

Abstract
Background
Severe combined immunodeficiency (SCID) is characterized by severely impaired T cell development and is fatal without treatment. Newborn screening (NBS) for SCID permits identification of affected infants before development of opportunistic infections and other complications. Substantial variation exists between treatment centers with regard to pre-transplant care and transplant protocols for NBS identified SCID infants, as well as for infants with other T lymphopenic disorders detected by NBS.
Methods
We developed approaches to management based on the study of infants identified by SCID NBS who received care at UCSF.
Results
From August 2010 through October 2016, 32 NBS SCID and leaky SCID cases from California and other states were treated and 42 NBS identified non-SCID T cell lymphopenia (TCL) cases were followed.
Conclusions
Our center’s approach supports successful outcomes; systematic review of our practice provides a framework for diagnosis and management, recognizing that more data will continue to shape best practices.
Introduction
Severe combined immunodeficiency (SCID) is a genetically heterogeneous group of inherited defects characterized by severely impaired T cell development combined with inability to make specific antibodies [1–4]. While fatal without treatment, SCID is treatable by allogeneic hematopoietic cell transplantation (HCT), or in certain genotypes by enzyme replacement (ERT) or gene therapy (GT). Unless diagnosed in the neonatal period, affected infants develop severe, opportunistic infections early in life. Avoidance of infection was the impetus for population-based SCID newborn screening (NBS) using newborn dried blood spots (DBS). All infants with SCID fail to generate a diverse repertoire of mature T cells, and consequently have absent or very low numbers of T cell receptor excision circles (TRECs), DNA byproducts of T cell receptor gene rearrangement [5,6]. An exception is late-onset adenosine deaminase (ADA) SCID where progressive loss of T cells occurs with time. Newborn screening for insufficient TRECs makes possible identification of SCID before infections occur, permitting optimal care of affected infants.
As SCID NBS has become widespread, and in conjunction with establishment of the Primary Immune Deficiency Consortium (PIDTC) funded by the National Institute of Allergy and Infectious Diseases and Office of Rare Diseases, National Center for Advancing Translational Sciences, NIH, new definitions for SCID have evolved for healthy-appearing affected infants [3, 4, 13]. In contrast to classical descriptions of SCID with infections [1], the new criteria are based upon laboratory parameters. Typical SCID cases have <300 autologous CD3 T cells/uL, <10% of the lower range of normal proliferation to the mitogen phytohemmaglutinin (PHA), and/or detectable maternal T cell engraftment as well as deleterious mutations in recognized SCID genes (Figure 1). In addition to typical SCID, TREC screening identifies “leaky” SCID due to hypomorphic mutations in known SCID genes; 26% of the SCID cases found by screening were leaky, as reported in an 11-program study of NBS for SCID in the US [1]. Leaky SCID cases have 300–1500 T cells/uL or more, but lack naïve CD4 T cells expressing CD45RA. Their T cells are functionally impaired and have limited diversity, and maternal cells are not detected. A subset of infants with leaky SCID have expansion of oligoclonal, dysregulated T cells leading to adenopathy, erythroderma with cutaneous and intestinal T cell infiltration, hepatosplenomegaly, eosinophilia, and highly elevated IgE, features collectively known as Omenn syndrome (OS) [3].
Figure 1.
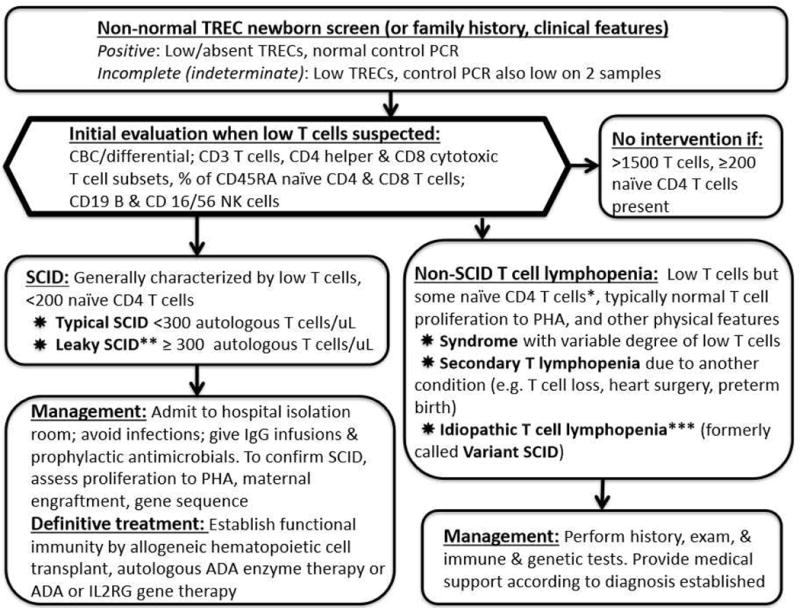
Identification of T cell immune defects by newborn TREC screening; primary immune defects may also be diagnosed due to a history affected family members or clinical features.
