

Abstract
Riboflavin is essential in numerous cellular oxidation/reduction reactions but is not synthesized by mammalian cells. Riboflavin absorption occurs through the human riboflavin transporters RFVT1 and RFVT3 in the intestine and RFVT2 in the brain. Mutations in these genes are causative for the Brown–Vialetto–Van Laere (BVVL), childhood-onset syndrome characterized by a variety of cranial nerve palsies as well as by spinal cord motor neuron (MN) degeneration. Why mutations in RFVTs result in a neural cell–selective disorder is unclear. As a novel tool to gain insights into the pathomechanisms underlying the disease, we generated MNs from induced pluripotent stem cells (iPSCs) derived from BVVL patients as an in vitro disease model. BVVL-MNs explained a reduction in axon elongation, partially improved by riboflavin supplementation. RNA sequencing profiles and protein studies of the cytoskeletal structures showed a perturbation in the neurofilament composition in BVVL-MNs. Furthermore, exploring the autophagy–lysosome pathway, we observed a reduced autophagic/mitophagic flux in patient MNs. These features represent emerging pathogenetic mechanisms in BVVL-associated neurodegeneration, partially rescued by riboflavin supplementation. Our data showed that this therapeutic strategy could have some limits in rescuing all of the disease features, suggesting the need to develop complementary novel therapeutic strategies.
Riboflavin (7,8-dimethyl-10-ribityl-isoalloxazine; vitamin B2) is a water-soluble group B vitamin and the precursor of the coenzymes flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN)1. They are essential cofactors in different metabolic processes, including carbohydrate, amino acid, and lipid metabolism and the electron transport chain2. Riboflavin cannot be readily synthesized in mammalian cells, so it must be absorbed from the diet via riboflavin transporters (RFVTs)3,4,5. RFVT1 and RFVT3 are predominantly expressed in the intestine and RFVT2 in the brain. The absence of riboflavin in the diet causes a range of developmental and growth disorders6. Furthermore, riboflavin supplementation is useful in the treatment of inborn errors of metabolism, such as mild multiple acyl-CoA dehydrogenation defect (MADD) and some mitochondrial diseases7,8.
In 2010, it was established that autosomal recessive mutations in the riboflavin transporter genes SLC52A2 (coding for RFT3, RFVT2) and SLC52A3 (alias C20orf54, coding for RFT2, RFVT3) are responsible for the neurodegenerative disorder previously identified as Brown–Vialetto–Van Laere (BVVL) or Fazio Londe (FL) syndrome9,10,11,12,13,14. RFVT1 does not seem to be associated with a human disease (or as an alternative lethal at embryonic stage), as no patients with this deficiency have been described.
BVVL syndrome is a rare neurodegenerative disease characterized by progressive ponto-bulbar palsy, bilateral sensorineural hearing loss, muscle weakness, and amyotrophy due to motor neuron (MN) degeneration13,14,15. The principal cause of death is respiratory failure due to diaphragm denervation9,10,11,12,16,17,18,19, and the FL clinical presentation is similar to BVVL, without the hearing loss. Therapy with high-dose oral riboflavin (B2) was efficacious in BVVL cases with significant clinical improvement1. Despite this advancement in the knowledge of riboflavin deficiency disorders, the causal relationship between riboflavin transporter mutations and neural/MN degeneration remains unknown. Addressing this question might contribute not only to increasing understanding of the disease but also to developing novel and more effective treatments for use in association with riboflavin replacement therapy.
In this study, we thus investigated several pathophysiological aspects in induced pluripotent (iPSC)-derived MNs from two BVVL patients (BVVL-MNs). Establishing in vitro patient-specific models allowed the identification of MN pathological features. Patient MNs exhibited a different transcriptional signature with respect to wild-type (WT) MNs (WT-MNs), as demonstrated by RNA sequencing analysis, suggesting impairment in cytoskeletal structure, axonogenesis, and neurotrophic factor metabolism. Indeed, we demonstrated a perturbation in mitochondrial function and in the autophagic pathway. These features represent emerging pathogenetic mechanisms in BVVL-associated neurodegeneration and possible targets for therapeutic intervention, in addition to riboflavin supplementation.
Results
Axonal elongation was significantly affected in MNs derived from BVVL patients
We reprogrammed primary fibroblasts from two BVVL patients (carrying the c.796C/c.9555C > T mutations in RFVT2 and the c.1555C > T/c.1255G > A mutations in the RFVT3 gene, respectively11; to generate iPSC lines, using a protocol based on the transfection of episomal reprogramming vectors20. Two WT iPSC lines reprogrammed from neonatal foreskin fibroblasts and unaffected adult skin fibroblasts were used as controls (Supplementary Table 1). After being reprogrammed, the colonies were selected for further expansion. At least three clones were isolated for each patient. The iPSCs could differentiate into cell types from all three embryonic germ layers, confirming their in vitro pluripotency. The cells expressed pluripotency markers such as NANOG and SSEA-3, as demonstrated by immunocytochemistry analysis (Fig. 1S).
Because MNs are selectively affected in riboflavin deficiency pathogenesis, we evaluated whether our BVVL iPSCs could be lineage-committed toward the MN fate in order to define the effect of RFVT mutations in this cell model. BVVL and WT iPSCs were differentiated into spinal MNs (BVVL-MNs and WT-MNs, respectively) with a multistage differentiation protocol previously developed for human pluripotent stem cells20 (Fig. 1A).
Figure 1. Characterization of BVVL iPSC-derived motor neurons (MNs).
