | 72.1 | 68.0 | 46.9 | 33.1 | 26.6 | 23.7 | 8.7 |
| Mean transcript length (bp) | 983 | 1,022 | 1,068 | 945 | 1,142 | 1,252 | 1,161 | 865 | 958 | 814 | 1,177 |
| Mean intergenic distance (bp) | 318 | 275 | 160 | 376 | 355 | 368 | 945 | 1,506 | 1,715 | 2,230 | 9,889 |
| overlapping genes | 325 | 281 | 283 | 202 | 385 | 92 | 298 | 333 | 132 | 197 | 193 |
The protein-coding gene statistics for Amphiamblys sp. were calculated in two variants: using all predicted genes, which includes the repetitive ORFs, or only the non-redundant set of genes
The 5.6 Mb of assembled sequences attributed to the genome of Amphiamblys sp. using genomic signature similarity and repeat finding account for 90% of the conserved microsporidian gene set (supplementary table S1, Supplementary Material online). Another 4% of genes from the set were found in the assembly but were failed to be included in the partial genome using either of the approaches and may represent predictions originating from contaminating sequences or false negatives of the genome identification method. Using BLASTp searches against the NCBI database, we have queried the predicted genes to reveal any potential cases of contamination or horizontal gene transfers (HGTs). Several genes in the partial genome were found to produce most significant hits against the prokaryotic proteins. Two genes match candidate HGTs of bacterial origin previously identified in other microsporidians: the bacterial type manganese superoxide dismutase (Xiang et al. 2010) and asparagine synthetase A (Heinz et al. 2012). Another three genes produce hits against uncharacterized prokaryotic proteins, and five more correspond to ORFs encoding full-length or partial SecA translocase-like proteins. Homologs of the typically bacterial SecA genes are found in a patchwork of diverse eukaryotes, including green plants, ciliates, trypanosomatids, insects and flatworms, but are uncommon in the fungal lineage. Finally, two uncharacterized genes in Amphiamblys sp. produce hits with an abnormally high identity (80% at the amino acid level) to the assembly of Capitella teleta, a close relative of the polychaete host for the hyperparasitic metchnikovellid. Although HGTs involving both prokaryotic and eukaryotic lineages appear to be commonplace in microsporidians (Corradi 2015), the metagenomic nature of the source material suggests contamination as the more likely explanation for the origin of these genes. Contigs containing most of the detected stray genes are relatively short and presently do not allow us to completely rule out misassembly errors or limitations of the contig classification method.
Amphiamblys sp. Genome Organization
The partial genome of Amphiamblys sp. shows large variation in size depending on the stringency of the genome identification method, with the repeat finding approach adding another 2.2 Mb of sequence to the 3.4 Mb identified using genomic signature similarity. Accordingly, a large fraction of the 5.6-Mb partial genome are repetitive sequences, which also account for 30% of the predicted 3,647 protein coding genes, leaving only 2,529 genes in the putative nonrepetitive set (table 1). Many repetitive ORFs originate from damaged copies of repeats and are frequently found as fragments of a single copy split by frameshift errors, further inflating the total gene count. We conclude that the genome of Amphiamblys sp. WSBS2006 is likely in the size range of 5–7 Mb and encodes around 2,800 nonrepetitive genes, with a more accurate estimation complicated by the prevalence of repetitive elements and uneven coverage resulting from the DNA amplification.
High incidence of repetitive sequences in the assembly is contrasted by genome compaction evident in the nonrepetitive regions. The genome compaction in Amphiamblys sp. is not as drastic as seen in Encephalitozoon spp. and their close relatives (Pombert et al. 2015), but puts it among the more gene dense microsporidian genomes (table 1). The mean intergenic distance for the nonrepetitive genes in the partial genome is 275 bp, more than half of intergenic regions are smaller than 100 bp and at least 140 gene pairs are predicted to overlap. The predictions cover 64% of the assembly and the average gene density is 0.66 genes/Kb in the 5.6-Mb partial genome. The 3.4-Mb partial genome, which has a lower proportion of repetitive sequence and larger average sequence length, has 74% coding sequence and an average gene density of 0.71 genes/Kb.
The genes in Amphiamblys sp. appear to be completely devoid of spliceosomal introns. This observation is supported by extensive reduction of the splicing machinery and lack of essential components of snRNP complexes. Notably, Amphiamblys sp. is missing genes for Sf3b1 and Prp8, which are invariably present in microsporidians with active splicing (Desjardins et al. 2015). The complete loss of spliceosomal introns has been reported previously in several microsporidians (Keeling et al. 2010; Cuomo et al. 2012; Desjardins et al. 2015) and is presumed to have occurred independently in multiple lineages. If confirmed by a complete genomic sequence, the absence of introns in an early diverging metchnikovellid would extend the list of these intronless lineages.
