

Summary
The gastrointestinal immune system plays a pivotal role in the host relationship with food antigens, the homeostatic microbiome and enteric pathogens. Here, we describe how to collect and process liver and intestinal samples to efficiently isolate and analyse resident immune cells. Furthermore, we describe a step‐by‐step methodology showing how to high‐dimensionally immunophenotype resident leucocytes using cytometry by time‐of‐flight, providing a well‐characterized antibody platform that allows the identification of every leucocyte subset simultaneously. This protocol also includes instructions to purify and cultivate primary murine hepatocytes, a powerful tool to assess basic cell biology and toxicology assays. Gut and liver samples from the same mouse can be collected, processed and stained in less than 6 hr. This protocol enables the recovery of several populations of purified and viable immune cells from solid and fibrous organs, preventing unwanted loss of adherent cells during isolation.
Keywords: cell isolation, cytometry by time of flight, lamina propria, liver, mass cytometry
Introduction
The gastrointestinal immune system (GIS), which includes the gastrointestinal tract, liver and associated lymphoid organs such as mesenteric lymph nodes, appendix (caecal patch in mice) and Peyer's patches, harbours one of the most complex populations of immune cells in our body.1, 2 Indeed, all leucocyte subsets can be found under homeostatic conditions within the intestines1, 3 or the liver.4, 5 These resident immune cells play a crucial role for tissue homeostasis from embryonic phase and throughout adulthood. GIS cells are involved in tissue remodelling and repair,6 immune tolerance to food antigens7, 8 and they sustain a healthy relationship with our commensal microbiota.9 For this, an exquisite network of immune cells organizes an efficient firewall against dissemination of gut‐derived bacteria, viruses and other pathogens within circulation. Notably, disruption of gastrointestinal homeostasis due to gut and liver diseases is associated with several infectious and inflammatory diseases, including sepsis,10, 11, 12 and autoimmune13, 14 and auto‐inflammatory diseases.13, 15 Therefore, it is becoming increasingly clear that expanding our knowledge on the immunobiology of the GIS may guide future clinical interventions and diagnoses, and also add to the development of more precise immune‐based therapies.
The gastrointestinal tract is composed of a unique vascular and parenchymal architecture that comprises a diverse array of tissue‐resident immune cells to mediate homeostatic functions of the organs. Taking into account that these cells are usually part of an intricate tissue network, their isolation for ex vivo studies can be challenging. Indeed, improper tissue processing will lead to inefficient recovery of leucocyte subtypes, which will not reflect the actual pool of resident cells. Moreover, cells with larger protrusions (i.e. macrophages or dendritic cells) or cells within the connective tissue that are firmly adhered to the extracellular matrix will be inadvertently discarded during the isolation procedure.16 Hence, reliable and efficient cell isolation procedures specifically designed for solid and fibrous organs are of broad interest.
Efforts to characterize the tissue‐resident immune cell populations in the GIS have to date mostly relied on flow cytometry‐based analysis, which has proven to be an invaluable tool to identify and assess the role of GIS‐resident immune cells in homeostasis and disease pathogenesis.17, 18 However, traditional flow cytometry has limitations that can make data interpretation difficult, these limitations include: a relatively small number of markers per panel and spectral overlap of fluorescent markers leading to compensation artefacts. These limitations are particularly evident in the characterizations of non‐lymphoid cell populations, (monocytes, macrophages, dendritic cells and granulocytes) in the liver and small intestine lamina propria, which harbour numerous unique tissue‐resident subsets that require several specific markers to study.
To overcome some of the limitations of flow cytometry, cytometry by time‐of‐flight (CyTOF), mass cytometry (Fig. 1) was recently developed and its use has become more widespread.19, 20, 21, 22, 23, 24 This technique uses a mass spectrometer to characterize single cells labelled with antibodies conjugated to metal isotopes instead of the fluorophores used in traditional flow cytometry. Currently, over 40 isotope‐conjugated markers can be analysed in a single panel without the need to compensate different isotope channels. The high‐dimensional data that are generated from using this many markers can be analysed straightforwardly using widely available algorithms on cytobank, these include viSNE,25 CITRUS26 and SPADE.27 One of the drawbacks of CyTOF is that the sample must comprise a purified cell population because the cells are vaporized during the run and gating for cells is based on DNA content, which will be compromised by samples with a high density of debris. This is an important consideration for the study of immune cells from the GIS, where the digestion of tissues such as the intestine and liver leads to loss of cell viability and generates a large amount of residual debris.
Figure 1.
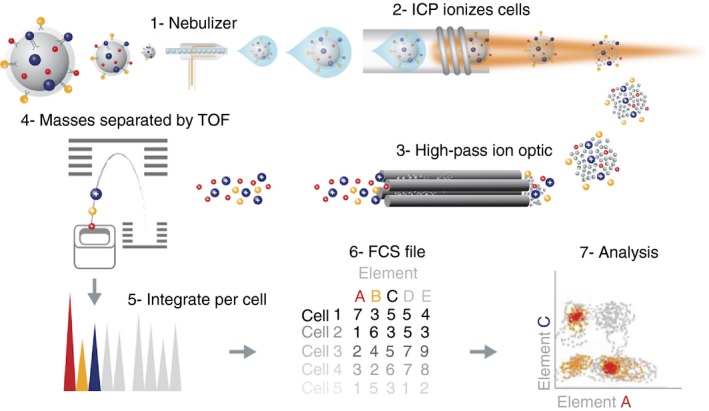
Schematic representation of basic principles of cytometry by time‐of‐flight (CyTOF). Cells are labelled with a panel of heavy‐metal‐conjugated probes using methods very similar to flow cytometry staining protocols. The single‐cell suspension is introduced into the nebulizer where it is aerosolized (step 1). It is critical to remove as much water as possible from the sample so that it can be efficiently ionized in the plasma. This is achieved first by aerosolizing the sample in the nebulizer followed by delivery to the plasma through the heated spray chamber. The single‐cell aerosol droplets that exit the spray chamber are transmitted to the inductively coupled plasma (ICP) source where they are vaporized, atomized and ionized in the plasma for subsequent mass analysis (step 2). This results in the formation of an ion cloud containing the ions derived from metal‐conjugated probes, endogenous cell components and argon. The ion cloud then passes through the plasma–vacuum interface. The purpose of the vacuum interface is to efficiently transport ions from the plasma at atmospheric pressure to the chambers that house the ion optics at less than 10−3 Torr (step 3). Mass cytometers use a three‐cone interface to transport the ion beam into a low‐pressure vacuum: sampler, skimmer and reducer. Low‐mass ions, non‐ionized particles and photons are removed in the high‐pass ion optics, resulting in a cloud of ions enriched for the probe isotopes. The ion clouds that exit the high‐pass ion optic consist of a mixture of high‐molecular‐weight probes in a randomly distributed array. These ions are sent to the TOF mass analyser, which separates the ions on the basis of their mass‐to‐charge ratio (step 4). The ion clouds, now enriched for high mass ions, enter through the entrance slit into the accelerator chamber of the TOF analyser. At 13‐microsecond intervals, or pushes (frequency of 76·8 kHz), a pulse is applied to the push‐out plate, accelerating the accumulated packet of ions orthogonally toward the reflector, which redirects the ions toward the detector. The electric fields in the accelerator and reflector are configured to focus ions into tight time‐resolved bands regardless of initial position or energy. Each packet of ions resolves into a series of bands, with the lightest probes reaching the detector first and each successively heavier mass reaching the detector at a later time. Each time‐resolved band of ions of mass M is separated from its M ± 1 neighbour by 20–25 ns. After the first packet of ions is pushed out and detected, a second pulse pushes out the next packet of ions for detection and the cycle repeats until data acquisition is complete. Data for each 13‐microsecond push are digitized sequentially and integrated to obtain ion counts for the channels selected for analysis (steps 5 and 6). The resulting record is processed according to cell event selection criteria set by the user. These criteria include a minimum signal threshold and a range for event duration consistent with single‐cell events. As a result, the data acquired contain the integrated number of total ion counts for each selected analyte on a per‐cell basis. These data are saved as text (.txt) and flow cytometry standard (.fcs) 3.0 format for data analysis in compatible software programs (step 7). [Colour figure can be viewed at wileyonlinelibrary.com]
Here, we describe a robust and simple protocol for the isolation, staining and CyTOF analysis of purified and viable leucocytes from the intestinal lamina propria and mesenteric limph nodes (Box 2 and 3, respectively) and the liver that generates high‐quality and reproducible high‐dimensional immunophenotyping data of the GIS. Additionally, using the same protocol, we also describe the procedures to isolate leucocytes from the mesenteric lymph nodes, and how to cultivate primary mouse hepatocytes. This may be of interest to groups studying liver biology and toxicology.