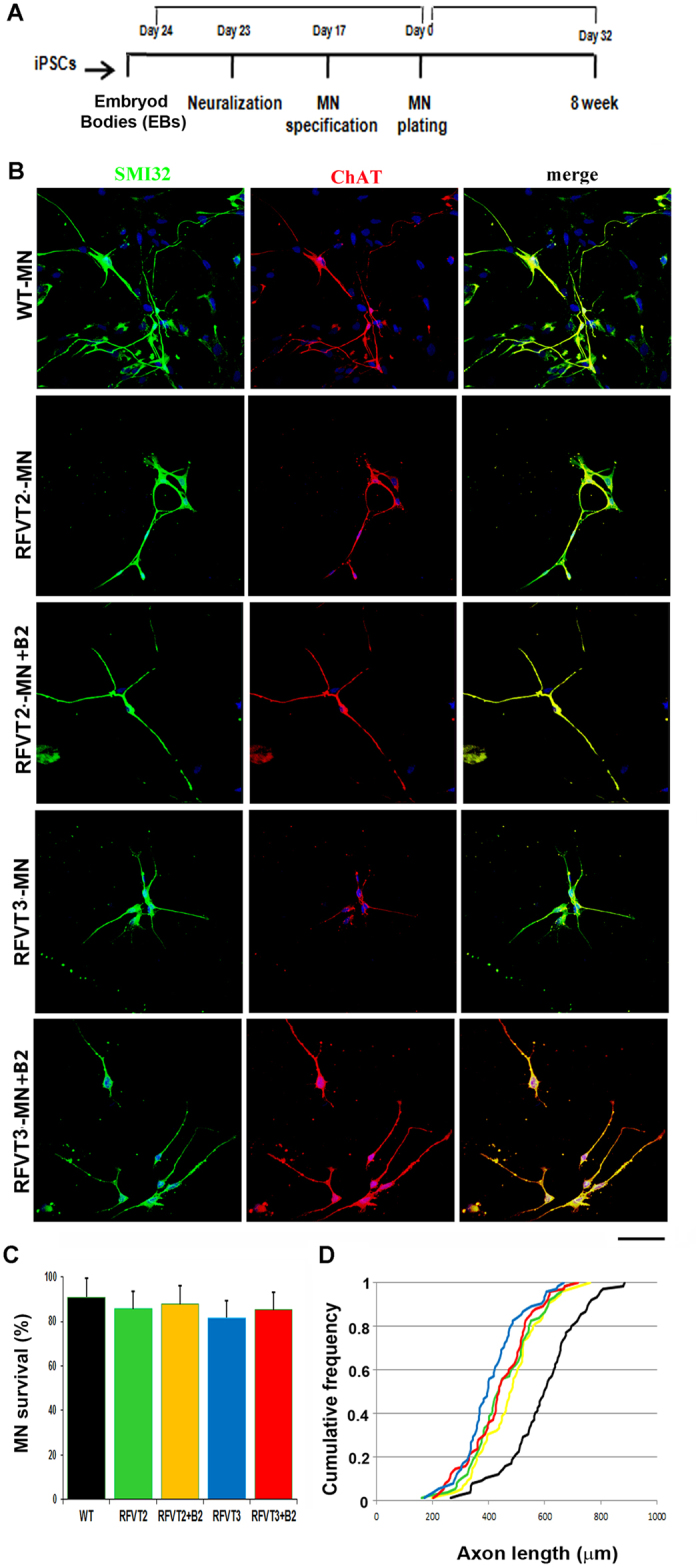
(A) Experimental outline for WT and BVVL-MN differentiation and characterization. (B) Immunocytochemistry of WT-MNs and BVVL-MNs (RFVT2-MNs, RFVT3-MNs). The cells were positive for typical MN markers: SMI32 (green) and ChAT (red) (merge, yellow signal). Nuclei are labeled with DAPI (blue). The analyses were performed in basal condition and after supplementation with riboflavin (B2). Scale bar: 75 μm. (C,D) Morphometric analysis of WT-MNs and BVVL-MNs. (C) Eight weeks after differentiation, BVVL-MN survival was not significantly reduced compared with WT-MNs (Student t test). Values represent means ± SD from five independent experiments performed in triplicate. (D) After 8 weeks of culture, BVVL-MNs exhibited a significantly reduced axon elongation with respect to WT cells. The improvement of axon elongation was observed after riboflavin supplementation, in particular in the patient with mutant RFVT3. Kolmogorov–Smirnov test. Five independent experiments performed in triplicate.
After 5 weeks under differentiation conditions, MNs were developed that expressed the pan-neuronal marker TUJ1 and the MN-specific transcription factor ISL1. Most of these TUJ1/ISLET1-positive neurons expressed choline acetyltransferase (ChAT) and were positive for the MN marker SMI32, indicating MN maturity (Fig. 1B). To evaluate the phenotype of BVVL, we included both basal and riboflavin-supplemented conditions (Fig. 1B).
The in vitro differentiation protocol generated a high proportion of MNs but also a mixed cell population containing non-MN cells. To further select MNs, we employed a physical strategy based on gradient centrifugation. This selection step allowed the enrichment of cells that were double-positive for ChAT and SMI32 (WT: 89.4 ± 6.3%; BVVL: 88.2 ± 8.6%). Less than 1% of iPSC-derived cells were positive for the astrocyte marker glial fibrillary acidic protein.
To reveal disease phenotypes associated with BVVL-MNs, we measured basal MN survival by counting SMI32-positive and ChAT-positive cells. No significant survival reduction was detected in either culture condition (Fig. 1C).
To investigate whether RFVT mutations impair axon stability and elongation, we performed morphological analyses. After 8 weeks of culture, BVVL-MNs exhibited a significantly reduced axon elongation that was improved but partially rescued by riboflavin supplementation, in particular in the patient with mutant RFVT3 (Fig. 1D). Overall, BVVL-MNs exhibited no major defects in cell survival but did show a significant axon elongation defect that riboflavin treatment ameliorated.
Dysregulated gene expression profiles in BVVL-MNs
To identify molecular pathways that are potentially relevant for BVVL pathogenesis, we applied massive parallel RNA sequencing to MNs derived from BVVL patients and controls in the basal condition and after riboflavin treatment.
RNA sequencing disclosed 537 differentially expressed transcripts (fold change ≥2 and diverge probability ≥0.8), 246 of which were up-regulated and 291 down-regulated in BVVL cells (Fig. 2A, Supplementary Table 2). As a measure of the transcripts most affected by RFVT mutation, we defined the identity of the 30 transcripts most increased and decreased in abundance at a false discovery rate (FDR) of 5% (Fig. 2B). To investigate the RNA-seq data for biological meaning, we used two bioinformatics tools that query for enriched gene ontology terms: gene-annotation enrichment analysis with DAVID21 and Gene Set Enrichment Analysis (GSEA)22.
Figure 2. Transcriptional analysis of BVVL-MNs vs WT-MNs by RNA-seq analysis shows dysregulation of specific mRNA subsets.

(A) Left: Graphical representation of the transcriptomic analysis of differentially expressed genes among BVVL-MNs vs WT-MNs. Right: A total of 537 differentially expressed genes (fold change ≥2 and diverge probability ≥0.8) were observed in BVVL-MNs compared to WT-MNs; 246 were up-regulated, and 291 were down-regulated in BVVL-MNs. (B) Top 30 genes (based on fold change) misregulated in BVVL-MNs vs WT-MNs with an FDR 5%. (C) Selected gene sets obtained using the GSEA algorithm. Gene sets that were enriched in BVVL-MNs include terms related to protein and RNA metabolism, while depleted genes were associated with the extracellular matrix and growth factors. (D) Reactome gene categories and selected genes dysregulated in BVVL-MNs. (E,F) Comparison between RNA-seq (E) and quantitative RT-PCR (F) data for a select number of transcripts involved in neurite outgrowth and axon guidance in BVVL-MNs with respect to WT-MNs. Error bars represent SEM. ***P < 0.001; **P < 0.001, *P < 0.05. (G) Western blot analysis of EPHA2 in BVVL and WT-MNs showing a reduced protein level in diseased cells, without significant modifications after riboflavin treatment. Numbers indicate protein levels normalized to actin.
Transcripts implicated in ‘cytoskeleton organization’, ‘transcriptional regulation’, and ‘protein translation’ as well as genes implicated in ‘mitochondrial function’ were the most dysregulated terms.