Analysis of intergenic regions in Amphiamblys sp. confirmed the presence of a microsporidia-specific triple-C motif associated with gene promoter regions (Cornman et al. 2009). The motif was identified upstream of 463 genes in the vicinity of the transcription start (supplementary fig. S5, Supplementary Material online). Variants of the triple-C motif were detected previously in diverse microsporidians (Peyretaillade et al. 2009; Cuomo et al. 2012; Heinz et al. 2012), and conservation of this feature in a metchnikovellid attests that it was established early in microsporidian evolution.
A number of intergenic regions in Amphiamblys sp. were found to carry a repetitive sequence reminiscent of a miniature inverted-repeat transposable element (MITE). The sequence consists of an imperfect 54-bp inverted repeat flanking the central region that varies in length from 15 to 180 bp (supplementary fig. S6, Supplementary Material online). We found no discernable target site duplication sequence characteristic of canonical MITEs, including the described microsporidian elements (He et al. 2015). No recognizable DNA transposons or transposase sequences, which could be implicated in MITE proliferation, were discovered in the partial genome. However, this negative result is inconclusive considering that the genomic sequence is incomplete, and that the applied genome identification method might be biased against sequences of transposable elements, as their compositional characteristics tend to deviate from the genomic norm (Jia and Xue 2009).
Repetitive Elements in the Genome of Amphiamblys sp
Survey of repetitive sequences revealed that a large fraction of ORFs in the assembly corresponds to either full or fragmentary repeats of roughly 4 different types of elements. One type of repetitive sequences are reverse transcriptase encoding ORFs, which can be further classified into LTR elements of Ty3/Gypsy family and non-LTR LINEs. Retrotransposons account for over 200 ORFs in the partial genome, mostly as pseudogene fragments, with a few putatively intact copies. The phylogeny of non-LTR elements supports close relationship of metchnikovellid sequences with sequences from other microsporidians (supplementary fig. S7, Supplementary Material online), reinforcing the suggested common ancestry of these elements in microsporidians (Heinz et al. 2012). The Ty3/Gypsy family retrotransposons and related integrase genes found in the metchnikovellid also appear in other microsporidians, but their phylogenetic relationship is less robust and more difficult to interpret (supplementary fig. S8, Supplementary Material online). The other three types of repeat elements in the genome of Amphiamblys sp. correspond to ORFs that show no similarity to any known mobile element or gene product in the NCBI’s nonredundant database. Each type of Amphiamblys-specific repeat element represents a novel protein family that shares no evident sequence similarity with other repeat types. Amphiamblys-specific repeat elements together account for more than 800 ORFs in the partial genome. We found no regularities in the organization of sequences flanking these ORFs that would be indicative of transposon activity, albeit two types of repetitive ORFs were found to be frequently organized in clusters of tandemly repeated sequences.
A peculiar feature of one of the repeat elements in Amphiamblys sp. appears to recapitulate the evolution of tandem repeats at a smaller scale. The ORF characterizing the repeat element is itself partly composed of a repeated unit encoding a 52–53 amino acid domain that is present in several consecutive copies in each ORF (supplementary fig. S9, Supplementary Material online). The number of domain copies in an ORF is typically close to 7, but varies even between repeats comprising the same tandem cluster. The tandem arrangement and repeat length variation point to a role of recombination in the dynamics of these repeat arrays. Presumably, the process generating this diversity in tandem clusters may also be responsible for the genome-wide distribution of repetitive ORFs.
In addition to these highly prolific ORFs, the survey detected an unusually expanded family of kinases. The family shows signs of aberrant amplification similar to Amphiamblys-specific repetitive elements (supplementary fig. S10, Supplementary Material online). The amplified kinase genes are present in over 40 ORFs and are also frequently found in tandem arrangements. They form a monophyletic group in the gene tree, and are likely to have originated from a member of CMGC group kinases. The amplified family kinases can be found in contigs up to 20 kb but are primarily located at the contig termini or in smaller contigs, which is common for repetitive sequences.