*Variable can be < 200 naïve CD4 T cells. **Omenn syndrome is a form of leaky SCID with rash; eosinophilia; autoreactive, oligoclonal T cells; and variable CD3 T cell count which can be >1500. ***Some infants never leave this group but some move out of this category when other diagnoses are made. These infants need to be followed over time.
NBS also identifies infants with low TRECs who do not have SCID, but who nonetheless have few T lymphocytes in the peripheral blood, termed T cell lymphopenia (TCL) [3,15]. While most of these infants have recognized conditions, such as DiGeorge syndrome, others have TCL with no apparent underlying cause [10,11]. Establishing a definitive diagnosis and managing these infants are new challenges for physicians, who must recognize the level of TCL that is medically significant, select diagnostic tests, and apply appropriate interventions.
We provide here our center’s approach to infants with SCID, leaky SCID, and non-SCID TCL identified by NBS, recognizing that individual state screening programs and providers currently employ a spectrum of approaches [3] and that more data are needed to identify best practices.
Approach to infants with TREC results that are not normal
In California, all infants with non-normal TREC tests immediately have a CBC, differential and flow cytometry analysis to distinguish the 43% of infants with confirmed TCL from the 57% with >1500 T cells/uL, for whom no further intervention is undertaken as long as naïve T cells are observed (Figure 1, right) [11,12,15]. Infants with <300 T cells/uL, or with more T cells/uL but <200 naïve CD4 T cells, representing ~16% of all those with non-normal TREC results (Figure 1, left), are immediately hospitalized due to the high likelihood of having typical or leaky SCID, respectively, or complete DiGeorge syndrome. The management of SCID cases at our center is discussed below. The remaining infants with 300–1500 T cells/uL, but with naïve CD4 T cells present (Figure 1, middle right), have multisystem syndromes, secondary, and undiagnosed or idiopathic TCL. Diagnosis and management of infants with non-SCID TCL is undertaken on a case-by-case basis, and generally includes (in addition to flow cytometry), proliferative responses to mitogens, serum immunoglobulin levels, and DNA arrays to reveal genomic copy number variation.
How we treat the infant with suspected SCID
Infants suspected to have SCID or leaky SCID based upon laboratory testing (and those identified by a family history or clinical features), are admitted to protective isolation in our hospital; any sign of infection or other abnormality is immediately pursued with cultures and treated (Table 1). Our immune evaluation is listed in Table 2. Early discussions between the family and the immunology and HCT teams present information about SCID, the diagnostic approach, and expectations for hospitalization. The HCT team works with the family to identify the optimal donor. A social worker versed in SCID is involved to identify needs for support. Infectious disease workup includes assessing for cytomegalovirus (CMV) by PCR, which may be transmitted via breast milk of CMV seropositive mothers prior to confirmation of TCL. Two infants in our 32-patient cohort (6%) were infected by CMV at baseline, with breast milk from a CMV seropositive mother the only identified potential source. Baseline lymphocyte subset confirmation and functional testing are obtained, along with HLA typing of the infant and family members to identify potential HCT donors. Evidence of maternal T cell engraftment is sought by analysis of maternal and infant DNA; maternal chimerism, detected in >50% of SCID cases [14], suggests that a maternal graft can be accepted without conditioning. An infant with CD45RO, but not CD45RA, T cells may have expansion of a limited repertoire of T cells of either autologous or maternal origin. A T-B-NK- lymphocyte profile suggests ADA deficiency, often accompanied by neutropenia; screening for ADA enzyme activity and metabolite levels is quickly accomplished before pursuing gene sequencing.
Table 1.
Approach to early management of new infant with lymphocyte subset profile consistent with SCID or leaky SCID.
|
See appendix 2 for prophylactic medication dosing
Table 2.