Taking advantage of GSEA, we identified gene sets that were significantly altered in BVVL-MNs (normalized enrichment score <1.5; Fig. 2C). Gene sets that were enriched in BVVL-MNs included terms related to “protein and RNA metabolism” while depleted genes were associated with “extracellular matrix and growth factors” (Fig. 2C,D). Several transcripts encoding for matrix metalloproteinases and metalloproteases of the ADAMS family, involved in tissue remodeling and dysregulated during neurodegeneration and in MN disease23, were specially downregulated in BVVL-MNs (Fig. 2D), supporting their also taking part in BVVL disease. Dysregulation of several growth factor transcripts related to IGF1 and TGF-β pathways was also observed in our in vitro model. One of the most represented gene sets is involved in neurite outgrowth and axon guidance and includes alteration of neurofilament (NF) mRNA, in particular in the NF heavy subunit (NFH), relevant constituents of the cytoskeletal structure, and molecules of the ephrin (EPHA) pathway that are important for axon guidance. Because NF imbalance has been associated with MN diseases24,25, we analyzed these events in-depth, as described below. Expression differences were validated by quantitative RT-PCR (Fig. 2E,F), confirming the downregulation of neuronal pentraxins (NPTX2), Palladin (PALLD), and Ephrin-A2 (EPHA2), as well as the up-regulation of SLC8A2 in BVVL-MNs in comparison with WT cells. Interestingly, all of these genes are related to neurite outgrowth and guidance. Given the potential relevance of EPHA2 in BVVL axonal defects, we also evaluated the protein level of EPHA2, confirming its downregulation in BVVL cells, without significant modifications after riboflavin treatment (Fig. 2G). Overall, our gene expression profile data indicate the presence of significant alterations in cytoskeletal organization and axonogenesis in the BVVL-MNs, suggesting that these could be key events in their degeneration.
Riboflavin treatment rescued a specific subset of genes related to axonal growth and metabolism
The comparison of treated versus untreated BVVL-iPSC MNs (BVVL-MNs B2 vs BVVL-MNs) revealed 765 differentially expressed transcripts (Fig. 3A, Supplementary Table 3). Remarkably, the genes that were most up-regulated in treated cells include some that were down-regulated in the comparison between BVVL-MNs and WT-MNs, suggesting rescue with a shift toward the WT gene expression profile. The direct comparison between the treated BVVL-MN and WT-MNs confirmed that riboflavin supplementation shifted the expression profile toward WT (Fig. S2).
Figure 3. Transcriptional analysis of BVVL-MNs before and after riboflavin treatment unravels the rescue of genes related to axonal growth and metabolism.

(A) Left: Graphical representation of the transcriptomic analysis of differentially expressed genes among BVVL-MNs before and after riboflavin (B2) treatment. Right: A total of 765 differentially expressed genes (fold change ≥2 and diverge probability ≥0.8) were observed in BVVL-MNs compared to B2-treated MNs (BVVL+B2). (B) Top 30 genes (based on fold change) differentially expressed in BVVL-MNs vs BVVL-MNs+B2 with an FDR 5%. (C) Selected gene sets obtained using the GSEA algorithm. Gene sets that were enriched in BVVL-MNs+B2 include terms related to protein, RNA, and mitochondrial metabolism, while depleted genes were associated with axon guidance. (D) Reactome gene categories and selected genes differentially expressed in BVVL-MNs include genes related to axon metabolism and growth as well as to mitochondrial function. (E,F) Comparison between RNA-seq (E) and quantitative RT-PCR (F) data for a select number of transcripts involved in neurite outgrowth and axon guidance in BVVL-MNs before and after treatment with B2 (BVVL-MNs+B2), in comparison with WT-MNs. B2 treatment improved the gene expression profile of DNAH10, INA, NCAN, and PSMB8, which became more similar to WT cells. Error bars represent SEM. ***P < 0.001; **P < 0.001, *P < 0.05.
To determine the most significant effects of riboflavin treatment in terms of transcriptional changes, we identified the top 30 transcripts most increased and decreased in abundance at a FDR of 5% (Fig. 3B). GSEA identified gene sets that were significantly modified in BVVL-MNs after riboflavin treatment (normalized enrichment score <1.5; Fig. 3C).
Gene sets that were modified after treatment in BVVL-MNs included terms related to protein and RNA metabolism, mitochondrial function, and autophagy (Fig. 3C). In particular, the reactome gene category that was significantly modified was related to axon guidance and mitochondrial metabolism (Fig. 3D).
Quantitative RT-PCR was used to validate the expression of some differentially expressed genes related to axon guidance, neuronal metabolism (DNAH10, INA, NCAN), and proteasome cell function (PSMB8), confirming that their expression pattern shifted toward the normal WT profile after riboflavin treatment (Fig. 3E,F). Overall, these data suggest that the therapeutic action of B2 supplementation also is exerted through the rescue of genes that govern axon growth and maintenance.
Alteration of neurofilament expression pattern in BVVL-MNs and its rescue after riboflavin treatment
RNA-seq analysis showed that a significant proportion of all transcripts involved in cytoskeletal and axon guidance were altered in BVVL-MNs (Fig. 2C–G). Perturbation of NF balance in in vitro and in vivo models results in phenotypes closely resembling human MN pathologies25,26 and has previously been suggested as a component of human ALS disease24,25.
Our RNA-seq data showed that the NFH was specifically up-regulated in our BVVL-MNs (Fig. 2D), as confirmed by real-time RT-PCR (Fig. 4A) and western blot analysis (Fig. 4B). In particular, the expression level of NF medium (NFM) and NF light (NFL) subunits was imbalanced with respect to NFH (Fig. 4B), suggesting that the altered proportion of NF subunits might have pathogenetic relevance, as already demonstrated in ALS25.
Figure 4. Neurofilament expression pattern is altered in BVVL MNs and is rescued by riboflavin treatment.

(A) Real-time RT-PCR showed the up-regulation of NFH mRNA in BVVL-MNs vs WT-MNs. This upregulation was significantly reduced in BVVL-MNs+B2. Error bars represent SEM. *P < 0.05. (B) Western blot analysis of NFH (200), NFM (medium) (168), and NFL (light) (68) in BVVL-MNs with respect to WT-MNs and BVVL-MNs+B2. Numbers indicate protein levels normalized to actin. NFH was upregulated in BVVL-MNs relative to WT-MNs. This up-regulation was reduced after B2 treatment. The expression level of NEFM and NEFL was imbalanced respect to NFH. (C) Immunofluorescent images of NFH (green) and ChAT+ (red) in WT-MNs, BVVL-MNs and BVVL-MNs+B2. NFH inclusions (green) were observed in the cell body and in neurites of BVVL-MNs. This feature was ameliorated by B2 treatment. (D) Quantification of NFH inclusion–containing in cell bodies and neuritis in WT-MNs, BVVL-MNs and BVVL-MNs+B2. Error bars represent SEM. *P < 0.05. Scale bar: 75 μm.
Furthermore, the NF immunofluorescent signal in MNs revealed that the expression levels of NFH were altered (Fig. 4C,D). In particular, progressively increased numbers of NFH inclusions in both cell bodies and neurites of BVVL-MNs was observed. Riboflavin treatment mitigated NFH aggregation and neurite degeneration (Fig. 4C,D). These results highlight that NF misregulation is a critical component of MN degeneration in BVVL.