Functional Annotation and Comparative Analyses
To verify the phylogenetic position of Metchnikovellidae obtained with rDNA sequences (fig. 1), we performed reconstructions using a multigene dataset. The multigene trees recover a monophyletic relationship between Microsporidia and Cryptomycota (Rozellomycota), placing the group sister to the rest of the fungal clade, and confirm early divergence of metchinkovellids within Microsporidia: maximum likelihood and Bayesian inference analyses place Amphiamblys sp. as the first branch of the microsporidian lineage following the divergence of a mitochondriate microsporidian Mitosporidium daphniae (fig. 3 and supplementary fig. S11, Supplementary Material online). In effect, Metchnikovellidae break up the long branch that separates highly derived microsporidians with a reduced organelle, mitosome, from their less derived mitochondriate relatives. The metchinkovellid genome holds evidence for the existence of a mitochondrial remnant: it encodes components of a mitochondrial translocase complex, present in a reduced but functional form in microsporidians (Waller et al. 2009), and mitochondrial iron–sulphur cluster assembly proteins, which were proposed to fulfil key function of microsporidian mitosomes (Goldberg et al. 2008). We found no evidence for mitochondrial genetic information processing in the partial genome, and lack of components for a functional tricarboxylic acid cycle or oxidative phosphorylation pathway. Therefore, we expect the organelle in Amphiamblys sp. to be functionally equivalent to a microsporidian mitosome, in contrast to the organelle in Mitosporidium (Haag et al. 2014).
Fig. 3.—

Phylogenetic analyses with a concatenated multigene dataset (300 genes). The tree topology, branch lengths and posterior probabilities were obtained using Bayesian inference (Phylobayes, CAT + Γ4); the bootstrap support values were calculated using maximum likelihood (RAxML, GTR + G) from 100 replicates. Support indexes for nodes with 1.00 posterior probability and 100% bootstrap support are omitted and the corresponding branches are marked with thick lines. See also supplementary figure S11, Supplementary Material online.
Functional annotation of the partial genome reveals a gene content broadly similar to that of highly derived microsporidians (fig. 4). The gene ontology terms in the metchnikovellid display typically microsporidian or intermediate gene counts for each major term, consistent with progressive loss of core function genes along the microsporidian stem (supplementary fig. S12, Supplementary Material online). The metabolic capacities are reduced to a level characteristic of derived microsporidians (Katinka et al. 2001), suggesting that they rely on the uptake of metabolites from their protozoan host in much the same way as the microsporidians parasitizing animals. The metchnikovellid retains the common core of microsporidian carbon metabolism, namely glycolysis, pentose phosphate pathway and trehalose biosynthesis (supplementary fig. S13, Supplementary Material online), but lacks the capacity for de novo biosynthesis of nucleotides, and is missing almost all enzymes for amino acid biosynthesis and fatty acid metabolism. Peculiarly, the genome retains two enzymes of the tricarboxylic acid cycle—malate dehydrogenase and citrate synthase. The predicted genes lack an evident targeting peptide sequence, and given the absence of an enzyme complement necessary to complete the TCA cycle, their function is unlikely to involve mitosome metabolism.
Fig. 4.—
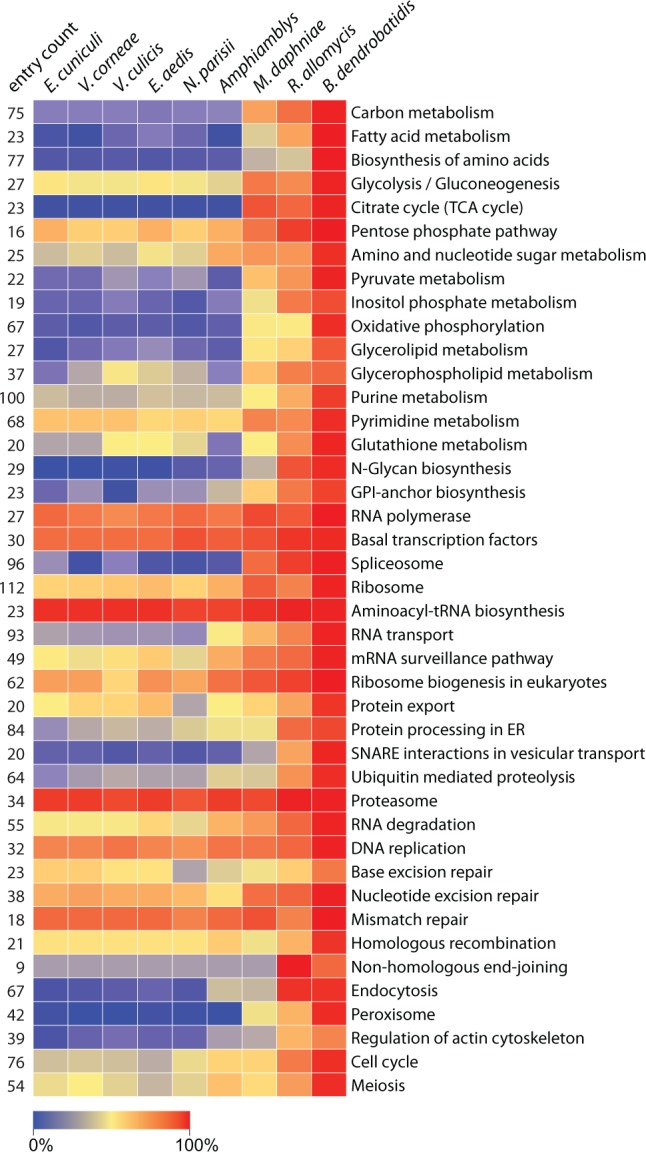
Heatmap illustrating conservation of pathways in microsporidian genomes. Reference pathways were selected from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways collection. The entry count is the total number of nonredundant pathway elements found in the surveyed genomes. Cell color represents the percentage of these entries present in the corresponding genome.