Box 2. Lamina propria cell isolation timing: ~ 3 hr 10 min.
Anaesthetize the mouse by intraperitoneal injection (see Reagent set‐up for dose). Wait about 10 min until mouse is under deep anaesthesia. Alternatively, these procedures can be run with a killed mouse.
Position the mouse on a surgical slate with the abdomen facing up, trapping the animal by the limbs using masking tape.
Apply 70% ethanol on the chest and abdomen skin of the mouse to keep the hair out of the way during surgery. Then, perform the midline incision in the abdomen from the pubis to the xiphoid process (Fig. 4a).
Dissociate the abdominal musculature from the skin to allow the visualization of blood vessels and to prevent disruption and possible bleeding. Perform lateral incisions in the skin and, subsequently, in the abdominal muscles along the costal margin to the mid axillary line (Fig. 4b).
Slide the abdominal viscera to the right side with the aid of a cotton swab (Fig. 4c).
Localize and remove mesenteric lymph nodes by finding the caecum localization (if desired) (Fig. 4c,d).
Localize stomach and separate it from oesophagus (Fig. 4e).
Bring stomach in a vertical direction all the way up from the mouse to detach the small intestine, caecum and large intestine (Fig. 4e,f).
Remove as much fat and mesothelium as possible and position the organs in order to separate the caecum and large intestine (Fig. 4g).
Place the small intestine on a paper in a ‘U’ shape. Identify the small intestine compartments and separate them accordingly (Fig. 4h).
Remove stomach.
Remove faeces from the small intestine squeezing the segment with scissors. This will help to identify Peyer's patches for further removal (Fig. 4j–l)
Transfer the collected intestine compartments into Petri dish in Buffer A to wash and remove mucus.
Place intestine compartments on a paper towel.
Clean out the faeces and mucus by opening up the intestines and shaking them in the buffer (Fig. 4m–p).
Place the intestines on a paper towel; re‐open them using forceps and scissors (Fig. 4q).
Cut the intestine into 1–2 cm pieces and transfer them to a 50 ml tube or T25 flasks containing 20 ml of Buffer B (Fig. 4r,s).
Shake or vortex once and discard the supernatant using metal strainer (pour Falcon tube content into a metal cell strainer placed in a beaker). Repeat this procedure until a clean supernatant is obtained.
Collect the pieces withheld on the cell strainer and transfer them into 50‐ml Falcon tubes containing 10 ml pre‐warmed Buffer C at 37°. Shake (200 rpm) for 20–30 min for epithelial removal.
Following incubation shake up and down using hands and vortex.
Remove the supernatant using metal strainer placed in a beaker.
Transfer the metal cell strainer to 1, 2, 3, 4 Petri dishes containing Buffer A (quick dry it in between washes using paper towel) (Fig. 5a).
Transfer the small pieces of intestine withheld on the metal strainer to a 50‐ml Falcon tube, fill up with 20 ml Buffer A, vortex and pour into the metal strainer. Repeat this at least twice (until supernatant is clean).
Transfer all tissue pieces to a 1·5‐ml Eppendorf tube, fill it up with Buffer A.
Spin 300 g for 30 seconds to 1 min (to pellet).
Remove supernatant.
Using microsurgery‐curved scissors, mince the intestine (Fig. 5b).
Fill it up with Buffer B.
Spin 300 g for 30 seconds to 1 min (to pellet)
Remove supernatant (Fig. 5c,d).
Add 1 ml of Buffer D solution containing DNAse and liberase TL into the tube containing minced intestine. Vortex to detach the pellet.
Pour into a six‐well plate or 50‐ml tube.
Wash the tube with Buffer D to collect all small pieces and transfer to the plate well.
Shake plates or tubes on a 37° shaker with Buffer C for 1 hr.
Vortex (twice) after every 20 min.
After incubation period carefully open plates or tubes.
Using a Pasteur pipette, transfer digested intestine into a 50‐ml Falcon tube containing Buffer A.
Centrifuge for 7 min 500 g and discard supernatant.
Fill tube with 10 ml Buffer A and vortex to detach pellet.
Pour Tube content into a new Falcon tube containing 100‐μm cell strainer
Use the back of 1‐ml syringe to gently help the content to pass through cell strainer, wash with Buffer A. Collect the filtrate.
Spin for 300 g for 7 min and remove the supernatant. Resuspend the pellet in Buffer B.
Pass tube content through 40‐μm cell strainer. Collect the filtrate.
Spin and remove the supernatant.
Make single cell suspension by re‐suspending the pellet in 450 μl of MACS beads solution. Transfer to a 15‐ml tube.
Add 50 μl of CD45 microbeads (up to five mice pooled). Use regular inversion to mix the cell suspension (do not vortex). For single sample use 25 μl of beads and 75 μl of MACS solution.
Incubate the cell suspension for 20 min at 4° (on ice).
Fill Tube with 9·5 ml of MACS solution.
Centrifuge for 7 min at 300 g.
Prepare a MACS column of appropriate size (see manufacturer's instructions). Columns for positive selection are ready to use [pre‐treat columns and cell strainer with 1 ml MACS solution (twice) while cells is centrifuging].
Resupend pellet in 1 ml MACS solution.
Pass the content to the beads column. Allow unlabelled cells to flow through the column.
Rinse column twice with 2 ml MACS solution.
Remove column from racks (magnetic field).
Fill the column with 5 ml buffer and elute the positive cells outside the magnetic field using the supplied plunger. Repeat this step twice.
Spin at 300 g for 7 min at 4° and discard the supernatant.
Resuspend pellet according to labelling protocols.
Note. Step 30: floating fat can be present on the supernatant and should be removed.
Box 3. Isolation of immune cells from mesenteric or intestinal lymph nodes timing: ~ 3 hr 30 min.
Position the mouse on a surgical slate with the abdomen facing up, trapping the animal by the limbs using masking tape.
Apply 70% ethanol on the skin of chest and abdomen to prevent hair from contaminating the surgery. Perform a midline incision in the abdomen from the pubis to the xiphoid process (Fig. 4a).
Dissociate the abdominal musculature from the skin to allow the visualization of blood vessels and to prevent disruption and possible bleeding. Perform lateral incisions in the skin and, subsequently, in the abdominal muscles along the costal margin to the mid axillary line (Fig. 4b).
Slide the abdominal viscera to the right side with the aid of a cotton swab (Fig. 4c).
Localize and remove mesenteric lymph nodes by finding the caecum localization (if desired) (Fig. 4c,d).
Carefully remove fat tissue from the mesenteric lymph node.
Place mesenteric lymph nodes on six‐well plates containing 70‐μm cell strainers and filled with 7 ml of complete media. The media will allow the organ to be constantly bathed, which may result in better cell survival.
Mash mesenteric lymph nodes using a 1‐ml syringe plunger.
Remove the cell strainers and transfer the media containing single cell suspension to a 15‐ml Falcon tube.
Centrifuge at 400 g for 5 min at 4°.
Discard the supernatant and resuspend the pellet with 500 μl of complete medium.
The integrated protocol, encompassing cell isolation to analysis of gastrointestinal immune cells, described here will expedite discoveries in uncovering the role of the GIS in health and disease. Indeed, high‐dimensional analysis can offer a more detailed characterization of the dynamic changes in resident cell populations during the disease courses of models of chronic inflammation (e.g. inflammatory bowel disease, hepatitis) or gastrointestinal cancers, and after perturbation to GIS homeostasis (e.g. changes in diet, microbiome). For example, we recently used viSNE to characterize the distinct temporal changes of novel myeloid cell clusters during the repopulation and seeding of liver phagocytes after depletion.23 Another emerging application of CyTOF analysis is the discovery of novel single cell‐based biomarkers, where the high‐dimensional nature of CyTOF data permits more rapid screening of targets than would be possible using traditional flow cytometry.21
Taking into account that this protocol deals with challenging solid and fibrous tissues, and the immunophenotyping methodology proposed here covers the majority of the leucocyte subsets, minimal modifications would allow the processing and analysis of different organs other than the gastrointestinal tract, which may include brain, lung, spleen and others.