Dysregulation of mitochondrial dynamics and autophagy in BVVL-MNs and improvement after riboflavin supplementation
To define whether the transcriptional changes in mitochondrial genes identified by RNA-seq in BVVL-MNs (Figs 2 and 3) were indicative of mitochondria disturbances, we analyzed whether a perturbation in mitochondrial dynamic and function was present at proteomic and biochemical level. Riboflavin is the precursor of the active coenzymes FMN and FAD, which are essential cofactors for the electron transport chain and the lipid metabolism2. Moreover, patients with BVVL syndrome present biochemical defects that mimic MADD syndrome, an inherited disease of mitochondrial fatty acid beta-oxidation that responds to riboflavin supplementation10. Thus, we determined whether a similar perturbation7 was detectable also in BVVL-MNs.
First, we assessed for the presence of an OXPHOS or fatty acid β-oxidation impairment in BVVL-MNs, as observed in MADD. Mitochondrial respiration and β-oxidation were not different in BVVL-MNs (with or without riboflavin treatment) with respect to control cells (Fig. 5A,B).
Figure 5. Mitochondrial function and autophagy in BBVL MNs before and after B2 treatment.

Respiratory chain activity (A) and β-oxidation activity (B) analysis in BVVL-MNs, BVVL-MNs+B2, and WT-MNs. No defect in respiratory chain and β-oxidation activity was observed in BVVL cells. The enzymatic activity of respiratory complexes I–IV (cI–IV) and acetyl CoA carboxylases (P CoA, P CoA, and B CoA) was determined and normalized to citrate synthase activity. Values are expressed as nmoles/min/mg protein. (C) Western blot analysis of MFN2 and DRP1 in BVVL-MNs, BVVL-MNs+B2, and WT-MNs. The MFN2/DRP1 ratio was reduced in BVVL-MNs relative to WT-MNs (*P < 0.05, ANOVA). (D) LC3BII/I and p62 expression in WT-MNs, BVVL2-MNs, and BVVL2-MNs+B2. The LC3B-II/I ratio was reduced whereas p62 expression was increased in BVVL-MNs with respect to WT (***P < 0.0001, *P < 0.05, ANOVA). After riboflavin treatment, LC3B-II/I ratio and p62 expression showed a shift towards a WT pattern. (E) Western blot analysis of Bnip3 and Parkin in WT-MNs, BVVL-MNs, and BVVL-MNs+B2 (**P < 0.001, *P < 0.05, ANOVA). The levels of both proteins were reduced in BVVL-MNs with respect to WT-MNs.
To then analyze whether a dysregulation of the fusion/fission mitochondrial events takes place in the BVVL-MNs, as occurs in MADD, we examined the expression level of mitofusin2 (MFN2), one of the key regulators of mitochondria fusion degraded during inhibition of fusion27, and dynamin-related protein 1 (DRP1), which mediates outer mitochondrial membrane fission (Fig. 5C). Our results showed that the MFN2/DRP1 ratio was decreased in BVVL-MNs when compared with control cells but that this pattern was not changed by riboflavin treatment (Fig. 5C). These findings are similar to what has been observed in MADD cells7.
We afterward investigated whether the fusion inhibition events in mitochondria correlate with changes in autophagy. To check for the induction of autophagy, we compared the expression of the autophagy markers LC3B and p62/SQSTM in BVVL-MNs and WT-MNs. A reduction in the LC3B-II/LC3B-I ratio and increase in autophagic substrate p62 demonstrated reduced autophagy in BVVL-MNs with respect to WT-MNs (Fig. 5D). After riboflavin treatment, the LC3B-II/LC3B-I ratio and p62 expression in BVVL-MNs shifted towards a WT pattern (Fig. 5D). Mitochondrial clearance by autophagy is mediated by the induction of proteins that accumulate on mitochondria before their removal28. We observed that protein levels of PARKIN and BNIP3 were negatively regulated in BVVL-MNs (Fig. 5E).
Taken together, our data suggest that a reduction of fusion and mitophagy was present in BVVL-MNs, supporting their limited capacity for renewing mitochondria. Riboflavin exposure normalized the mitophagy rate. These findings showed that pathogenetic pathways in BVVL involve mitochondria turnover but not their functionality. Overall, mitochondrial turnover could represent key pathogenetic components and potential therapeutic targets of BVVL.
Discussion
BVVL and FL are rare genetic MN diseases caused by mutations in RFVT2/3 genes; however, the mechanisms by which mutant RFVT proteins induce MN degeneration are largely unknown1. Riboflavin (B2) supplementation seems to ameliorate the clinical phenotype even if the exact mechanisms of action on MNs have not yet clarified1.
Patient-specific iPSCs represents a novel tool for in vitro disease modelling and therapeutic discovery in neurodegenerative diseases that involve a specific neuronal population29. As an innovative platform to gain insights into the pathogenetic mechanisms underlying BVVL, in this study, we generated for the first time an in vitro BVVL disease model using human patient–specific iPSC-derived MNs. Because surface specific MN markers have not yet been established, we isolated MNs with gradient centrifugation, confirming the generation of pure MNs by immunocytochemical and molecular marker expression.
This system allowed analysis of the impact of RFVT2/3 variants on MN phenotype and to study the effects of riboflavin supplementation. Our studies demonstrated that the BVVL-MNs showed a significant reduction in their axonal length, improved by riboflavin treatment. This phenotype is similar to that observed in MN disease such as SMA20,30,31, and this neuropathological feature could be responsible for BVVL neurological motor signs. In fact, several studies have demonstrated a reduction in axonal growth in the MNs derived from SMA-iPSCs relative to control iPSCs20,32.
Considering the cytoskeletal-dependent neuronal pathology observed in BVVL in a broader context, we can group BVVL with those pathologies in which cytoskeletal affect neuropathological features: these defects could be responsible for the neurological motor signs of BVVL. In general, the cytoskeleton is important in a broad series of events regulating neurogenesis and maintenance of neuronal function; therefore, alterations in genes controlling cytoskeletal dynamics can easily lead to severe neurologic diseases33, particularly in combination with defects in the developing nervous system. Microtubules dysfunction has been observed in a model of Parkinson’s disease (PD) in which inhibition of the mitochondrial Complex I leads to accumulation of reactive oxygen species34,35. Moreover, a recent study using progressive motor neuronopathy mice carrying a missense loss-of-function mutation in tubulin binding cofactor E (TBCE) highlighted the potential contribution of cytoskeletal defects to sensory neuropathy in human MN disease36,37. The cytoskeletal alterations observed in many neurological disorders may be primary or secondary factors in neurological motor signs, but regardless, identifying therapies that can correct these microtubule abnormalities could improve the clinical manifestations. We hope that future research will target specific proteins or pathways affecting the cytoskeleton and ways to shift the status of the altered cytoskeleton back toward normal, thus leading to effective therapies for a wide spectrum of neurodegenerative diseases.
The improvement in RNA sequencing technologies has allowed identification of specific transcriptional changes in human MN diseases such ALS and SMA, contributing to defining possible disease hallmarks and pathways associated with differential MN vulnerability, thus presenting a number of striking targets for future therapeutic strategy38,39. In particular, Eggan and colleagues observed a specific transcriptional signature indicative of increased oxidative stress, reduced mitochondrial function, altered subcellular transport, and activation of the ER stress in ALS-MNs39. Similar changes in hyper-activation of the ER stress pathway have been described in SMA-MNs38 in association with RNA splicing abnormalities20 leading to the hypothesis that they could be mechanisms of selective MN death.