A possible role for the predicted malate dehydrogenase (MDH) is suggested by its primary structure. Although the gene clusters with a group of mitochondrial MDHs, an active site arginine residue crucial for substrate specificity is replaced with tyrosine in Amphiamblys sp. (supplementary fig. S14, Supplementary Material online). Substitutions at this site in the enzyme family were shown to effectively shift specificity between oxaloacetate/malate and pyruvate/lactate substrates (Wilks et al. 1988). We hypothesize that the MDH enzyme in Amphiamblys sp. may instead function as a lactate dehydrogenase, similarly to MDH-derived lactate dehydrogenases of trichomonads (Wu et al. 1999) and apicomplexans (Boucher et al. 2014). Conversion of pyruvate to lactate may then constitute the concluding step of anaerobic energy metabolism in the metchnikovellid by balancing the reducing potential of glycolysis. From the partial genome data the fate of glycolysis by-products is otherwise perplexing, since we have not found any components of the pyruvate dehydrogenase complex, glycerol-3-phosphate shuttle, or an alternative oxidase (supplementary fig. S13, Supplementary Material online), which were proposed to keep the microsporidian energy metabolism sustainable (Dolgikh et al. 2009; Williams et al. 2010).
One of the defining features of microsporidian metabolism is the ability to import ATP directly from the host using a family of horizontally acquired nucleotide transport proteins (Tsaousis et al. 2008; Heinz et al. 2014). The presence of the transporter family is expected in metchnikovellids: the family is shared by all derived microsporidians and is also found in their cryptomycote relative, Rozella allomycis (James et al. 2013). However, we failed to find any genes similar to the microsporidian nucleotide transporters in the partial genome or the initial assembly. We cannot claim that the family is absent from the complete genome, but if true it may give further evidence to the transient role of these transporters in the early stages of microsporidian evolution, suggested by their absence from the genome of Mitosporidium (Haag et al. 2014). Interestingly, the partial genome of Amphiamblys sp. contains a member of the mitochondrial carrier family (MCF). The MCF gene is similar to inorganic phosphate carriers from Saccharomyces, and does not group with the only other MCF member in derived microsporidians, the ADP/ATP carrier from Antonospora locustae (Williams et al. 2008) (supplementary fig. S15, Supplementary Material online). It is compelling to suggest that the MCF gene in Amphiamblys sp. has evolved for nucleotide transport, in parallel to the gene in Antonospora, and may have a role in supporting the mitosomal metabolism.
It was noted in previous studies that the DNA repair and recombination complexes have experienced degeneration in microsporidians (Gill and Fast 2007; Haag et al. 2014). We found that while many of the repair and recombination pathways in Amphiamblys sp. have lost some components and display similar gene counts next to other microsporidians, the nucleotide excision repair (NER) pathway in the metchnikovellid appears to be missing in its entirety (supplementary table S2, Supplementary Material online). The NER performs recognition and excision of DNA lesions, followed by DNA synthesis to restore the undamaged molecule, and is carried out by orthologs of the mammalian XPA, XPC, XPG, XPF, ERCC1 proteins and the transcription factor IIH (TFIIH) complex. The participating NER-specific proteins are almost universally conserved in fungi and microsporidians, but none of them are found in the partial genome. Furthermore, the XPD subunit of TFIIH whose major function is associated with NER is highly divergent in Amphiamblys sp. in contrast to its microsporidian orthologs. The presence of a divergent XPD gene may be a result of its requirement as a scaffolding protein in the normal transcription initiation combined with the loss of helicase activity, which is essential only for repair (Kuper et al. 2014). It is unclear whether the loss of NER components in the metchnikovellid is complemented by another repair mechanism or whether it is able to forego NER entirely, but this example further illustrates the plasticity of reductive processes in the microsporidian lineage.