Early clinical and laboratory assessment for new infant with suspected SCID
|
microcephaly and bird-like facial appearance; infants with Navajo background
Special considerations
Iatrogenic anemia
Iatrogenic anemia is a concern in small infants with SCID. Blood draws are planned to minimize discomfort and use the smallest possible volume for each test. Transfusions are avoided to minimize risks of infection and allosensitization. If transfusions are needed, only CMV negative, irradiated, leukoreduced packed red cells are given. A standard operating procedure developed with our clinical laboratory uses specific minimum volumes for every test (Appendix 1).
Adenosine deaminase (ADA) deficiency
ADA-SCID, diagnosed in 6 of our initial 32 SCID infants (19%), is a systemic metabolic disorder in which elevated concentrations of purine intermediates are particularly toxic to lymphocytes, but affect all tissues. Early complications of ADA deficiency included neutropenia, seen in 4 of our 6 ADA infants, and pulmonary alveolar proteinosis (PAP) seen in 2, one of whom required intubation for respiratory distress. Two others with tachypnea at diagnosis were suspected to have PAP. Bronchoscopy to obtain cultures as well as histology is required to distinguishable PAP from infection [16]. Elevations in liver function tests can occur with ADA deficiency, but also can be a sign of a viral infection. Prompt administration of ERT with PEG-ADA has led to resolution of abnormalities over several days in our patients. Therefore, unless there is an HLA matched sibling, we start ERT immediately at full doses of 30 units/kg or more to achieve rapid detoxification. After T cells appear, the infants are cared for at home with intramuscular PEG-ADA injections (125 units) twice weekly. Prior to HCT without other conditioning, we discontinue ERT, monitoring rising metabolites until approaching pre PEG-ADA levels. One of our ADA deficient patients received a matched sibling HCT, but 5 others who lacked a matched related or unrelated donor received ERT for several months. After 6 months of age, 4 infants enrolled in the experimental gene therapy (GT) protocol of Dr. Donald Kohn at UCLA, all with successful development of functional gene corrected lymphocytes. GT is planned for a fifth patient.
Leaky SCID and Omenn Syndrome (OS)
Infants with defects in any SCID gene that permit residual protein function can have more T cells than those with typical SCID; without screening the diagnosis may be delayed many months or years. Although nearly all are identified by TREC NBS, the mildest cases may not have low TREC numbers at birth. OS, due to hypomorphic mutations in RAG1, RAG2, requires immunosuppression while awaiting HCT, which should be implemented as swiftly as possible [3]. Aggressive autoreactive oligoclonal T cell expansion occurring within a few days to weeks of life can result in a severe scaly rash, diarrhea, and failure to thrive. Systemic corticosteroids; calcineurin inhibitors, and/or anti-thymocyte globulin or anti-CD52 monoclonal antibody treat this condition effectively until an HCT can be done.
Radiation Sensitive SCID (RS-SCID)
RS-SCID includes deficiencies of Artemis, DNA Ligase IV, DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Cernunnos-XLF, and nibrin (the protein associated with Nijmegen breakage syndrome). Infants presenting with abnormal NBS generally have a T- (or T low) B-NK+ phenotype as the process of recombination of T and B cell antigen receptor genes require non-homologous end-joining by the DNA repair complex [17]. While RS-SCID is treatable with HCT, donor selection and conditioning must be adjusted because of increased sensitivity to typically used alkylating agents, such as busulfan, melphalan, thiotepa or cyclophosphamide [17,18]. T-B-NK+ lymphocyte profiles and developmental abnormalities, such as microcephaly and bird-like facial appearance, suggest heightened radiation sensitivity, prompting radiation sensitivity testing of patient fibroblasts, Western blots and/or gene sequencing (e.g. LIG4, NHEJ1) to establish the diagnosis. Although these take several weeks to perform, they must be obtained prior to HCT if conditioning is planned. Despite this delay, it is our practice to hospitalize these infants while the workup is performed. While no absolute contraindication exists for clinically indicated X-rays, exposure should be minimized. Ultrasound and magnetic resonance imaging are safe alternatives.
Unique psychosocial vulnerability of parents with SCID-affected infants diagnosed by NBS
The combination within the first month of life of the postpartum period and the infant’s hospitalization for SCID treatment present a situation of compounded vulnerability with the potential to destabilize family functioning and parental ability to provide care during and after HCT. At our institution, care providers create an early, open dialogue about psychosocial health impacts in this unique situation. Social workers interact extensively with the families during the hospitalization and after discharge.