In this study, for the first time, by applying RNA sequencing analysis to our model in the basal condition and after riboflavin treatment, we identified specific BVVL gene expression changes in RNA/protein metabolism, axonal and cytoskeletal structure, and mitochondrial/oxidative function. Novel candidate genes, with significantly altered expression in BVVL-MNs compared to WT-MNs were selected. In particular, we detected the significant downregulation of neuronal pentraxin 2 (NPTX2), palladin (PALLD), and EPH receptor A2 (EPHA2), all genes related to neurite outgrowth and axon guidance. NPTX and their corresponding receptors have been studied as synapse-associated proteins in the nervous system and are relevant in promoting neuronal growth and circuitry organization. NPTX2 downregulation can negatively affect these events40. PALLD is a critical component of the developing nervous system, with an important role in axonal extension41. A reduction in the expression of PALLD, as observed in our BVVL-MNs, can result in a failure of neurite outgrowth. The significantly altered expression of the axon guidance protein EPHA2, observed in our model, could have an important scientific relevance based on its critical function during MN axon outgrowth to the hindlimb with potential roles in pathfinding42. Indeed, the perturbation of axon guidance protein, which guide growing axons during development and control the structural plasticity of synaptic connections in adults, has been implicated in several neurological diseases43 such as autism spectrum disorders and epilepsy44 and MN disorders, in particular ALS45, even if the molecular mechanisms are poorly understood. For several neurological diseases, different studies have identified causative mutations in genes for axon guidance, but for other disorders such as BVVL, additional studies are needed to demonstrate the role of these proteins in the pathogenetic mechanisms and to identify new targets for therapeutic intervention.
The comparison of B2-treated versus untreated BVVL-MNs allowed us to identify differentially expressed transcripts. Interestingly, the genes that were most up-regulated in treated cells include some that were down-regulated in the comparison between BVVL-MNs and WT-MNs, supporting a rescue of gene expression profile with a shift toward the WT pattern. Among the transcripts that were significantly modified towards a WT pattern, we identified DNAH10 (Dynein axonemal heavy chain 10) and INA (alpha-internexin), as well as Neurocan (NCAN), a proteoglycan that modulates axonal outgrowth. DNAH10 is a microtubule-associated protein and key component of MN axons46. Interestingly, mutations in DNAH10 have been recently associated with motor-sensory neuropathies (Charcot–Marie–Tooth disease)46. Alpha-internexin (INA) is a neuronal intermediate filament protein that plays a similar role to light NF47. Its expression precedes that of NF light during neuronal development47. NCAN is an important nervous tissue–specific chondroitin sulfate proteoglycan that modulates axonal outgrowth48. In general, these findings support the hypothesis that the therapeutic action of B2 supplementation is elicited mainly through the normalization of the expression of key genes that control axonal elongation and function.
Furthermore, our data showed an alteration in the delicate NF stoichiometric balance, a key pathogenetic event that has been previously suggested to be central in several MN diseases including ALS24,25. NF aggregation has been detected in ALS patients and transgenic animals49,50,51, and transgenic alteration of NFs in neurons, especially overexpression, can result in ALS24,52,53,54. These data supported the idea that the altered stoichiometry of NF subunits generated by variation in NF subunit expression results in their pathological aggregation25,55,56,57,58. Indeed, ALS spinal MNs displayed NF aggregation followed by neurite degeneration in association with a decreased stability of NFL mRNA and altered protein proportion of NF subunits. Of importance, conditional expression of NFL in ALS MNs corrects the NF subunit proportion, mitigating NF aggregation and neurite degeneration25.
In agreement with this observation in ALS-MN, our BVVL-MNs presented an increased level of NFH with aggregation that is likely responsible for neurite degeneration. The substantially higher NF content in MNs demands tight regulation; thus, its deregulation can account for the selective MN degeneration present in BVVL. This finding confirms the potential of targeting NF regulation for therapeutic intervention in MN diseases, but also in BVVL.
As noted, riboflavin is the precursor of the active coenzymes FMN and FAD that are essential cofactors for the electron transport chain and the lipid metabolism2, leading us to investigate these pathways. We identified perturbation in the expression of genes related to mitochondrial pathways as well as in the mitochondrial dynamic demonstrated by RNA sequencing and western blot studies, while their functionality in terms of respiratory chain and β-oxidation activity was not altered. Our proteomic data suggest the presence of an altered mitochondria turnover characterized by inhibition of fusion events leading to damage of the mitochondria and block of autophagy and mitophagy in BVVL-MNs, resulting in a reduced capability of removing dysfunctional mitochondria. The wellness of the mitochondrial network represents a requisite for neuronal health and survival. A timely elimination of aged and dysfunctional mitochondria through mitophagy, a selective and regulated form of autophagy, is an essential protective mechanism even more relevant in non-mitotic cells such as neurons, in which damaged cellular components cannot be diluted through recurring cell division cycles59,60. In fact, accumulation of large defective mitochondria has been associated with cell senescence/ageing and with PD, in fact brain belonging to PD patients present reduced expression of autophagy protein61,62. Some forms of MN disorders are characterized by autophagy impairment, which reduces their capacity to maintain protein homeostasis, and results in eventual neuronal cell loss63,64, although a causal relationship between autophagic dysfunction and disease has not been fully established. As showed in other MN disease, our data indicated that one possible pathogenetic mechanism in BVVL-MN degeneration is their incapacity to effectively cope with increased damage to the mitochondria, leading to cell apoptosis/necrosis.
In conclusion, iPSC patient-derived MNs allowed us to unravel novel molecular and cellular pathogenetic events that represent possible targets for therapeutic intervention, in addition to riboflavin supplementation for BVVL and other MN diseases. The strategy of using iPSCs obtained from patients to differentiate MNs offers the possibility to study the cell type of interest and to dissect the cellular (morphological) and molecular properties of the affected cells. Moreover, this approach allowed us to perform detailed analyses following riboflavin supplementation. Despite these advantages, limitations in understanding the pathogenesis of MN diseases remain. We analyzed the MN cultures in the commonly used 2D in vitro context, but the physiological environment obviously is 3D and characterized by a tight communication among different cell types (i.e., neurons with glial cells). Moreover, the in vitro system allows direct access to the cell of interest whereas in the human body, similar access may be much more challenging. Despite these limitations of our approach, we still consider it to be a useful model for unraveling new aspects of human pathologies, especially in cases where tissue accessibility is hard to achieve. We aim to continue our investigations of BVVL pathogenesis using iPSC-derived MNs in 3D culture systems and co-culturing of glial cells and neurons.
Materials and Methods
Derivation of human fibroblasts and reprogramming into iPSCs
The studies involving human samples were conducted in compliance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) and with national legislation and institutional guidelines. Human fibroblast cell lines were obtained from Eurobiobank with informed consent (ethical committee approved at the IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico). Fibroblasts were generated from dermal biopsies following informed consent as described previously65. Generation of iPSCs was performed by non-viral transduction with six reprogramming factors (OCT4, SOX2, NANOG, LIN28, c-Myc, and KLF4)66. iPSC colonies with embryonic stem cell–like morphology were expanded and successively detached for the analysis. iPSCs were grown on Matrigel (BD Biosciences) with E8 media (Invitrogen). All cell cultures were maintained at 37 °C, 5% CO2.