One of the current limitations of CyTOF is the low sample acquisition rate of the mass cytometer (200–600 events per second) compared with a traditional flow cytometer (i.e. BD FACS Aria; BD, Franklin Lakes, NJ; up to 20 000 events per second). To circumvent this low acquisition rate, individual samples can be bar‐coded and pooled together for a single run. However, the low acquisition rate will still make it difficult to analyse very rare populations, as it would take 20–100 times longer than flow cytometry to collect enough events for proper statistical testing. One solution for analysing very rare populations by mass cytometry is to first enrich for these populations using bead or flow‐sorting‐based techniques before staining and running on the mass cytometer.
Materials and methods
Reagents
Common use of reagents
Mice: 18–25 g C57BL/6. Other mouse backgrounds are acceptable. All experiments involving animals must conform to all relevant governmental and institutional regulations.
Ethanol, 70% [volume/volume (vol/vol)] in distilled water
Commercial ethyl alcohol absolute 99·5% (Merk Millipore, Frankfurt, Germany, cat. no. 100990)
Sterile saline solution, 0·9% [weight (wt)/vol] (NaCl)
Fetal bovine serum (FBS; Gibco, Grand Island, NY, ref.12657‐029)
Anaesthetic solution (see Reagent set‐up)
Reagents for liver cells isolation
Hydrochloric acid 1 m (Synth Diadema ‐ SP, Brazil, – cod. SC 1002‐1)
Penicillin–streptomycin (Sigma‐Aldrich, St Louis, MO, P4333)
Collagenase from Clostriduim histolyticum (Sigma‐Aldrich, C2139‐1G)
Milli‐Q water
Type I collagen from rat tail (Sigma‐Aldrich, C3867)
Trypan Blue Solution 0·4% (Thermo Fisher Scientific, Waltham, MA, cat. number 15250061)
Sodium chloride – NaCl (Synth, Diadema ‐ SP, Brazil cod. 01C1060·01.AH)
Potassium chloride – KCl (Synth, cod. 01C1058·01.AH)
Potassium dihydrogen phosphate – KH2PO4 (Synth, cod. 01F2002·01.AG)
Sodium phosphate dibasic anhydrous – Na2HPO4 (Sigma‐Aldrich, cod. V000317)
Sodium bicarbonate – NaHCO3 (Synth, cod. 01B20007·01.AG)
Magnesium sulphate heptahydrate – MgSO4·7H2O (Dinâmica Química Contemporânea, Sao Paulo, Brazil, cod. 1159)
Magnesium chloride hexahydrate – MgCl2·6H2O (Dinâmica Química Contemporânea, cod. 1055)
Calcium chloride dihydrate – CaCl2·2H2O (Synth, cod. 01C2013·01.AH)
Sodium hydroxide – NaOH (Sigma‐Aldrich, S5881)
RPMI‐1640 medium (Cultilab, Campinas ‐ SP, cod. 0010 – see Reagent set‐up)
Williams’ E medium (Sigma‐Aldrich, W4125 – see Reagent set‐up)
Hanks’ Medium A (see Reagent set‐up)
Hanks’ Medium B (see Reagent set‐up)
PBS 10 × (see Reagent set‐up)
PBS 1 × (see Reagent set‐up)
Reagents for lamina propria cells isolation
X‐VIVO™ 15 chemically defined, serum‐free haematopoietic cell medium (Lonza, Basel, Switzerland 04‐418Q)
Iscove's modified Dulbecco's medium (GlutaMAX™; Gibco, cat. no. 31980‐030)
Liberase TL (Roche Molecular Biochemicals, Indianapolis, IN USA, 05401020001)
Deoxyribonuclease I from bovine pancreas (Sigma‐Aldrich, D5025)
EDTA (Sigma‐Aldrich, E6758)
1,4‐Dithiothreitol (Sigma‐Aldrich, D9760)
Bovine serum albumin – (Sigma‐Aldrich, A5123)
Lamina propria buffer A (Reagent set‐up)
Lamina propria buffer B (Reagent set‐up)
Lamina propria Buffer C (Reagent set‐up)
Lamina propria buffer D (Reagent set‐up)
MACS buffer (Reagent set‐up)
HEPES (Sigma‐Aldrich, 7365‐45‐9)
EDTA stock solution – 0·5 m (Reagent set‐up)
CyTOF staining reagents
CD45 MicroBeads, mouse (Miltenyi Biotec, Bergisch Gladbach, Germany, 130‐052‐301)
CyTOF‐PBS (Low‐barium PBS): PBS Ca2+‐ and Mg2+‐free (Gibco, cat. no. 10010‐023).
Bovine serum albumin, protease‐free (Sigma‐Aldrich, A3059)
Sodium azide (Sigma‐Aldrish, 71289)
MaxPar® Fix and Perm Buffer (Fluidigm/DVS Sciences, South San Francisco, CA, USA, cat. no. 201067)
MaxPar® Intercalator Ir 500 μm (Fluidigm/DVS Sciences, cat. no. 201192B)
Cell‐ID™Cisplatin – Natural Abundance Platinum (Fluidigm/DVS Sciences, cat. no. 201064)
EQ™ Four‐Element Calibration Beads (Fluidigm/DVS Sciences, cat. no. 201078)
Cell staining buffer – CSB (see Reagent set‐up)
Intercalator solution (see Reagent set‐up)
MaxPar® Multi‐Metal Labelling Kit (Fluidigm/DVS Sciences)
Reagent set‐up
Anaesthetic solution: Mix 10% (wt/vol) ketamine solution (2·5 ml) with 2% (wt/vol) xylazine solution (2·0 ml) and 1 × PBS (5·5 ml). Inject 50 μl intraperitoneally per 10 g of body weight (ketamine: 60–80 mg/kg and xylazine: 8–15 mg/kg). Anaesthetic solution should be freshly made.
Solutions for liver cell isolation – non‐parenchymal cells
RPMI‐1640 medium: In a beaker, dilute the contents of a sachet of RPMI‐1640 in 800 ml of distilled water. Add 2·2 g of sodium bicarbonate and 10 ml of penicillin–streptomycin (100 ×). Complete up to a final volume of 1000 ml, keeping the solution under constant stirring with a magnetic stirrer.
Digestion solution 1 – liver non‐parenchymal cell isolation: Dilute 10 mg of collagenase (1 mg/ml) in 9 ml of RPMI‐1640 medium supplemented with 1 ml of FBS (10% vol/vol).
Solutions for liver cell isolation – hepatocytes
Williams’ medium E: In a beaker, dilute the contents of a sachet of RPMI‐1640 in 800 ml of distilled water. Add 2·2 g of sodium bicarbonate and 10 ml (1/100 vol/vol) of Pen strep (100 ×). Complete for a final volume of 1000 ml keeping the solution under constant stirring with a magnetic stirrer. Measure the pH, filter into sterile receptacle and store at 4°.
Stock solutions to prepare Hanks’ A and B (for hepatocytes isolation): Solution 1: In a beaker, dilute 35·1 g of sodium chloride (NaCl – 1·2 m), 1·9 g of potassium chloride (KCl – 50 mm), 0·27 g of potassium dihydrogen phosphate (KH2PO4 – 4 mm) and 0·14 g of disodium phosphate (Na2HPO4 – 2 mm) in 400 ml of distilled water keeping the solution under constant stirring with a magnetic stirrer. Complete for a final volume of 500 ml, filter and store at 4°.
Solution 2: In a beaker, dilute 26·25 g of sodium bicarbonate (NaHCO3 – 625 mm) in 400 ml of distilled water keeping the solution under constant stirring with a magnetic stirrer. Complete for a final volume of 500 ml, filter and store at 4°.
Solution 3: In a beaker, dilute 1·23 g of magnesium sulphate heptahydrate (MgSO4·7H2O – 10 mm), 1·27 g of magnesium chloride hexahydrate (MgCl2·6H2O – 12·5 mm) and 5·51 g of calcium chloride dihydrate (CaCl2·2H2O – 75 mm) in 400 ml of distilled water keeping the solution under constant stirring with a magnetic stirrer. Complete to a final volume of 500 ml, filter and store at 4°.