Post-Partum Depression
Given the high stress experienced by these new parents, special efforts are made to recognize post-partum depression (PPD) and post-traumatic stress disorder (PTSD). PPD is a significant health concern affecting 10–15% of US women after normal births [19]. PPD has also been reported in new fathers [19]. The onset of PPD is typically 2 weeks after childbirth, coinciding with the diagnosis of SCID by NBS. Mothers who initiate or maintain breastfeeding are at lower risk for PPD, and interruption of breastfeeding may contribute to PPD [20]. Thus, interrupting breastfeeding while serologic status for CMV is tested adds to the stress during the postpartum period.
Post-Traumatic Stress Disorder
Parents learning of an abnormal metabolic newborn screen result have reported shock as if a death in the family occurred [21]. Likewise, the emotional distress experienced by parents of newborns with SCID may stem from shock, grief, guilt, and apprehension regarding losing the child. During the first weeks of hospitalization, confirmation of the SCID diagnosis and characterization of the genotype are sought through genetic testing. Additional testing for HLA typing is conducted on siblings and parents. Genetic testing is highly personal and is often associated with anxiety, fear and guilt for passing on an inherited condition [22, 23]. Our pilot study in which parents were interviewed revealed the majority of mothers and fathers caring for infants with SCID suffer from PPD and PTSD (M. Dorsey, unpublished data). Thus involvement of experienced social workers and nurses is a critical component of overall care for SCID families, with early introduction to available services and support serving as a key to decreasing stress. In addition, routine screening for PPD and PTSD can identify families in need of referral to mental health providers.
Treatment
Transplant: Allogeneic and Autologous Gene-Modified Hematopoietic Stem Cells (HSC)
Historically, single centers reported transplant outcomes for SCID in which the contributions of specific gene defects, infections, and variations in treatment protocols could not be isolated. The PIDTC is addressing these issues by enrolling large numbers of patients from centers across North America [4,14]. In a retrospective report, Pai et al. (2014) showed that infants who received transplants before 3.5 months of age had a 5-year survival rate of 94%, as did infants older than 3.5 months but with no history of infection (90%) or whose infections had resolved by the time of HCT (82%) [4]. Children over 3.5 months of age with active infection at the time of HCT and no HLA matched sibling had the lowest survival (50%). HLA-matched sibling donors resulted in the best outcomes regardless of age or infection. While reduced intensity (RIC) or myeloablative (MAC) conditioning versus no conditioning or serotherapy alone had a higher likelihood of full T cell reconstitution and no need for immunoglobulin replacement therapy, long term effects of chemotherapy remain to be addressed [4]. For patients infected at the time of transplant at any age, the use of haploidentical donors without conditioning had the best 5-year survival. In a prospective PIDTC study, Dvorak et al. (2013) reported that the time needed for an unrelated adult donor (URD) or umbilical cord blood (UCB) search delayed HCT by approximately a month compared to using a mismatched related donor (MMRD) [14]. Additional studies will determine whether earlier HCT produces superior outcomes, or if closer HLA-matching in unrelated donors justifies the prolonged time to transplant in uninfected infants.
Gene correction of autologous HSC has been successful for children with ADA and X-linked (IL2RG) SCID [24–31]. The initial success in IL2RG SCID was tempered by the development of T cell leukemia due to retroviral vector insertional mutagenesis (IM) in 5 of the initial 20 recipients [26,27]. Current trials use self-inactivating retroviral or lentiviral vectors, which appear to have a significantly lower risk of IM [28]. Initial gene therapy attempts without conditioning for ADA SCID failed; when low dose busulfan was added as conditioning T and sometimes B cell immunity was reconstituted [29]. In early IL2RG SCID gene therapy trials without conditioning, T cell reconstitution occurred, but B and NK cell reconstitution were limited [28, 30]. Results from an initial trial using low dose busulfan for IL2RG SCID suggest that B and NK cells will be reconstituted [31].