MN differentiation
MN differentiation was performed as previously described65,66. Briefly, cells were plated with neuronal medium DMEM-F12 (Life Technologies) supplemented with MEM nonessential amino acids solution (Life Technologies), N2 (Life Technologies), and 2 mg/ml heparin (Sigma-Aldrich). After 10 days, 0.1 μM retinoic acid (RA; Sigma-Aldrich) was added for neural caudalization. On day 17, posteriorized neuroectodermal cells were collected and suspended for a week in the same medium with 0.1 μM RA and 100–200 ng/ml Sonic hedgehog (R&D Systems), followed by the addition of 10 ng/ml brain-derived neurotrophic factor (PeproTech) and 10 ng/ml glial derived neurotrophic factor (PeproTech) on day 24. A centrifugation gradient was employed to maximize MN enrichment20,67,68.
The BBVL cells were cultured at two riboflavin concentrations: 5 nmol/l (identified as the basal condition) and 1 μmol/l (identified as the therapeutic condition). The high concentration of riboflavin (1 μmol/l) was chosen because this concentration is above the level of riboflavin observed in plasma from individuals treated with riboflavin7,69. Riboflavin 1 μmol/l is a standard concentration present in culture media. The low concentration of riboflavin (5 nmol/l) was chosen because it represents a level close to that for riboflavin observed in plasma in BVVL patients7,10.
Immunocytochemistry and phenotypic analysis
Immunocytochemistry of iPSCs and MNs
Cells were fixed in 4% paraformaldehyde for 10 min, permeabilized with Triton 0.25%, and then blocked with 10% bovine serum albumin in 1× phosphate-buffered saline and 0.3% Triton X-100 for 1 h at room temperature. We incubated the cells with primary antibodies to NANOG (1:100, Abcam), SSEA-3 (1.100, Covance), ChAT (1:200, Chemicon & Millipore), SMI32 (Covance, 1:500), and Neurofilament-200 (1:1000 Sigma) overnight, and then with anti-rabbit or anti-mouse or -goat Alexa Fluor 488 or 594 (1:400; Life Technologies) secondary antibody for 1.5 h at room temperature.
Morphometric analysis
Phenotypic analysis was performed as previously described65,66,70. We quantified MNs by determining cells positive for MN marker and counting 10 randomly selected fields/well (3 wells/condition/experiment in four to five experiments). The morphometric axon length was determined using soma diameter and the distance between two points. For all imaging, we used a confocal LEICA SP2 and Nikon microscope.
mRNA-seq library preparation, sequencing, and gene expression analysis
Total RNA was isolated from pooled cells using the PicoPure RNA Isolation Kit (Arcturus), which allows the recovery of RNA from a small amount of cells, and quantified using Bioanalyzer (Agilent). A TruSeq RNA sample preparation Kit v2 (Illumina) was used to obtain cDNA libraries from total RNA, and the Nextera DNA sample preparation kit (Illumina) was employed to prepare the sequencing library. Libraries were sequenced on an Illumina HiSeq 2000 employing the standard Illumina pipeline software on an Illumina platform (performed at BGI). Two biological replicates for patients and controls, each comprising four pooled samples, were analyzed. Each replicate was analyzed separately and then pooled. Raw data were processed using TopHat version 2.0.9 and Cufflinks to align genome reads, assemble the data into transcripts, and define expression levels. The aligned mRNA reads were used to identify differentially expressed genes.
Bioinformatics analysis
For NGS data analysis, we used two bioinformatics tools that query for enriched gene ontology terms: gene-annotation enrichment analysis with DAVID21 and GSEA22. Enrichment for up-regulated or down-regulated sets of genes from the REACTOME pathway and GO term database was computed by running GSEA against the test statistic-ranked list of genes in the experiment. Ranking was based on the Cuffdiff 2-derived test statistic. All REACTOME and GO term gene sets with >15 members in the MSigDB package “c2.all.v3.0.symbols. gmt” and “c5.all.v4.0.symbols.gmt,” respectively, were downloaded from ftp://gseaftp.broadinstitute.org/.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen). Concentrations were measured with a Nanodrop spectrophotometer. Only samples with ratios between 1.8 and 2.0 were further analyzed. A total of 1 μg of total RNA was reverse-transcribed using the Ready-To-Go kit (GE Healthcare). Reverse-transcribed material (5 ng for each sample) was amplified using the TaqMan Universal PCR Master Mix (Applied Biosystems) and the appropriate primers to evaluate gene expression (probe ID available upon request) in the 7500 Real Time PCR System. The expression levels for all genes were normalized to the average levels of the housekeeping gene ACTB and referred to the relevant control samples.
Biochemical studies
The specific activities of individual respiratory-chain complexes and β-oxidation were evaluated in MNs, as previously described71,72. The specific activity of each complex was normalized to that of citrate synthase.
Protein studies
Proteins extracted from MNs were separated by SDS-PAGE. The primary antibodies used in these experiments were anti EphA2 (1:1000, Abcam), anti-NFs (1:1000, Sigma), MFN2 (Sigma, 1:1000), DRP1 (Sigma, 1:1000), p62 (Millipore, 1:1000), LC3 I/II (Millipore, 1:500), PARKIN (Abcam, 1:2000), and BNIP3 (Abcam, 1:1000) using the manufacturer instructions. The following secondary antibodies were used: polyclonal anti-rabbit (1:2700, Dako) and polyclonal anti-mouse (1:3200, Dako). The nitrocellulose membrane was stripped and re-probed with anti-actin monoclonal antibody as a loading control. Densitometry was performed using ImageJ software.
Statistical analysis
Stats Direct software was used for all statistical analyses. All counting data from immunocytochemical analyses and cell survival were expressed as mean ± SD or SEM. Differences between two means were analyzed using the Student’s t-test (two-tailed), and differences among more than two means were analyzed using one- or two-way ANOVA. When ANOVA revealed significant differences, pair-wise comparisons between means were performed using Tukey’s post-hoc test.
Additional Information
How to cite this article: Rizzo, F. et al. Genome-wide RNA-seq of iPSC-derived motor neurons indicates selective cytoskeletal perturbation in Brown–Vialetto disease that is partially rescued by riboflavin. Sci. Rep. 7, 46271; doi: 10.1038/srep46271 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The contribution of Italian Ministry of Health to SC and EB (GR-2010-2309463) is gratefully acknowledged. We thank Associazione Amici del Centro Dino Ferrarri for their support.
Footnotes
The authors declare no competing financial interests.
Author Contributions F.R., A.R., C.C., and S.C. performed in vitro experiments; V.M. performed molecular biology experiments; S.S. and A.B. performed proteomic analysis; F.F. performed biochemical assay; F.R., A.R., and S.C. analyzed Rna-seq data; A.N. contributed to in vitro experiments and manuscript writing for the revision. N.B., G.P.C., M.N., F.R., E.B., and S.C. contributed ideas and supported the work. F.R., A.R., and S.C. contributed in the manuscript writing.