Hanks’ A solution: In a beaker, dilute 0·5 g of d‐glucose (0·1% wt/vol), 0·095 g of EGTA (0·5 mm), 50 ml of the stock solution 1, 50 ml of the stock solution 2 and 10 ml of penicillin–streptomycin (100 ×) in 400 ml of distilled water keeping the solution under constant stirring with a magnetic stirrer. Complete for a final volume of 500 ml, set pH to 7–7·4, filter and store at 4°.
Hanks’ B solution: In a beaker, dilute 0·5 g of d‐glucose (0·1% wt/vol), 50 ml of the stock solution 1, 20 ml of the stock solution 2, 20 ml of the stock solution 3 and 10 ml of penicillin–streptomycin (100 ×) in 400 ml of distilled water keeping the solution under constant stirring with a magnetic stirrer. Complete for a final volume of 500 ml, set pH to 7–7·4, filter and store at 4°.
Digestion solution 2 – hepatocyte isolation: Dissolve 20 mg of collagenase in 25 ml of Hanks’ B to obtains a 0·8 mg/ml collagenase solution.
Sodium hypochlorite solution: Dilute 10 ml of sodium hypochlorite in 90 ml of distilled water.
Solutions for lamina propria cell isolation
EDTA stock solution: Dissolve 116·9 g of EDTA in H2O to obtain a 0·5 m stock solution. Stir vigorously on a magnetic stirrer. Adjust the pH to 8·0 with NaOH. Dispense into aliquots and sterilize by autoclaving.
Lamina propria buffer A: Mix 25 ml of FBS and 12·5 ml of HEPES (1 m) in 462·5 ml of Hanks’ balanced salt solution (HBSS). Sterilize the solution by filtration using a 0·22‐μm syringe filter, and store it for up to 1 month at 4°.
Lamina propria buffer B: Mix 2 ml of EDTA stock solution (0·5 m) and 12·5 ml of HEPES (1 m) in 485·5 ml of HBSS. Sterilize the solution by filtration using a 0·22‐μm syringe filter, and store it for up to 1 month at 4°.
Lamina propria buffer C: Mix 5 ml of EDTA stock solution (0·5 m), 50 ml of FBS and 0·5 ml of HEPES (1 m) in 437·5 ml HBSS and then dissolve 0·77 g of dithiothreitol to obtain a 1 mm solution. Sterilize the solution by filtration using a 0·22‐μm syringe filter, and store it for up to 1 month at 4°.
Lamina propria buffer D: Dissolve 5 mg of liberase TL and 450 μl of DNAse stock solution in 30 ml of Iscove's or X‐Vivo medium.
DNAse stock solution: Dissolve 20 mg of DNAse in 10 ml of sterile saline solution, 0·9% (wt/vol) to obtain a 2 mg/ml (4000 U/ml) stock solution. Store the solution in 1 ml aliquots at −20° for up to 1 month.
MACS buffer: Dissolve 50 g of BSA (0·5% – wt/vol) in 100 ml of PBS 1 × and store up to 1 week at 4°.
Solutions for CyTOF staining
Cell staining buffer – CSB: Dissolve 2·5 g of BSA and 100 mg of sodium azide in 500 ml low‐barium PBS. Store in a Nalgene polypropylene bottle that has never been washed with detergent. Prepare up to 1 month in advance and store at 4°.
Intercalator solution: MaxPar Intercalator‐Ir 500 μm 1 : 4000 dilution in MaxPar Fix and Perm Buffer.
Procedures for hepatocyte culture
The culture of hepatocytes (see hepatocutes isolatin in BOX 1) is performed in 6‐well and 24‐well plates with or without cover slip, depending on the purpose of the experiment.
Box 1. Hepatocyte isolation timing: ~ 3 hr 30 min.
Perfuse (see Fig. 2) the liver with 50 ml of Hanks’ A (see Reagent set‐up) previously warmed at 37°. Keep the solution in the water bath at 37° during perfusion.
Perfuse the liver with the digestion solution 2 for hepatocyte isolation (see Reagent set‐up). Keep the solution in the water bath at 37° during the entire process. Critical: it is not necessary to stop the perfusion to change tubes containing different solutions, simply pour digestion solution 1 into the tube when Hanks’ solution is almost over.
When the digestion solution 1 ends, carefully remove the catheter from the hepatic portal vein.
Carefully remove the liver and transport it to the laminar flow in a covered Petri dish with cold William's E medium supplemented with 10% FBS (vol/vol).
In the laminar flow, open the Petri dish and tear liver's capsule using two forceps to release the cells. Gently shake the liver to help spread its contents on the Petri dish. Repeat this until there are no large pieces left.
Use a previously autoclaved mesh to filter all supernatant from the Petri dish. Use a sterile beaker to collect all supernatant. Gently squeeze the mesh against the beaker wall to help with filtration process.
Transfer the filtrate to a 50‐ml tube and complete to a final volume of 50 ml with cold William's E medium supplemented with 10% FBS (vol/vol). Critical: Use the medium to wash the Petri dish before filtration and the beaker after filtration.
Centrifuge the tube at 60 g for 3 min at 4°.
Discard the supernatant and resuspend the pellet in 50 ml cold William's E medium supplemented with 10% FBS.
Centrifuge the tube at 60 g for 3 min at 4°.
Remove the supernatant and resuspend the pellet in 10 ml of cold William's E medium supplemented with 10% FBS (vol/vol).
Mix 10 μl of cells with 10 μl of Trypan Blue and count the cells in a Neubauer chamber (see Reagent set‐up).
Add the appropriate volume of cells in each well and complete to 2 ml with William's E medium supplemented with 10% FBS (vol/vol).
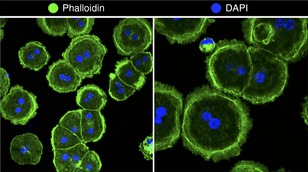
Incubate the cells at 37° with 5% CO2 for 2 hr to initial adherence. Critical: Cells will remain viable for 6–8 hr (Fig. B1).
Figure B1. Hepatocyte isolation protocol. Representative image of primary murine hepatocytes stained with Alexa‐fluor 488 conjugated phalloidin (in green) and DAPI. [Colour figure can be viewed at wileyonlinelibrary.com]
Suggested hepatocyte density for different plates.
| Surface | Number of cells | Final volume (ml) |
|---|---|---|
| Six‐well plates | 5 × 105 | 2 |
| Cover slip for six‐well plates (22 × 22 mm) | 3 × 105 | 2 |
| 24‐well plates | 2 × 105 | 1 |
| Coverslip for 24‐well plates (13 mm diameter) | 1 × 105 | 1 |
Preparation of the cover slips: Using a small forceps, wash each cover slip individually, according to the following sequence: distilled water (five times), absolute ethanol (five times), hydrochloric acid (HCl, 1 m) for 1 hr, distilled water (five times), absolute ethanol (five times). Place it to dry in the laminar flow on a sterile filter paper for approximately 30 min, keeping the UV light on. Subsequently, turn the cover slips so that they stay under UV light for over 30 min. Place one coverslip inside each well.
Preparation of the plates: 1 day before the hepatocytes isolation experiment, treat the plates and the cover slips with type I collagen (see Reagent set‐up) to enhance cell adhesion. Prepare the solution of type I collagen at a concentration of 5 μg/cm2 and pipette it into the wells. In wells with cover slip, pipette slowly and carefully, so that the solution can form a big drop just on the surface of the cover slip without draining to the bottom of the wells. Let it dry overnight in laminar flow under UV light. The plates and cover slips treated with type I collagen can be sealed and stored at 4° for a week. Before use, hydrate collagen with Williams’ E medium for 30 min. Remove medium before adding the required amount of cell suspension.
Required amount of type I collagen depending on the plate design:
| Surface | Area (cm2) | Type I collagen – stock solution (µl) (5 mg/ml) |
|---|---|---|
| Six‐well plates | 9·6 | 1200 |
| Cover slip for six‐well plates (22 × 22 mm) | 4·9 | 600 |
| 24‐well plates | 1·9 | 200 |
| Coverslip for 24‐well plates (13 mm diameter) | 1·33 | 150 |
Procedure
Cannulation of the portal vein timing: ~ 1 hr
-
1
Clean the perfusion catheter with sodium hypochlorite solution (see Reagent set‐up) for 5 min at fast flow.
-
2
Wash the perfusion catheter with distilled water for 10 min at fast flow.
-
3
Calibrate the water bath to 37° to preheat the solutions before perfusion. The liver should be perfused with Hanks’ A and Hanks’ B for hepatocyte isolation, and with PBS 1 × for hepatic non‐parenchymal cell isolation.