Protein Family Gain and Loss Analysis
To get a systematic view on the evolution of the ancestral microsporidian proteome, we plotted gain and loss of protein families on the microsporidian phylogeny. In agreement with earlier reports of massive gene loss at the base of Microsporidia (Heinz et al. 2012; Nakjang et al. 2013), the reduction of ancestral proteome dominates family dynamics up to the radiation of highly derived microsporidians (fig. 5). Very few families specific to the microsporidian lineage appear to be shared by either Amphiamblys or Mitosporidium, resulting in a meager number of gains preceding their divergence. Notably, none of the previously identified microsporidian-specific core protein families (Nakjang et al. 2013) are found in Amphiamblys sp. The enrichment analysis shows at least two major transitions in the functional complement of the ancestral proteome, one marked by the efflux of genes associated with mitochondria and respiration in the common ancestor of metchnikovellids and derived microsporidians (supplementary table S3A, Supplementary Material online), and the other by a prominent reduction of endocytic components in the microsporidian stem lineage following the divergence of metchnikovellids (supplementary table S3B, Supplementary Material online). The latter transition is noteworthy, as metchnikovellids may consequently represent an intermediate step in the evolution of an avesicular trans-Golgi network seen in microsporidians (Beznoussenko et al. 2007). Specifically, the genome of Amphiamblys sp. encodes a number of proteins required for clathrin vesicle formation that were lost by derived microsporidians, including clathrin heavy chain, Sla2/Hip1R and Las17/WAS homologs, Epsin and Sla1 homology domain containing proteins, and several subunits of the Arp2/3 complex. The lack of corresponding protein domains (PF13838, PF00637, PF07651, PF01608, PF00568, PF02205, PF00786, PF03983, PF01417, PF04045, PF04062, PF05856) in derived microsporidians is further confirmed by a more sensitive Pfam search (supplementary table S4, Supplementary Material online). This indicates that the molecular machinery for clathrin vesicle-mediated transport might be conserved in metchnikovellids, which would constitute one of the key distinctions from their highly derived relatives.
Fig. 5.—

Gain and loss of protein families in the evolution of the microsporidian lineage. The gains (green) and losses (red) of protein families were calculated using the Dollo parsimony principle on the basis of OrthoMCL clustered orthologous groups. The number 5,541 is the estimated count of ancestral protein families at the Fungi/Cryptomycota divergence, i.e., the number of orthologous groups conserved in at least one member of the fungal lineage and one member of the cryptomycote + microsporidian lineage, or at least one member of any of these two lineages and the holozoan lineage. The Venn diagram depicts the number of orthologous groups shared by the corresponding lineages and groups exclusive to Mitosporidium or Amphiamblys; the taxon Microsporidia (5 genomes) in the diagram does not include the genomes of Mitosporidium and Amphiamblys, while the taxon Fungi includes the genome of Rozella (11 genomes total). The discrepancy between the number of gains for Mitosporidium and Amphiamblys in the tree and the number of unique orthologous groups in the Venn diagram are due to groups shared with holozoans Capsaspora and Monosiga, which are not featured in the Venn diagram.
Conclusions
With phylogenomic analysis of Amphiamblys sp., we confirm that the unusual microsporidian group Metchnikovellidae occupies basal position in the tree, branching off the microsporidian stem after the divergence of Mitosporidium. The early branching position of metchnikovellids allows us to refine the timeline of genomic changes associated with the emergence of the microsporidian lineage. The gene-dense but repetitive genome of Amphiamblys sp. is generally consistent with the idea that major gene loss and intron loss occurred early in microsporidian evolution. The comparative analyses with the partial genome show early convergence of microsporidian core metabolism to glycolysis, pentose phosphate and trehalose biosynthesis pathways, along with the expense of mitochondrial respiration and amino acid biosynthesis pathways. The presented results also highlight several features that separate the metchnikovellid from its highly derived relatives, such as retention of two TCA cycle enzymes, conservation of clathrin vesicle transport machinery, loss of nucleotide excision repair components, in all demonstrating nonuniformity of reductive processes in Microsporidia. The metchnikovellids are therefore instrumental in reconstructing the ancestral microsporidian proteome, but the case may yet be intractable, as it is unclear to what extent the perceived shared losses in microsporidian lineages are a product of parallel evolution rather than ancestral traits. Hopefully, further genomic data from early microsporidian lineages and cryptomycote microsporidia-like lineages (Corsaro et al. 2014, 2016) will provide the necessary basis to differentiate between these evolutionary modes and accurately reconstruct the early evolution of Microsporidia.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank the staff of the White Sea Biological Station of Lomonosov Moscow State University for assistance with collecting samples of polychaete hosts; S.A. Karpov and A.A. Kobseva for providing fresh water enrichment cultures containing unidentified species of Metchnikovellidae; M.D. Logacheva and A.A. Penin, Laboratory of Evolutionary Genomics of Lomonosov Moscow State University for genome sequencing; V.A. Yudina for ribosomal rRNA gene sequencing. The material collection, sequencing and rRNA analyses were supported by the 15-34-20065 and 15-29-02601 grants from the Russian Foundation for Basic Research and the MK-7436.2015.4 grant from the Council for Grants of the President of the Russian Federation. The phylogenetic and comparative genomic analyses were supported by the Russian Scientific Foundation grant 14-50-00029.