References
- Jaeger B. & Bosch A. M. Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. Journal of inherited metabolic disease 39, 559–564, doi: 10.1007/s10545-016-9924-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhart W. D., Gudipati V. & Macheroux P. The human flavoproteome. Archives of biochemistry and biophysics 535, 150–162, doi: 10.1016/j.abb.2013.02.015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M. et al. Functional characteristics of the human ortholog of riboflavin transporter 2 and riboflavin-responsive expression of its rat ortholog in the small intestine indicate its involvement in riboflavin absorption. The Journal of nutrition 140, 1722–1727, doi: 10.3945/jn.110.128330 (2010). [DOI] [PubMed] [Google Scholar]
- Yao Y. et al. Identification and comparative functional characterization of a new human riboflavin transporter hRFT3 expressed in the brain. The Journal of nutrition 140, 1220–1226, doi: 10.3945/jn.110.122911 (2010). [DOI] [PubMed] [Google Scholar]
- Yonezawa A., Masuda S., Katsura T. & Inui K. Identification and functional characterization of a novel human and rat riboflavin transporter, RFT1. American journal of physiology. Cell physiology 295, C632–641, doi: 10.1152/ajpcell.00019.2008 (2008). [DOI] [PubMed] [Google Scholar]
- Powers H. J. Riboflavin (vitamin B-2) and health. The American journal of clinical nutrition 77, 1352–1360 (2003). [DOI] [PubMed] [Google Scholar]
- Cornelius N., Corydon T. J., Gregersen N. & Olsen R. K. Cellular consequences of oxidative stress in riboflavin responsive multiple acyl-CoA dehydrogenation deficiency patient fibroblasts. Human molecular genetics 23, 4285–4301, doi: 10.1093/hmg/ddu146 (2014). [DOI] [PubMed] [Google Scholar]
- Carrozzo R. et al. Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency. The chaperon-like effect of vitamin B2. Mitochondrion 18, 49–57, doi: 10.1016/j.mito.2014.09.006 (2014). [DOI] [PubMed] [Google Scholar]
- Green P. et al. Brown–Vialetto–Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. American journal of human genetics 86, 485–489, doi: 10.1016/j.ajhg.2010.02.006 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch A. M. et al. Brown—Vialetto—Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. Journal of inherited metabolic disease 34, 159–164, doi: 10.1007/s10545-010-9242-z (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccolella M. et al. Brown–Vialetto–Van Laere and Fazio-Londe overlap syndromes: a clinical, biochemical and genetic study. Neuromuscular disorders: NMD 22, 1075–1082, doi: 10.1016/j.nmd.2012.05.007 (2012). [DOI] [PubMed] [Google Scholar]
- Ciccolella M. et al. Riboflavin transporter 3 involvement in infantile Brown–Vialetto–Van Laere disease: two novel mutations. Journal of medical genetics 50, 104–107, doi: 10.1136/jmedgenet-2012-101204 (2013). [DOI] [PubMed] [Google Scholar]
- Francis D. A., Ponsford J. R., Wiles C. M., Thomas P. K. & Duchen L. W. Brown–Vialetto–Van Laere syndrome. Neuropathology and applied neurobiology 19, 91–94 (1993). [DOI] [PubMed] [Google Scholar]
- Foley A. R. et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain: a journal of neurology 137, 44–56, doi: 10.1093/brain/awt315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosemberg S., Lancellotti C. L., Arita F., Campos C. & de Castro N. P. Jr. Progressive bulbar paralysis of childhood (Fazio-Londe disease) with deafness. Case report with clinicopathologic correlation. European neurology 21, 84–89 (1982). [DOI] [PubMed] [Google Scholar]
- Brown M. Variations in the Position and Development of the Kidneys. Journal of anatomy and physiology 28, 194–208 191 (1894). [PMC free article] [PubMed] [Google Scholar]
- Van Laere J. E. [A new case of chronic progressive bulbo-pontine paralysis and deafness]. Revue neurologique 133, 119–124 (1977). [PubMed] [Google Scholar]
- Sathasivam S. Brown–Vialetto–Van Laere syndrome. Orphanet journal of rare diseases 3, 9, doi: 10.1186/1750-1172-3-9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. O., Gibbs J. R., Van Maldergem L., Houlden H. & Singleton A. B. Exome sequencing in Brown–Vialetto–Van Laere syndrome. American journal of human genetics 87, 567–569; author reply 569-570, doi: 10.1016/j.ajhg.2010.05.021 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti S. et al. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Science translational medicine 4, 165ra162, doi: 10.1126/scitranslmed.3004108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B. T. & Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57, doi: 10.1038/nprot.2008.211 (2009). [DOI] [PubMed] [Google Scholar]
- Subramanian A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg G. A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. The Lancet. Neurology 8, 205–216, doi: 10.1016/S1474-4422(09)70016-X (2009). [DOI] [PubMed] [Google Scholar]
- Cote F., Collard J. F. & Julien J. P. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 73, 35–46 (1993). [DOI] [PubMed] [Google Scholar]
- Chen H. et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell stem cell 14, 796–809, doi: 10.1016/j.stem.2014.02.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Chalabi A. et al. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Human molecular genetics 8, 157–164 (1999). [DOI] [PubMed] [Google Scholar]
- Kubli D. A. & Gustafsson A. B. Mitochondria and mitophagy: the yin and yang of cell death control. Circulation research 111, 1208–1221, doi: 10.1161/CIRCRESAHA.112.265819 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G. & Schwarz T. L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell death and differentiation 20, 31–42, doi: 10.1038/cdd.2012.81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini E. et al. Pluripotent stem cell-based models of spinal muscular atrophy. Molecular and cellular neurosciences 64, 44–50, doi: 10.1016/j.mcn.2014.12.005 (2015). [DOI] [PubMed] [Google Scholar]
- Faravelli I. et al. Motor neuron derivation from human embryonic and induced pluripotent stem cells: experimental approaches and clinical perspectives. Stem cell research & therapy 5, 87, doi: 10.1186/scrt476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizzardo M. et al. Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Scientific reports 5, 11746, doi: 10.1038/srep11746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X. et al. Modeling the differential phenotypes of spinal muscular atrophy with high-yield generation of motor neurons from human induced pluripotent stem cells. Oncotarget, doi: 10.18632/oncotarget.14925 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnucci C., Piemonte F., Sferra A., Piermarini E. & Bertini E. The cytoskeletal arrangements necessary to neurogenesis. Oncotarget 7, 19414–19429, doi: 10.18632/oncotarget.6838 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y., Liu W., Jiang H., Jiang Q. & Feng J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. The Journal of biological chemistry 280, 34105–34112, doi: 10.1074/jbc.M503483200 (2005). [DOI] [PubMed] [Google Scholar]