Treatment outcomes at UCSF
Twenty-six California NBS identified SCID and leaky SCID cases and 6 from other states were treated at UCSF between September 2010 and October 2016. Twenty-nine (94%) are alive and well, 19 of them >2 years post-HCT. All of 26 who are >6 months post-HCT have reconstituted T cells and 13/26 (50%) have B cell reconstitution. Only 4 (14%) had a recognized family history of SCID. Gene diagnoses were: 7 IL2RG, 6 ADA, 5 DCLRE1C, 4 IL7R, 4 RAG1, 2 RAG2, 1 JAK3, 1 RMRP, 1 BCL11B [31]. One infant with unknown genotype (despite family exome and genome sequencing) died of refractory CMV infection present upon admission. One of 5 Artemis-deficient Navajo infants died of refractory HHV6 infection. Four ADA patients underwent busulfan-conditioned autologous HCT with gene corrected CD34+ cells; all are reconstituted with T cells and 2/4 who are >6 months post-GT have B cell reconstitution.
Our approach to selection of the best donor and conditioning regimen for patients with typical SCID is summarized in Figure 2. The guiding principles are to (1) use the donor associated with the lowest risk of graft vs. host disease (GvHD); (2) minimize the duration of pre-HCT lymphopenia and risk for infections; and (3) use the least amount of chemotherapy possible to ensure donor T-cell engraftment but minimize the risk of short-and long-term toxicity. Unless there is an urgent need to proceed with HCT, such as a CMV infection, busulfan is not given to infants <3 months old. This approach accepts that booster HCT may be needed and B cell immunity will typically not be restored when a donor other than a matched sibling is used, except in patients with intrinsically normal B cells, e.g., IL7R SCID, leaving many patients dependent upon gammaglobulin replacement. In many patients with typical SCID, irrespective of donor type, no chemotherapy is required, though use of serotherapy may be beneficial when utilizing T-cell replete stem cell sources or when maternal chimerism is present and the mother is the donor [33]. For patients with leaky SCID or Omenn syndrome, a conditioning regimen is required. When using busulfan, we carefully monitor pharmacokinetics, which is highly variable in small infants, and modify divided doses to achieve the target cumulative exposure predicted to allow stem cell engraftment [34]. GvHD prophylaxis is aggressive, since there is no benefit to alloreactivity. Stem cells from haploidentical donors currently undergo CD34-selection [35], though in the near future, alternate ex vivo T-cell depletion strategies are planned. With the identification of maternal engraftment as a risk factor for the development of GvHD despite CD34-selection [33], low-dose rabbit anti-thymocyte globulin (rATG) serotherapy has also been successfully utilized in the most recent patients. For unrelated donors, we also employ pre-HCT serotherapy [36]. Serotherapy may abrogate an early (<100 days) wave of homeostatic donor T-cell expansion, but this typically can be tolerated in an uninfected patient identified by NBS, and naïve T-cell generation occurs at around 3 months post-HCT.
Figure 2.

Donor selection and conditioning for typical SCID. 1Excludes Omenn syndrome and leaky SCID, both classified as atypical SCID for transplant purposes and requiring at least some conditioning; also excludes radiation sensitive SCID (DCLREC1, LIG4, etc.), for which donor selection and conditioning are individualized to balance risks of rejection vs. chemotherapy toxicity. 2Gene therapy clinical trials should be considered for X-linked or ADA SCID when there is no HLA matched sibling. 3Based on availability, CMV status, donor age, and other variables. 4Non-radiation-sensitive T-B-NK+ SCID generally requires chemotherapy plus serotherapy for unrelated donor transplant. 5For X-linked and JAK3 SCID a maternal donor with serotherapy alone is preferred over a 9/10 unrelated or 6–7/8 cord donor. 6Consider chemotherapy conditioning for enhanced B and/or T cell reconstitution and to prevent rejection.
Post-transplant Management
Before hospital discharge, close attention must be given to availability of immediate medical care as well as T cell immunity and infection risk at home. If there are no siblings or if siblings are attending grade school, SCID patients are considered for discharge when they have >50×106/L CD4+ cells and >10% of the lower limit of normal CD45+ lymphocyte proliferation to PHA. If preschool-age siblings are in the home, they should not attend daycare or group activities, and patient T-cell counts and proliferation need to be higher before discharge is considered.
Immune reconstitution is monitored with lymphocyte evaluation including naïve and memory T cell, NK cell and B cell subsets every 4 weeks until normal then every 3 months for the first year. Vaccines are initiated in patients who have B cell reconstitution when IgG replacement has been discontinued. At our institution, this is defined as total CD19 B cells >150 cells/uL; normal IgA and IgM for age; and IgM isohemagglutinin titers ≥1:8. If one of these criteria is not met, normal percentages of CD27+IgM-IgD- B cells is considered. Baseline titers against vaccine antigens are obtained 3 months after the last IgG infusion and 3–4 weeks following administration of the primary series of inactivated vaccines. If adequate responses are demonstrated, including a normal lymphocyte proliferation to tetanus toxoid, live attenuated vaccines are given.