Literature Cited
- Abeel T, Van Parys T, Saeys Y, Galagan J, Van de Peer Y. 2012. GenomeView: a next-generation genome browser. Nucleic Acids Res. 40:e12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi DE, et al. 2009. Genomic survey of the non-cultivatable opportunistic human pathogen, Enterocytozoon bieneusi. PLoS Pathog. 5:e1000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Williams N, Misleh C, Li WW. 2006. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34:W369–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Eddy SR. 2002. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12:1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beznoussenko GV, et al. 2007. Analogs of the Golgi complex in microsporidia: structure and avesicular mechanisms of function. J Cell Sci. 120:1288–1298. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher JI, Jacobowitz JR, Beckett BC, Classen S, Theobald DL. 2014. An atomic-resolution view of neofunctionalization in the evolution of apicomplexan lactate dehydrogenases. eLife 3:e02304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caullery M, Félix M. 1897. Sur un type nouveau (Metchnikovella n.g.) d’organismes parasites des Grégarines. C R Séances Soc Biol Fil. 49:960–962. [Google Scholar]
- Conesa A, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. [DOI] [PubMed] [Google Scholar]
- Cornman RS, et al. 2009. Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathog. 5:e1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi N. 2015. Microsporidia: eukaryotic intracellular parasites shaped by gene loss and horizontal gene transfers. Annu Rev Microbiol. 69:167–183. [DOI] [PubMed] [Google Scholar]
- Corradi N, Keeling PJ. 2009. Microsporidia: a journey through radical taxonomical revisions. Fungal Biol Rev. 23:1–8. [Google Scholar]
- Corradi N, Pombert JF, Farinelli L, Didier ES, Keeling PJ. 2010. The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat Commun. 1:77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsaro D, et al. 2014. Microsporidia-like parasites of amoebae belong to the early fungal lineage Rozellomycota. Parasitol Res. 113:1909–1918. [DOI] [PubMed] [Google Scholar]
- Corsaro D, et al. 2016. Molecular identification of Nucleophaga terricolae sp. nov. (Rozellomycota), and new insights on the origin of the Microsporidia. Parasitol Res. 115:3003–3011. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csűrös M. 2010. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26:1910–1912. [DOI] [PubMed] [Google Scholar]
- Cuomo CA, et al. 2012. Microsporidian genome analysis reveals evolutionary strategies for obligate intracellular growth. Genome Res. 22:2478–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins CA, et al. 2015. Contrasting host-pathogen interactions and genome evolution in two generalist and specialist microsporidian pathogens of mosquitoes. Nat Commun. 6:7121.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgikh VV, et al. 2009. Heterologous expression of pyruvate dehydrogenase E1 subunits of the microsporidium Paranosema (Antonospora) locustae and immunolocalization of the mitochondrial protein in amitochondrial cells. FEMS Microbiol Lett. 293:285–291. [DOI] [PubMed] [Google Scholar]
- Eddy SR. 2011. Accelerated Profile HMM Searches. PLoS Comput Biol. 7:e1002195.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 2005. PHYLIP (Phylogeny Inference Package) version 3.6. Seattle: Department of Genome Sciences, University of Washington; Available from: http://evolution.genetics.washington.edu/phylip.html. [Google Scholar]
- Finn RD, et al. 2014. Pfam: the protein families database. Nucleic Acids Res. 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd R, Abebe E, Papert A, Blaxter M. 2002. Molecular barcodes for soil nematode identification. Mol Ecol. 11:839–850. [DOI] [PubMed] [Google Scholar]
- Gill EE, Fast NM. 2007. Stripped-down DNA repair in a highly reduced parasite. BMC Mol Biol. 8:24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AV, et al. 2008. Localization and functionality of microsporidian iron-sulphur cluster assembly proteins. Nature 452:624–628. [DOI] [PubMed] [Google Scholar]
- Goujon M, et al. 2010. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38:W695–W699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Mercier R, Pavé A. 1980. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 8:r49–r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag KL, et al. 2014. Evolution of a morphological novelty occurred before genome compaction in a lineage of extreme parasites. Proc Natl Acad Sci U S A. 111:15480–15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B, DeRego T, Geib S. 2014. GAG: the Genome Annotation Generator (Version 1.0). Available from: http://genomeannotation.github.io/GAG. [DOI] [PMC free article] [PubMed]
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98. [Google Scholar]
- Han Y, Wessler SR. 2010. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38:e199.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Ma Z, Dang X, Xu J, Zhou Z. 2015. Identification, Diversity and Evolution of MITEs in the Genomes of Microsporidian Nosema Parasites. PLoS One 10:e0123170.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath LS, Pati A. 2007. Genomic signatures in De Bruijn chains In: Giancarlo R, Hannenhalli S, editors. Algorithms in Bioinformatics: 7th International Workshop, WABI 2007, Philadelphia, PA, USA, September 8–9, 2007. Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; p. 216–227. [Google Scholar]
