
Fig. 1.
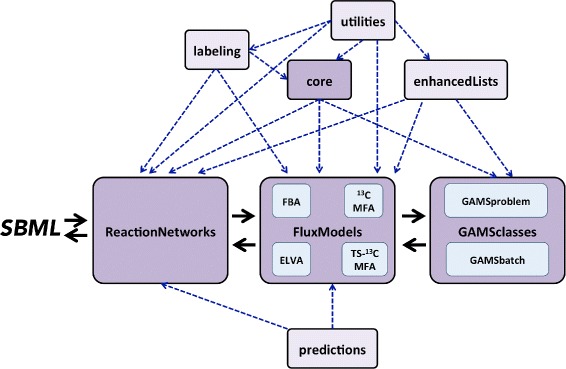
Diagram of jQMM library module relationships and typical flow for flux analysis. Flux calculations typically start with information stored in an SBML (Systems Biology Markup Language) file and translated into a reaction network. That reaction network is enclosed in a flux model of the appropriate type for the desired method (FBA, 13C MFA, 2S-13C MFA, ELVA). The flux model instance uses the GAMSclasses module in order to solve the appropriate optimization problem, and turns it back to the flux model instance, which stores the information as a reaction network or an SBML file. The core, labeling, utilities and enhancedLists modules are used by these “main workflow” modules (in darker shade). Predictions use information from reaction networks and flux models to make flux predictions for genetic modifications