Non-SCID TCL workup and management
For infants with 300 – 1500 T cells/uL, reduced but present naïve CD4 T cells (can be variable and <200 cells/uL), and no maternal cells (Figure 1, right panel; Figure 3), initial immune evaluation is similar to that for SCID, but hospitalization may not be required if immunodeficiency is not profound.
Figure 3.
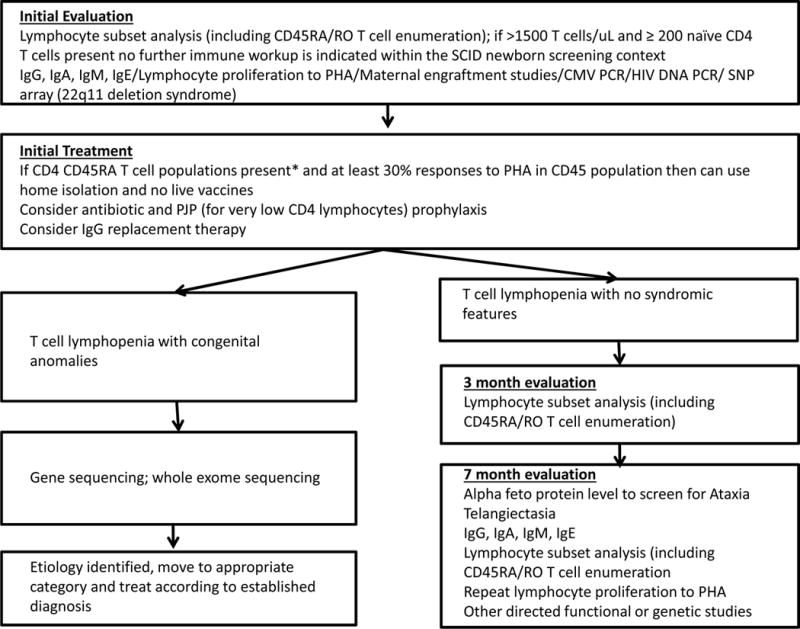
Evaluation and management of non-SCID T cell lymphopenia (persistently <1500 CD3 T cells); continued follow-up is needed to determine degree and persistence of immune impairment, whether prophylactic therapy is needed, and whether an underlying etiology can be found.
*Variable can be <200 naïve CD4 T cells
Non-SCID TCL conditions evaluated by UCSF immunologists (aside from 10 cases of TCL due to extreme preterm birth alone, which resolved in survivors) included multisystem syndromes in which T cells are variably reduced. Of 28 syndromic infants, diagnoses were: 17 DiGeorge syndrome/22q11.2 deletion or TBX1 intragenic mutation (61% of syndromic TCL); 2 each of ataxia telangiectasia (AT), CHARGE syndrome and trisomy-21; and 1 each of Noonan syndrome, Kabuki makeup syndrome, CLOVES syndrome, Fryns syndrome, and immuno-osseous dysplasia. Of these, 1 each with CHARGE, CLOVES, Fryns and Noonan syndromes died of severe congenital anomalies. Nine infants have had secondary TCL, in which T cell generating capacity is normal but circulating T cells were diminished. Causes included 2 cases of hydrops, 3 of severe congenital heart disease; and 1 each of chylothorax, neonatal leukemia and maternal immunosuppressive medication (fingolomod) taken during pregnancy for multiple sclerosis. An additional 5 infants have had idiopathic lymphopenia with no underlying cause identified [11, 12, 14], although the syndromic infants with AT and immuno-osseous dysplasia and maternal medication were also in this category at initial presentation. Resolution of idiopathic TCL occurred in 1 infant, a fraternal twin, while 2 continue to be followed with low, but functional T cells, and 2 have been lost to follow-up. Long-term follow up is important in identifying underlying diagnoses when TCL persists (Figure 3).
It is important to remember that not all serious disorders of T cells are identified by TREC screening; combined immunodeficiencies (CIDs) that are associated with intact T cell development beyond the point of TCR gene recombination in the thymus, including Zap-70 deficiency and MHC class I and II non-expression, may have normal numbers of TRECs even though T cell function is severely impaired [37].