-
4
Calibrate the perfusion pump to medium flow of 12·5 ml/min.
-
5
Anaesthetize the mouse by intraperitoneal injection (see Reagent set‐up for dose). Wait about 10 min until mouse is under deep anaesthesia.
-
6Position the mouse on a surgical stage with the abdomen facing up, taping the animal by the limbs (Fig. 2a).
Figure 2.
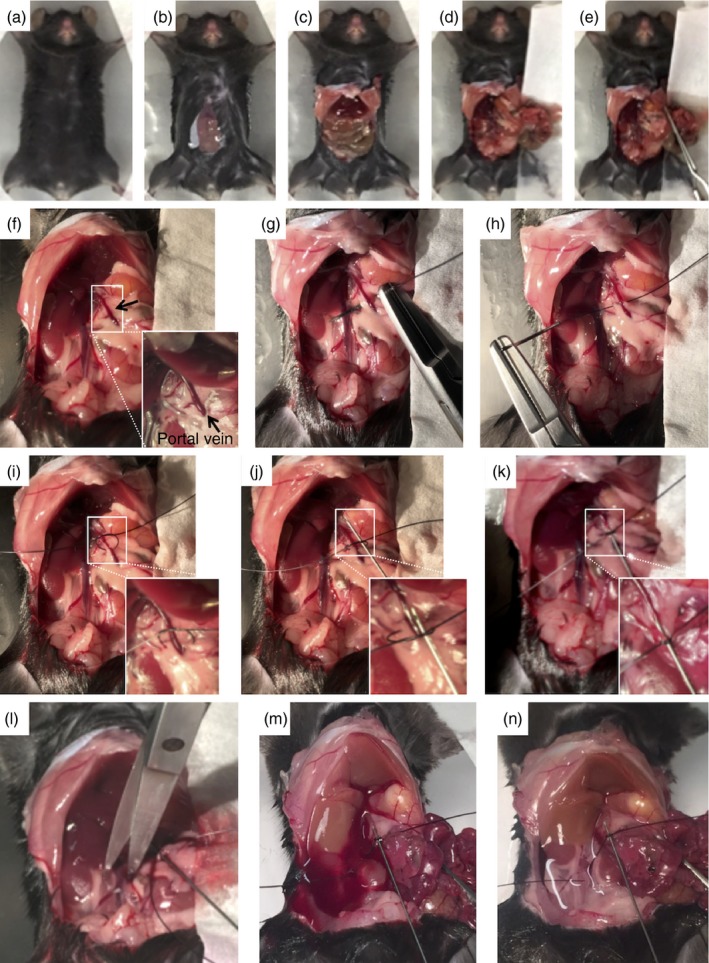 Surgical procedures for portal vein cannulation and liver perfusion. (a–e) Midline laparotomy and viscera dislocation to visualize the branch of portal vein. (f) Higher magnification imaging showing the anatomical location of the portal vein. (g, h) A suture wire is trespassed under the portal vein in the median portion, perforating the associated adipose tissue and mesentery, without lacerating the portal vein. (i, j) A loose knot will receive the cannulation catheter (k) that after portal vein perforation will firmly retain the catheter in a stable position. (l–n) After confirmation of cannulation patency, the vena cava is cut to allow liver perfusion. Note that liver colour changes from wine red to brown after perfusion. [Colour figure can be viewed at wileyonlinelibrary.com]
Surgical procedures for portal vein cannulation and liver perfusion. (a–e) Midline laparotomy and viscera dislocation to visualize the branch of portal vein. (f) Higher magnification imaging showing the anatomical location of the portal vein. (g, h) A suture wire is trespassed under the portal vein in the median portion, perforating the associated adipose tissue and mesentery, without lacerating the portal vein. (i, j) A loose knot will receive the cannulation catheter (k) that after portal vein perforation will firmly retain the catheter in a stable position. (l–n) After confirmation of cannulation patency, the vena cava is cut to allow liver perfusion. Note that liver colour changes from wine red to brown after perfusion. [Colour figure can be viewed at wileyonlinelibrary.com] -
7
Apply 70% ethanol on the chest and abdominal skin of the mouse. This will avoid small pieces of fur entering the peritoneal cavity. Perform the midline incision in the abdomen from the pubis to the xiphoid process (Fig. 2b).
-
8
Dissociate the abdominal musculature from the skin to allow the visualization of blood vessels and to prevent disruption and possible bleeding. Perform lateral incisions in the skin and, subsequently, in the abdominal muscles along the costal margin to the mid axillary line (Fig. 2c).
-
9
Slide abdominal viscera to the right side using a cotton swab (Fig. 2d).
-
10
Stabilize abdominal viscera with self‐closing forceps to visualize the portal vein (Fig. 2e).
-
11
Locate the hepatic portal vein (white arrow), and move the forceps and the organs until the portal vein remains straight to be cannulated (Fig. 2f).
-
12
Using a Mayo–Hegar needle holder with a 20 mm needle and 5‐0‐nylon filament, trespass the suture under (dorsally) the hepatic portal vein (Fig. 2g,h).
-
13
Make a loose knot with the nylon filament around the hepatic portal vein without tying the blood vessel (Fig. 2i).
-
14
Insert the catheter inside the loose knot made with the suture thread so that, after cannulating the hepatic portal vein the knot will hold the catheter and keep it immobilized during perfusion (Fig. 2j).
-
15
Carefully and with the bevel of the catheter facing up, cannulate the hepatic portal vein at the middle portion of its exposed part. To do this, position the catheter parallel to the hepatic portal vein and with a small angle (˜ 5°) insert the needle approximately 5 mm into the vein.
-
16
After cannulating, require the help of a second person to tight the knot around the portal vein and the catheter (Fig. 2k). Keep the catheter immobilized until the knot is tight. Release the catheter with extreme caution to prevent rupture of the blood vessel or untie the knot.
-
17
Start the perfusion for 1–2 seconds to check whether the vein was properly cannulated and then stop perfusion, the liver should change its colour to pale/beige and no leakage should occur.
-
18
Cut inferior vena cava using a scissor to release the blood and start the perfusion immediately (Fig. 2l).
-
19
The liver changes rapidly to its original colour after the blood is washed by the perfusion solution (Fig. 2m,n).
-
20
Wait until the perfusion is over and then carefully remove the liver. For liver non‐parenchymal cells isolation follow next steps. However, this protocol can also be used for hepatocyte isolation (Box 1).
Isolation of liver non‐parenchymal cells timing: ~ 1 hr 30 min
-
After cannulation (Fig. 2), perfuse the liver with 50 ml of PBS 1 × (see Reagent set‐up) previously warmed at 37°. Keep the solution in the water bath during perfusion.
-
Carefully remove the liver from the peritoneal cavity and wash it in a Petri dish containing PBS once (Fig. 3a). This will remove blood and clots from liver surface.
Figure 3.
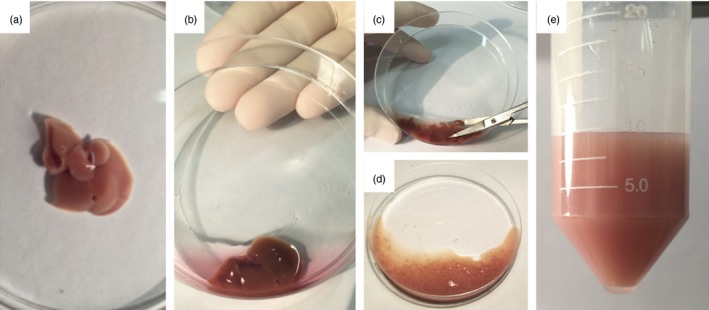 Liver processing for isolation of non‐parenchymal cells. (a) Liver aspect after perfusion in vivo. (b) Initial digestion in a Petri dish using collagenase‐based solution. (c, d) Liver is minced to very small fragments using scissors to enhance digestion by collagenase. (e) Final aspect after digestion protocol. [Colour figure can be viewed at wileyonlinelibrary.com]
Liver processing for isolation of non‐parenchymal cells. (a) Liver aspect after perfusion in vivo. (b) Initial digestion in a Petri dish using collagenase‐based solution. (c, d) Liver is minced to very small fragments using scissors to enhance digestion by collagenase. (e) Final aspect after digestion protocol. [Colour figure can be viewed at wileyonlinelibrary.com] -
Transfer to another Petri dish with 3 ml of the digestion solution 1 (see Reagent set‐up) for hepatic non‐parenchymal cell isolation (Fig. 3b).