- Choi W. S., Palmiter R. D. & Xia Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. The Journal of cell biology 192, 873–882, doi: 10.1083/jcb.201009132 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer M. K. et al. Sensory neuropathy in progressive motor neuronopathy (pmn) mice is associated with defects in microtubule polymerization and axonal transport. Brain pathology, doi: 10.1111/bpa.12422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sferra A. et al. TBCE Mutations Cause Early-Onset Progressive Encephalopathy with Distal Spinal Muscular Atrophy. American journal of human genetics 99, 974–983, doi: 10.1016/j.ajhg.2016.08.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S. Y. et al. Genome-wide RNA-Seq of Human Motor Neurons Implicates Selective ER Stress Activation in Spinal Muscular Atrophy. Cell stem cell 17, 569–584, doi: 10.1016/j.stem.2015.08.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E. et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell stem cell 14, 781–795, doi: 10.1016/j.stem.2014.03.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini A. et al. The Neuronal Pentraxin-2 Pathway Is an Unrecognized Target in Human Neuroblastoma, Which Also Offers Prognostic Value in Patients. Cancer research 75, 4265–4271, doi: 10.1158/0008-5472.CAN-15-0649 (2015). [DOI] [PubMed] [Google Scholar]
- Boukhelifa M. et al. A role for the cytoskeleton-associated protein palladin in neurite outgrowth. Molecular biology of the cell 12, 2721–2729 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart J. et al. Expression of EphA4, ephrin-A2 and ephrin-A5 during axon outgrowth to the hindlimb indicates potential roles in pathfinding. Developmental neuroscience 22, 237–250, doi: 17446 (2000). [DOI] [PubMed] [Google Scholar]
- Van Battum E. Y., Brignani S. & Pasterkamp R. J. Axon guidance proteins in neurological disorders. The Lancet. Neurology 14, 532–546, doi: 10.1016/S1474-4422(14)70257-1 (2015). [DOI] [PubMed] [Google Scholar]
- Sbacchi S., Acquadro F., Calo I., Cali F. & Romano V. Functional annotation of genes overlapping copy number variants in autistic patients: focus on axon pathfinding. Current genomics 11, 136–145, doi: 10.2174/138920210790886880 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoecke A. et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nature medicine 18, 1418–1422, doi: 10.1038/nm.2901 (2012). [DOI] [PubMed] [Google Scholar]
- Braathen G. J. et al. Variants in the genes DCTN2, DNAH10, LRIG3, and MYO1A are associated with intermediate Charcot-Marie-Tooth disease in a Norwegian family. Acta neurologica Scandinavica 134, 67–75, doi: 10.1111/ane.12515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan M. P., Chin S. S., Fliegner K. H. & Liem R. K. Alpha-internexin, a novel neuronal intermediate filament protein, precedes the low molecular weight neurofilament protein (NF-L) in the developing rat brain. The Journal of neuroscience: the official journal of the Society for Neuroscience 10, 2735–2748 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N. Proteoglycans and neuronal migration in the cerebral cortex during development and disease. Frontiers in neuroscience 9, 98, doi: 10.3389/fnins.2015.00098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S. Proximal axonal enlargement in motor neuron disease. Neurology 18, 841–851 (1968). [DOI] [PubMed] [Google Scholar]
- Hirano A., Donnenfeld H., Sasaki S. & Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. Journal of neuropathology and experimental neurology 43, 461–470 (1984). [DOI] [PubMed] [Google Scholar]
- Bruijn L. I. et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854 (1998). [DOI] [PubMed] [Google Scholar]
- Xu Z., Cork L. C., Griffin J. W. & Cleveland D. W. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell 73, 23–33 (1993). [DOI] [PubMed] [Google Scholar]
- Wong P. C. et al. Increasing neurofilament subunit NF-M expression reduces axonal NF-H, inhibits radial growth, and results in neurofilamentous accumulation in motor neurons. The Journal of cell biology 130, 1413–1422 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney M. E. et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994). [DOI] [PubMed] [Google Scholar]
- Bergeron C. et al. Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. Journal of neuropathology and experimental neurology 53, 221–230 (1994). [DOI] [PubMed] [Google Scholar]
- Kong J. & Xu Z. Overexpression of neurofilament subunit NF-L and NF-H extends survival of a mouse model for amyotrophic lateral sclerosis. Neuroscience letters 281, 72–74 (2000). [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S. et al. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proceedings of the National Academy of Sciences of the United States of America 95, 9626–9630 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J., Couillard-Despres S., Jacomy H., Gravel C. & Julien J. P. Extra neurofilament NF-L subunits rescue motor neuron disease caused by overexpression of the human NF-H gene in mice. Journal of neuropathology and experimental neurology 58, 1099–1110 (1999). [PubMed] [Google Scholar]
- Ventruti A. & Cuervo A. M. Autophagy and neurodegeneration. Current neurology and neuroscience reports 7, 443–451 (2007). [DOI] [PubMed] [Google Scholar]
- Komatsu M. et al. Constitutive autophagy: vital role in clearance of unfavorable proteins in neurons. Cell death and differentiation 14, 887–894, doi: 10.1038/sj.cdd.4402120 (2007). [DOI] [PubMed] [Google Scholar]
- Blagosklonny M. V. Program-like aging and mitochondria: instead of random damage by free radicals. Journal of cellular biochemistry 102, 1389–1399, doi: 10.1002/jcb.21602 (2007). [DOI] [PubMed] [Google Scholar]
- Tain L. S. et al. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nature neuroscience 12, 1129–1135, doi: 10.1038/nn.2372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassif M. et al. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 10, 1256–1271, doi: 10.4161/auto.28784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S., Yamashita T. & Kwak S. Autophagy in spinal motor neurons of conditional ADAR2-knockout mice: An implication for a role of calcium in increased autophagy flux in ALS. Neuroscience letters 598, 79–84, doi: 10.1016/j.neulet.2015.05.025 (2015). [DOI] [PubMed] [Google Scholar]
- Rizzo F. et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot-Marie-Tooth 2A motor neurons. Human molecular genetics, doi: 10.1093/hmg/ddw258 (2016). [DOI] [PubMed] [Google Scholar]
- Corti S. et al. Direct reprogramming of human astrocytes into neural stem cells and neurons. Experimental cell research 318, 1528–1541, doi: 10.1016/j.yexcr.2012.02.040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras M., Gagnon V., Minotti S., Durham H. D. & Berthod F. Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. Journal of neuroscience methods 163, 111–118, doi: 10.1016/j.jneumeth.2007.02.024 (2007). [DOI] [PubMed] [Google Scholar]
- Wada T. et al. Highly efficient differentiation and enrichment of spinal motor neurons derived from human and monkey embryonic stem cells. PloS one 4, e6722, doi: 10.1371/journal.pone.0006722 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempleni J. Determination of riboflavin and flavocoenzymes in human blood plasma by high-performance liquid chromatography. Annals of nutrition & metabolism 39, 224–226 (1995). [DOI] [PubMed] [Google Scholar]
- Allodi I. et al. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Scientific reports 6, 25960, doi: 10.1038/srep25960 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresolin N. et al. Fatal infantile cytochrome c oxidase deficiency: decrease of immunologically detectable enzyme in muscle. Neurology 35, 802–812 (1985). [DOI] [PubMed] [Google Scholar]
- Crugnola V. et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Archives of neurology 67, 849–854, doi: 10.1001/archneurol.2010.128 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.