Alpha-fetoprotein (AFP) levels obtained before the age of 6 months are generally not helpful in the diagnosis of AT and should be interpreted with caution. It is known that AFP may be elevated in healthy infants but trends down with age unlike AT where AFP levels will remain elevated [38]. Similarly, functional antibody responses cannot reliably be assessed before completion of the primary series of killed vaccines. Those infants with <1500 T cells/mL are considered immune deficient and advised to avoid live rotavirus vaccination, due to the risk of acquiring diarrheal disease from the vaccine strain rotavirus [39]. Need for home isolation, prophylactic antibiotics, and gammaglobulin infusions are made on a case-by-case basis.
Conclusion
Early detection of SCID through TREC NBS creates the opportunity to provide immune reconstitution while infants are healthy and uninfected. While NBS optimizes the chances of successful definitive treatment, newborns with SCID present new issues in management, including the parental emotional difficulties as well as defining best treatment regimens. Early detection of non-SCID TCL also poses new challenges. Further experience and multi-center studies will establish best practices.
What is Known on This Subject
In the new era of severe combined immunodeficiency newborn screening establishing a diagnosis and managing these infants are new challenges for pediatricians.
What This Study Adds
This paper offers the first systematic approach to infants with severe combined immunodeficiency, leaky severe combined immunodeficiency and T cell lymphopenia identified by newborn screening.
Acknowledgments
Rare Diseases Clinical Research Network (RDCRN), California Department of Public Health (CDPH). Thanks to Mica Muskat, PNP and Alan Nguyen, MB BCh at UCSF. Thanks to Robert Currier, Fred Lorey and members of the California Newborn Screening Program, California Department of Public Health; UCSF Providers, patients and their families.
Funding source: Drs. Cowan, Dvorak, and Puck: Primary Immunodeficiency Treatment Consortium (PIDTC) NIH NIAID and NCATS ORD U54 AI082973 and R13 AI094943. Dr. Puck: NIH NIAID RO1 AI105776; NIH NCATS UCSF CTSI (UL1 TR000004).
Abbreviations
- SCID
Severe Combined Immunodeficiency
- NBS
Newborn Screening
- TCL
T cell lymphopenia
- TRECs
T cell receptor excision circles
- PIDTC
Primary Immune Deficiency Consortium
- HCT
Hematopoietic cell transplant
- ADA
Adenosine deaminase
- HLA
Human leukocyte antigen
- MMRD
Mismatched related donor
- MUD
Matched unrelated donor
- RIC
Reduced intensity conditioning
- MAC
Myeloablative conditioning
- CMV
Cytomegalovirus
- PPD
Post partum depression
- PTSD
Post traumatic stress disorder
Appendix 1. Workup Outline for Infants with SCID to Prepare for Hematopoietic Cell Transplant
| Blood tests for infant | Amount of blood* | Tube type, notes |
|---|---|---|
| CBC w/diff | 0.25 mL | EDTA |
| Lymphocyte subsets and lymphocyte T cells subsets, naïve and memory (CD45 RA/RO) | 0.5 ml | Purple microtainer, ordered as 2 separate tests |
| IgM, IgA, IgG, IgE | 2 ml (1 ml without IgE) | SST |
| Blood chemistries, albumin, total bilirubin, LFTs | 0.5 ml | Light green microtainer |
| HLA typing (prefer buccal swab if maternal engraftment suspected) | (2.6 mL if blood) | 2 Buccal swabs or small ACD yellow top tube |
| Maternal engraftment by STR markers on CD3 selected cells | 2.6 mL | 2.6 ml (small) ACD |
| Serum CMV PCR | 1.5 mL | EDTA |
| Lymphocyte proliferation to PHA (and other mitogens) | 5 mL | Na heparin |
| ABO, Rh, Antibody screen | Microtainer | EDTA |
| SCID Gene molecular diagnosis (sequencing) | 1 ml | Blood or Buccal swabs |
| ADA and PNP enzyme studies (if appropriate SCID) | 1 mL | EDTA |
| Infectious Disease Markers (Should be done within 30 days of admission to BMT) | ||
| HIV DNA PCR- immunology lab | 1 ml | EDTA |
| HBsAg | 0.6 mL | SST |
| EBV PCR | 1.5 mL | EDTA |
| HCV DNA PCR | 2 ml | EDTA |
| Other Pre HCT procedures | ||
| Cardiac Echo | ||
| Audiology Exam | ||
| Other tests and procedures to consider | ||
| Central line placement for infusions and blood draws | ||
| BAL only if indicated, to be sent for PCP, respiratory virus panel, cultures for bacteria, fungi, AFB, CMV; pertussis PCR | ||
| Stool for cultures, rotavirus antigen, Norovirus, C. difficile if diarrhea present, O&P | ||
| Respiratory virus PCR panel | ||
Minimum amounts by special arrangement with our laboratories. Suggest working with local lab to achieve acceptable minimum amounts that are institution specific.