-
Mince the liver into small pieces (Fig. 3c,d) using scissors.
-
Transfer the minced liver to a 50‐ml tube. Collect all the small liver pieces with the rest of digestion solution (˜ 7 ml).
-
Incubate for 30 min at 37° under constant stirring.
-
Filter the homogenate (Fig. 2e) using a 70 μm cell strainer and transfer the filtrate to a 50‐ml tube.
-
Complete the tube with cold RPMI medium 10% FBS (vol/vol) to a final volume of 50 ml.
-
Centrifuge at 4° for 5 min at 300g. This step will retrieve all cells in suspension.
-
Discard the supernatant and resuspend the pellet in 15 ml of cold RPMI medium 10% FBS (vol/vol).
-
Transfer the samples to 15‐ml tubes and centrifuge at 4° for 3 min at 60 g. This step will eliminate hepatocytes and possible lumps.
-
Collect the supernatant and transfer it to a 15‐ml tube. Centrifuge at 4° for 3 min at 60 g. This step will eliminate hepatocytes.
-
Collect the supernatant; transfer it to a 15 ml tube. Centrifuge at 4° for 5 min at 300g. This step will recover all the cells in suspension, with the exception of hepatocytes.
-
Discard supernatant.
-
Resuspend the pellet in 200 μl of cold RPMI medium 10% FBS (vol/vol). Count the cells (Boxes 1, 2, 3).
Figure 4.
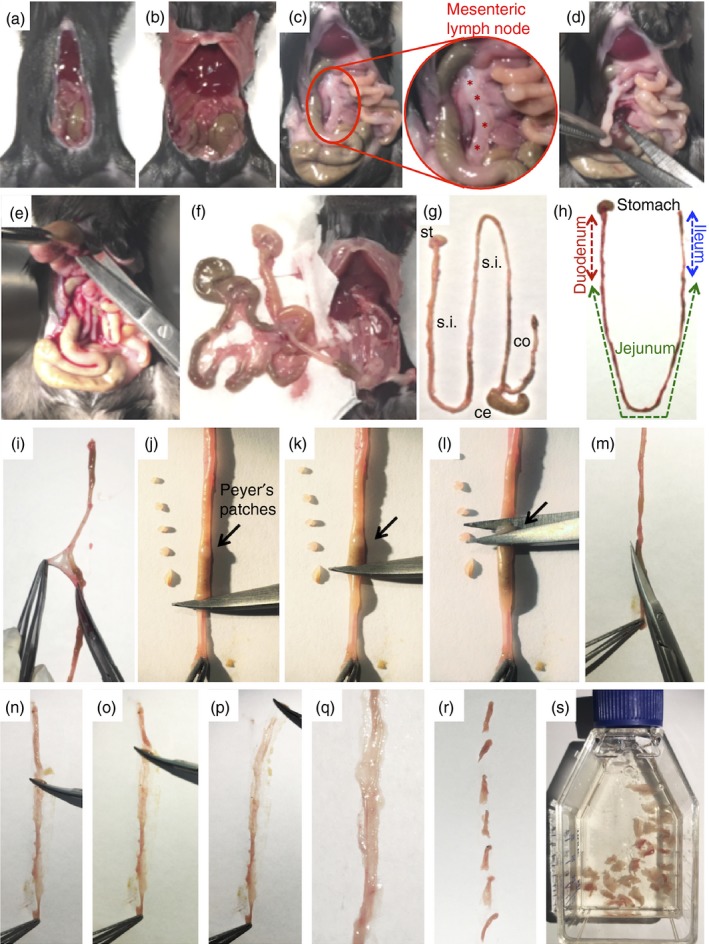
Experimental procedures to remove and process different portions of gastrointestinal tract. (a,b) Midline laparotomy and surgical approach to visualize visceral organs. (c) Four mesenteric lymph nodes (indicated by red stars) can be visualized and (d) removed for further analysis. (e) Stomach is carefully detached from the liver and adjacent organs and (f) all the viscera are removed from the peritoneal cavity. (g) Final aspect of stomach (st), small intestine (s.i.), caecum (ce) and colon (co). (h) Schematic representation of different portions of small intestine. (i, j, k, l) Removal of visceral adipose tissue, mesentery and Peyer's patches. (m) Longitudinal incision to expose intestinal lumen. (n, o, p ,q) Removal all intestinal contents. (r) Intestine is minced into smaller pieces. (s) Initial cleaning/digestion procedures. [Colour figure can be viewed at wileyonlinelibrary.com]
Procedures for CyTOF analysis timing: ~ 3–7 hr
Cell‐surface CyTOF staining protocol:
-
1
Prepare single cell suspensions. In general, try to start with 1·5 × 106 to 3 × 106 cells in 100 μl. Mouse liver (in 5 ml complete Media) used 100–200 μl suspension; mouse lamina propria (in 1 ml complete media) used 100–200 μl.
-
2
Pipette cells into 96‐well polypropylene plates and centrifuge cells for 3 min at 500 g.
-
3
Wash once with 200 μl CyTOF CSB, spin 3 min at 500 g.
-
4
Cell Viability Step: Add 20 μl of cisplatin solution (cisplatin diluted 1 : 1000 in CSB) to wells. Incubate for 5 min at room temperature. Add 200 μl of CSB to solution to dilute out cisplatin. Centrifuge cells for 3 min at 500 g.
-
5
Wash cells with 200 μl CSB, spin 3 min at 500 g. For many samples there can be a residue left in the wells from the culture medium and/or viability stain. To improve cell pelleting, move all samples to fresh wells during this CSB washing step.
-
6
Add 20 μl Fc‐Block antibodies (rat‐anti mouse CD16/32, BD clone 2.4G2) per well (1 : 100 in CSB); incubate for 10 min at room temperature.
-
7
Add 20 μl of metal‐coupled surface antibody cocktail to each well (1 : 100 dilution of each surface antibody in CSB). Incubate at room temperature for minimum of 30 min. Note: Staining antibodies used (see Supplementary material, Table S1 and S2) were coupled to selected lanthanide metals using the MaxPar® Multi‐Metal Labeling Kit according to the manufacturer's protocol.
-
8
Wash twice with 200 μl CSB, spin with 500 g for 3 min.
-
9
Resuspend cells in 200 μl of Intercalator solution.
-
10
Incubate at room temperature for 1 hr OR leave overnight at 4°.
-
11
Centrifuge cells during 3 min at 500 g.
-
12
Wash twice with 200 μl CSB, centrifuge cells during 3 min at 500 g.
-
13
Wash twice with 200 μl MilliQ water (low barium); centrifuge cells during 3 min at 500 g.
-
14
Resuspend cells in 200 μl MilliQ water containing 1 : 10 dilution of EQ beads (for normalization).
-
15
Count cells using a haemocytometer to get a final cell count of 5 × 105 cells/ml.
-
16
Transfer cell suspension to 5‐ml filter cap tubes.
-
17
Add additional EQ Bead MilliQ water to get final volume to ˜ 500 μl.
-
18
Samples are ready for run.
Running CyTOF on helios platform:
-
Acquire samples on a CyTOF2 (Fluidigm, Inc.) mass cytometer operating on the helios platform (software version 6.5.358) using dual count detection.
-
Calibrate the instrument daily, no more than 8 hr before acquisition with the automated full tuning protocol and validate with 4‐element EQ‐beads run at stock concentration.
-
Deliver the filtered samples to the DFCI CyTOF Core diluted in water with 1 : 10 4‐element EQ beads at a concentration at or below 5 × 105 cells/ml and process at a flow rate of 30 μl/min, event rate of 200–600 events per second, and software default settings for threshold and event duration. The length of acquisition is dependent on the final sample volume.
-
After acquisition, normalize samples to the bead signal with the helios software normalizer. Perform normalization in 100‐second intervals with a minimum of 50 beads per interval. Exclude intervals with fewer than 50 beads from the data.
-
Export normalized data as flow cytometry standard (.fcs) files.
Analysis of CyTOF data using cytobank:
-
Import normalized data .fcs files for liver and lamina propria samples into a new experiment file in cytobank.