- Heger A, Holm L. 2000. Rapid automatic detection and alignment of repeats in protein sequences. Proteins 41:224–237. [DOI] [PubMed] [Google Scholar]
- Heinz E, et al. 2012. The genome of the obligate intracellular parasite Trachipleistophora hominis: new insights into microsporidian genome dynamics and reductive evolution. PLoS Pathog. 8:e1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz E, et al. 2014. Plasma membrane-located purine nucleotide transport proteins are key components for host exploitation by microsporidian intracellular parasites. PLoS Pathog. 10:e1004547.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TY, et al. 2013. Shared signatures of parasitism and phylogenomics unite cryptomycota and microsporidia. Curr Biol. 23:1548–1553. [DOI] [PubMed] [Google Scholar]
- Jia J, Xue Q. 2009. Codon usage biases of transposable elements and host nuclear genes in Arabidopsis thaliana and Oryza sativa. Genomics Proteomics Bioinformatics 7:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katinka MD, et al. 2001. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414:450–453. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, et al. 2010. The reduced genome of the parasitic microsporidian Enterocytozoon bieneusi lacks genes for core carbon metabolism. Genome Biol Evol. 2:304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuper J, et al. 2014. In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS Biol. 12:e1001954.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson JIR. 2000. The hyperparasitic microsporidium Amphiacantha longa Caullery et Mesnil, 1914 (Microspora: Metchnikovellidae) - description of the cytology, redescription of the species, emended diagnosis of the genus Amphiacantha and establishment of the new family Amphiacanthidae. Folia Parasitol (Praha). 47:241–256. [DOI] [PubMed] [Google Scholar]
- Larsson JIR, Køie M. 2006. The ultrastructure and reproduction of Amphiamblys capitellides (Microspora, Metchnikovellidae), a parasite of the gregarine Ancora sagittata (Apicomplexa, Lecudinidae), with redescription of the species and comments on the taxonomy. Eur J Protistol. 42:233–248. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Rodrigue N, Stubbs D, Richer J. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 62:611–615. [DOI] [PubMed] [Google Scholar]
- Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney JO. 1998. GCUA: general codon usage analysis. Bioinformatics 14:372–373. [DOI] [PubMed] [Google Scholar]
- Mitchell A, et al. 2015. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43:D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35:W182–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Mérida A, Viguera E, Claros MG, Trelles O, Pérez-Pulido AJ. 2014. Sma3s: a three-step modular annotator for large sequence datasets. DNA Res. 21:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakjang S, et al. 2013. Reduction and expansion in microsporidian genome evolution: new insights from comparative genomics. Genome Biol Evol. 5:2285–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi H, Park J, Takagi T. 2006. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34:5623–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paccanaro A, Casbon JA, Saqi MA. 2006. Spectral clustering of protein sequences. Nucleic Acids Res. 34:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23:1061–1067. [DOI] [PubMed] [Google Scholar]
- Pati A, Heath LS, Kyrpides NC, Ivanova N. 2011. ClaMS: a classifier for metagenomic sequences. Stand Genomic Sci. 5:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyretaillade E, et al. 2009. Identification of transcriptional signals in Encephalitozoon cuniculi widespread among Microsporidia phylum: support for accurate structural genome annotation. BMC Genomics 10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrung M, et al. 2011. TopiaryExplorer: visualizing large phylogenetic trees with environmental metadata. Bioinformatics 27:3067–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombert JF, Haag KL, Beidas S, Ebert D, Keeling PJ. 2015. The Ordospora colligata genome: evolution of extreme reduction in microsporidia and host-to-parasite horizontal gene transfer. mBio 6:e02400–e02414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. 2005. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1):i351–i358. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35:D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotari YM, Paskerova GG, Sokolova YY. 2015. Diversity of metchnikovellids (Metchnikovellidae, Rudimicrosporea), hyperparasites of bristle worms (Annelida, Polychaeta) from the White Sea. Protistology 9:50–59. [Google Scholar]
- Roure B, Rodriguez-Ezpeleta N, Philippe H. 2007. SCaFoS: a tool for selection, concatenation and fusion of sequences for phylogenomics. BMC Evol Biol. 7(Suppl 1):S2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Bränden CI, Ernberg I, Cöster J. 2003. Quantifying the species-specificity in genomic signatures, synonymous codon choice, amino acid usage and G+C content. Gene 311:35–42. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R. 2008–2015. RepeatModeler Open-1.0. Available from: http://www.repeatmasker.org.