-
Gating Strategy: first, exclude normalization beads (Fig. 6a), then gate samples on DNA1 and DNA2 double‐positive cells to exclude debris and select for cells with appropriate DNA content levels (Fig. 6b). From the DNA1/2 double‐positive gate, gate live immune cells as Cisplatin‐negative (195P_Viability y‐axis, Fig. 6a) and CD45‐positive (165Ho_CD45 x‐axis, Fig. 6c). Myeloid cells were gated on CD45+ CD3− CD19− cells (Fig. 7d).
-
ViSNE Analysis: analyse gated CD45+ (Fig. 6d,e) live cells and CD45+ CD3− CD19− myeloid cells (Fig. 7e,f) from the liver and lamina propria using the viSNE algorithm in cytobank. Analyse a minimum of 10 000 cells for liver and lamina propria control samples using every marker in the panel for clustering (se Supplementary materials, Tables S1 and S2, for the panels used for the liver and lamina propria).
-
After viSNE analysis, manually define clusters of different cell populations in tSNE1 and tSNE2 dot plots for total CD45+ live cells (Fig. 6d,e) and CD45+ CD3− CD19− myeloid cells (Fig. 7e,f) from lamina propria and liver samples according to surface marker expression. Assign demarcating colours to each population using overlaid visualization in the cytobank. See Fig. 7(e,f) for detailed marker expression levels of myeloid subsets in the liver and lamina propria.
Figure 6.
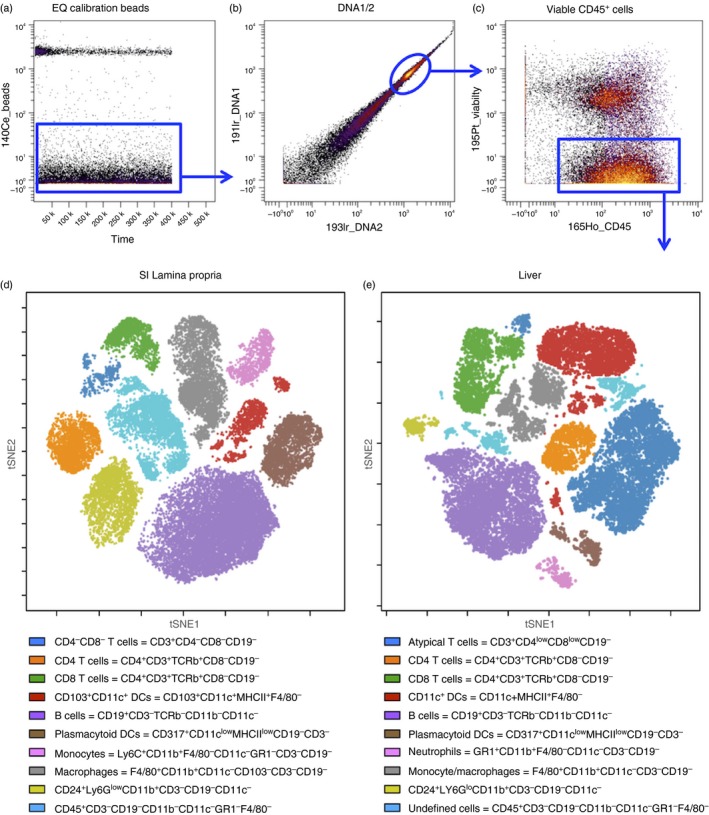
ViSNE analysis of small intestine lamina propria and liver CD45+ immune cells. (a) Dot plot of EQ calibration beads (y‐axis) versus time (x‐axis); (b) Dot plot of 191lr_DNA1 (y‐axis) versus 193lr_DNA2 (x‐axis) on gated bead‐excluded cells. (c) Dot plot of 195P‐Viability (y‐axis) and 165Ho‐CD45 (x‐axis) gated on DNA1/2 double‐positive cells. (d, e) Dot plots depict tSNE1 (y‐axis) versus tSNE2 (x‐axis) of clustering after viSNE analysis for a representative sample from (d) small intestine lamina propria and (e) liver. Undefined cells in liver may include natural killer cells, innate lymphoid cells and others. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 7.
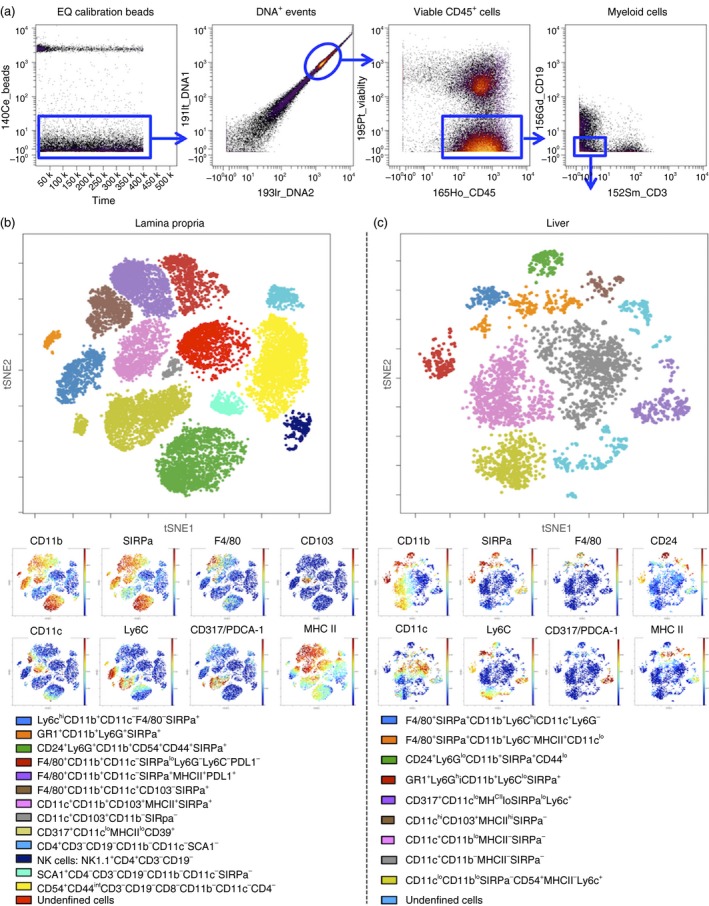
ViSNE analysis of small intestine lamina propria and liver myeloid cells. (a) Dot plot of EQ calibration beads (y‐axis) versus time (x‐axis). (b) Dot plot of 191lr_DNA1 (y‐axis) versus 193lr_DNA2 (x‐axis) on gated bead‐excluded cells. (c) Dot plot of 195P‐Viability (y‐axis) and 165Ho‐CD45 (x‐axis) gated on DNA1/2 double‐positive cells. (d) Gating strategy to obtain live CD45+ CD3− CD19− myeloid cells for viSNE analysis. (e) Dot plot depicts tSNE1 (y‐axis) versus tSNE2 (x‐axis) of clustering after viSNE analysis for a representative sample from small intestine (SI) lamina propria with major myeloid populations labelled according to lineage marker expression (see key). Dot plots depict expression levels (red = high expression, blue = low expression) selected markers: CD11b, CD11c, SIRPa (CD172), Ly6C, F4/80, CD317/PDCA‐1, CD103 and MHC class II. (f) Dot plot depicts tSNE1 (y‐axis) versus tSNE2 (x‐axis) of clustering after viSNE analysis for a representative liver sample with major myeloid populations labelled according to lineage marker expression. Dot plots depict expression levels (red = high expression, blue = low expression) selected markers: CD11b, CD11c, SIRPa (CD172), Ly6C, F4/80, CD317/PDCA‐1, CD24 and MHC class II. [Colour figure can be viewed at wileyonlinelibrary.com]
Anticipated results (Results and discussion)
The number of cells recovered during this protocol will vary according to the mouse strain, age, gender, and mainly due to experimental model. For example, models for inflammatory diseases usually allow for a higher number of leucocytes. However, under certain conditions (i.e. irradiation, chemical ablation or immunosuppression protocols), mouse leucocyte populations can be dramatically reduced throughout the body. Also, any experimental conditions that induce liver injury (drugs, chemicals, infections, hepatectomy) will decrease the number of viable hepatocytes recovered using this protocol. Under these circumstances, polling samples to achieve sufficient cell density to further staining and analysis may be necessary. Under baseline conditions, an adult mouse (8–12 weeks old, weighing around 20–25 g) yields up to 1 × 107 to 5 × 107 liver non‐parenchymal cells, 1 × 107 to 3 × 107 hepatocytes, 5 × 105 to 10 × 105 lamina propria leucocytes and 1 × 107 to 2 × 107 mesenteric lymph node cells (pool of four lymph nodes). Cell viability after these procedures is usually > 90%. Regarding the identification and immunophenotyping of leucocyte populations, different antibody panels will allow for a high variable number of cell clusters. In this protocol, and based on previous studies,24, 28 we could identify 20 populations of leucocytes within the liver and lamina propria (Fig. 6). Further gating on myeloid cells (CD45+ CD3− CD19− events) revealed at least 10 different populations in each organ (Fig. 7).
Figure 5.
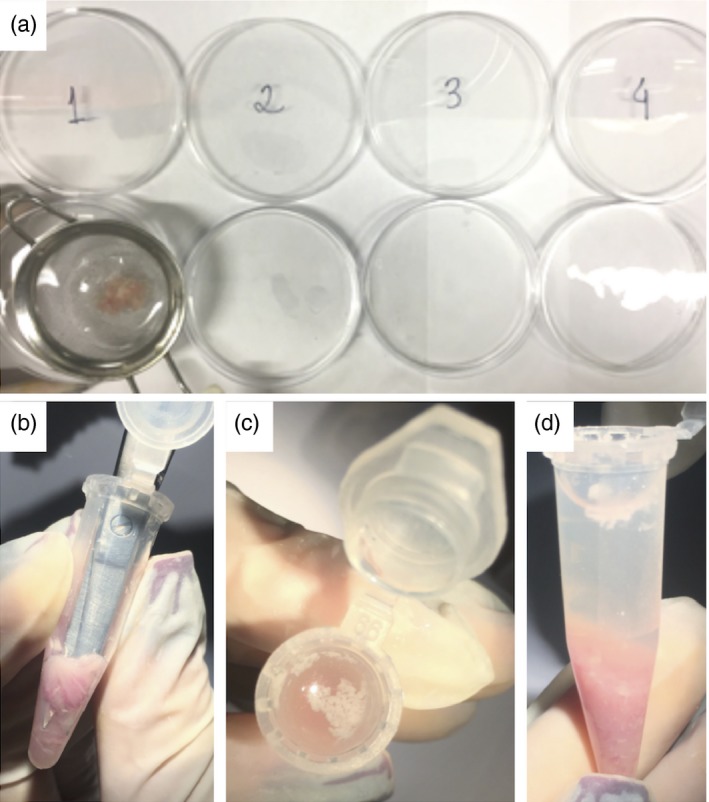
Digestion of intestinal fragments. (a) Initial washing procedures. (b) Intestinal fragments are minced inside a centrifuge tube to enhance digestion. (c, d) Note that all visceral fat in the supernatant should be discarded. [Colour figure can be viewed at wileyonlinelibrary.com]
It is worth mentioning that the proposed protocol might be applied not only in a plethora of immune cell characterization under homeostasis,22, 24 but also during inflammatory processes.23, 29 In fact, using the same isolation protocol and CyTOF analysis, we recently showed that during massive liver macrophage depletion by clodronate injection (CLL), an overt inflammatory response was observed. Using a multiplexed cytokine array, we demonstrated that several different cytokines, including MIG, interleukin‐9, CXCL1, and others were significantly increased after macrophage depletion. Concomitantly, CyTOF analysis revealed that 2 days after CLL injection, all the clusters defined as liver macrophages and dendritic cells were significantly reduced. Interestingly, CyTOF was also useful to monitor their replenishment dynamics over time.23 In addition, due to changes in the expression of the surface of intracellular markers used for CyTOF staining throughout the experimental protocol, minor or major changes in clustering position may be observed, which may require a deeper and more complex analysis.
Disclosures
The authors declare that there are no conflicts of interest.
Supporting information
Table S1. List of monoclonal antibodies used in this study – liver non‐parenchymal cells
Table S2. List of monoclonal antibodies used in this study – small intestine lamina propria
Acknowledgements
The authors would like to thank Dr Michelle Poulin and Dr Gary Impey (Fluidigm) for their help in the preparation of CyTOF figures and description. Also, we would like to thank CAPES, FAPEMIG and CNPq (Brazil) for financial support.
References
- 1. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014; 14:667–85. [DOI] [PubMed] [Google Scholar]
- 2. Perez‐Lopez A, Behnsen J, Nuccio SP, Raffatellu M. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol 2016; 16:135–48. [DOI] [PubMed] [Google Scholar]
- 3. Herbrand H, Bernhardt G, Forster R, Pabst O. Dynamics and function of solitary intestinal lymphoid tissue. Crit Rev Immunol 2008; 28:1–13. [DOI] [PubMed] [Google Scholar]
- 4. Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ et al Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol 2005; 3:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol 2013; 14:996–1006.24048121 [Google Scholar]
- 6. Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev 2014; 260:102–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol 2012; 5:232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faria AM, Weiner HL. Oral tolerance: therapeutic implications for autoimmune diseases. Clin Dev Immunol 2006; 13:143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 2016; 16:341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McDonald B, Kubes P. Neutrophils and intravascular immunity in the liver during infection and sterile inflammation. Toxicol Pathol 2012; 40:157–65. [DOI] [PubMed] [Google Scholar]
- 11. Nesseler N, Launey Y, Aninat C, Morel F, Malledant Y, Seguin P. Clinical review: the liver in sepsis. Crit Care 2012; 16:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bernal W, Wendon J. Acute liver failure. N Engl J Med 2013; 369:2525–34. [DOI] [PubMed] [Google Scholar]
- 13. Targan SR, Karp LC. Defects in mucosal immunity leading to ulcerative colitis. Immunol Rev 2005; 206:296–305. [DOI] [PubMed] [Google Scholar]
- 14. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009; 9:313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Podolsky DK. The current future understanding of inflammatory bowel disease. Best Pract Res Clin Gastroenterol 2002; 16:933–43. [DOI] [PubMed] [Google Scholar]
- 16. Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High‐yield and high‐purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 2015; 10:305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Breton G, Lee J, Liu K, Nussenzweig MC. Defining human dendritic cell progenitors by multiparametric flow cytometry. Nat Protoc 2015; 10:1407–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lian ZX, Okada T, He XS, Kita H, Liu YJ, Ansari AA et al Heterogeneity of dendritic cells in the mouse liver: identification and characterization of four distinct populations. J Immunol 2003; 170:2323–30. [DOI] [PubMed] [Google Scholar]
- 19. Yao Y, Liu R, Shin MS, Trentalange M, Allore H, Nassar A et al CyTOF supports efficient detection of immune cell subsets from small samples. J Immunol Methods 2014; 415:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Unen V, Li N, Molendijk I, Temurhan M, Hollt T, van der Meulen‐de Jong AE et al Mass cytometry of the human mucosal immune system identifies tissue‐and disease‐associated immune subsets. Immunity 2016; 44:1227–39. [DOI] [PubMed] [Google Scholar]
- 21. Bendall SC, Simonds EF, Qiu P, el Amir AD, Krutzik PO, Finck R et al Single‐cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011; 332:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S et al Unsupervised high‐dimensional analysis aligns dendritic cells across tissues and species. Immunity 2016; 45:669–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB et al Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology 2016; 151:1176–91. [DOI] [PubMed] [Google Scholar]
- 24. Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KW et al High‐dimensional analysis of the murine myeloid cell system. Nat Immunol 2014; 15:1181–9. [DOI] [PubMed] [Google Scholar]
- 25. el Amir AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC et al viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013; 31:545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bruggner RV, Bodenmiller B, Dill DL, Tibshirani RJ, Nolan GP. Automated identification of stratifying signatures in cellular subpopulations. Proc Natl Acad Sci U S A 2014; 111:E2770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu P, Simonds EF, Bendall SC, Gibbs KD Jr, Bruggner RV, Linderman MD et al Extracting a cellular hierarchy from high‐dimensional cytometry data with SPADE. Nat Biotechnol 2011; 29:886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G et al Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol 2013; 13:145–9. [DOI] [PubMed] [Google Scholar]
- 29. Lutz C, Mozaffari M, Tosevski V, Caj M, Cippa P, McRae BL et al Increased lymphocyte apoptosis in mouse models of colitis upon ABT‐737 treatment is dependent upon BIM expression. Clin Exp Immunol 2015; 181:343–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of monoclonal antibodies used in this study – liver non‐parenchymal cells
Table S2. List of monoclonal antibodies used in this study – small intestine lamina propria