- Smit AFA, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. Available from: http://www.repeatmasker.org.
- Sokolova YY, Paskerova GG, Rotari YM, Nassonova ES, Smirnov AV. 2013. Fine structure of Metchnikovella incurvata Caullery and Mesnil 1914 (microsporidia), a hyperparasite of gregarines Polyrhabdina sp. from the polychaete Pygospio elegans. Parasitology 140:855–867. [DOI] [PubMed] [Google Scholar]
- Sokolova YY, Paskerova GG, Rotari YM, Nassonova ES, Smirnov AV. 2014. Description of Metchnikovella spiralis sp. n. (Microsporidia: Metchnikovellidae), with notes on the ultrastructure of metchnikovellids. Parasitology 141:1108–1122. [DOI] [PubMed] [Google Scholar]
- Sprague V. 1977. Classification and phylogeny of the Microsporidia In: Bulla LA, Cheng TC, editors. Comparative pathobiology: volume 2 systematics of the Microsporidia. Boston (MA: ): Springer US; p. 1–30. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2):ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Stothard P. 2000. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1102–1104. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. 2008. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18:1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomarat F, Vivarès CP, Gouy M. 2004. Phylogenetic analysis of the complete genome sequence of Encephalitozoon cuniculi supports the fungal origin of microsporidia and reveals a high frequency of fast-evolving genes. J Mol Evol. 59:780–791. [DOI] [PubMed] [Google Scholar]
- Tsaousis AD, et al. 2008. A novel route for ATP acquisition by the remnant mitochondria of Encephalitozoon cuniculi. Nature 453:553–556. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium. 2015. UniProt: a hub for protein information. Nucleic Acids Res. 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S. 2000. Graph clustering by flow simulation. [PhD thesis]: University of Utrecht. [Google Scholar]
- Vávra J, Lukeš J. 2013. Microsporidia and ‘the art of living together’. Adv Parasitol. 82:253–319. [DOI] [PubMed] [Google Scholar]
- Vivier E. 1975. The microsporidia of the protozoa. Protistologica 11:345–361. [Google Scholar]
- Vossbrinck CR, Debrunner-Vossbrinck BA. 2005. Molecular phylogeny of the Microsporidia: ecological, ultrastructural and taxonomic considerations. Folia Parasitol (Praha). 52:131–142. [DOI] [PubMed] [Google Scholar]
- Vossbrinck CR, Woese CR. 1986. Eukaryotic ribosomes that lack a 5.8S RNA. Nature 320:287–288. [DOI] [PubMed] [Google Scholar]
- Waller RF, et al. 2009. Evidence of a reduced and modified mitochondrial protein import apparatus in microsporidian mitosomes. Eukaryot Cell 8:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks HM, et al. 1988. A specific, highly active malate dehydrogenase by redesign of a lactate dehydrogenase framework. Science 242:1541–1544. [DOI] [PubMed] [Google Scholar]
- Williams BA, et al. 2010. A broad distribution of the alternative oxidase in microsporidian parasites. PLoS Pathog. 6:e1000761.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BA, Haferkamp I, Keeling PJ. 2008. An ADP/ATP-specific mitochondrial carrier protein in the microsporidian Antonospora locustae. J Mol Biol. 375:1249–1257. [DOI] [PubMed] [Google Scholar]
- Williams BA, Hirt RP, Lucocq JM, Embley TM. 2002. A mitochondrial remnant in the microsporidian Trachipleistophora hominis. Nature 418:865–869. [DOI] [PubMed] [Google Scholar]
- Wu G, Fiser A, ter Kuile B, Šali A, Müller M. 1999. Convergent evolution of Trichomonas vaginalis lactate dehydrogenase from malate dehydrogenase. Proc Natl Acad Sci U S A. 96:6285–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts J, Van de Peer Y, De Wachter R. 2001. Distribution of substitution rates and location of insertion sites in the tertiary structure of ribosomal RNA. Nucleic Acids Res. 29:5017–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, et al. 2010. A tandem duplication of manganese superoxide dismutase in Nosema bombycis and its evolutionary origins. J Mol Evol. 71:401–414. [DOI] [PubMed] [Google Scholar]
- Xu Y, Weiss LM. 2005. The microsporidian polar tube: a highly specialised invasion organelle. Int J Parasitol. 35:941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.