Appendix 2. Prophylactic medications for infants with SCID
| Medication | Dose | Notes |
|---|---|---|
| IVIG | Initial loading dose 1 gm/kg. Get trough IgG at 1–2 weeks. After loading give at least 0.5 gm/kg every 3–4 wks (use full vial, e.g. 2.5 or 5g) | Maintain IgG trough >800 mg/dL, usually 0.5 gm/kg every 3 wk |
| TMP/SMX for PJP prophylaxis* | 2.5 mg/kg TMP component BID PO 2 days/week | Start when >30 days old; monitor bilirubin and CBC |
| Leukovorin (folinic acid) | 1.25 mg TMP/SMX 2 days/week with TMP/SMX | Minimizes TMP/SMX neutropenia |
| Atovaquone for PJP prophylaxis | 30 mg/kg PO once daily, 1–3 mo 45 mg/kg PO once daily, 4 mo – 2 yr |
Alternative to TMP/SMX |
| Dapsone | 4 mg/kg PO weekly | Alternative to TMP/SMX |
| Acyclovir for herpesvirus prophylaxis | 12.5 mg/kg PO BID or 8 mg/kg IV q12hr (≤12kg), 400mg/m2 PO BID or 250mg/m2 IV q12hr (>12kg) | |
| Fluconazole for fungal prophylaxis | 6 mg/kg/q48 hours (full term <1 month) 6mg/kg/day ≥ 1 month | Ensure normal LFTs prior to starting; follow LFTs |
| Palivizumab | Per institutional protocol, q 4 weeks | During RSV season |
| PEG-ADA (Adagen) | At least 30 units/kg (up to half of a vial) twice weekly; start with full dose | Start for ADA deficient patients, while awaiting HCT or gener therapy |
Inhaled pentamidine is not recommended in children ≤ 5 years of age. There are no recommendations for IV pentamidine for prophylaxis in infants and would avoid this in the newly diagnosed babies given the risk of nephrotoxicity and possible hepatoxicity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributors’ Statement: Dr Dorsey contributed to concept of study, acquired data, interpreted data, and wrote the paper. Drs. Dvorak, Cowan, Puck contributed to concept of study, provided intellectual content and critically revised the manuscript. All authors provided patient care; all approved the final version and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement: The authors have no conflicts of interest relevant to this article to disclose.
References
- 1.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–55. doi: 10.1146/annurev.immunol.22.012703.104614. [DOI] [PubMed] [Google Scholar]
- 2.Ochs HD, Puck JM, editors. Primary Immunodeficiency diseases: a molecular and genetic approach. Oxford University Press; 2013. [Google Scholar]
- 3.Shearer WT, Dunn LD, Notarangelo G, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol. 2014;133(4):1092–8. doi: 10.1016/j.jaci.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pai SY, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371(5):434–46. doi: 10.1056/NEJMoa1401177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115(2):391–8. doi: 10.1016/j.jaci.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 6.Morinishi Y, Imai K, Nakagawa N, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. The Journal of Pediatrics. 2009;155(6):829–833. doi: 10.1016/j.jpeds.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 7.Comeau AM, Hale JE, Pai SY, et al. Guidelines for implementation of population-based newborn screening for severe combined immunodeficiency. J Inherit Metab Dis. 2010;33:S273–81. doi: 10.1007/s10545-010-9103-9. [DOI] [PubMed] [Google Scholar]
- 8.Kwan A, Hu D, Gomes H, et al. Successful newborn screening for SCID in the Navajo Nation. Clin Immunol. 2015;158(1):29–34. doi: 10.1016/j.clim.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chase NM, Verbsky JW, Routes JM. Newborn screening for SCID: three years of experience. Ann N Y Acad Sci. 2011;1238:99–105. doi: 10.1111/j.1749-6632.2011.06241.x. [DOI] [PubMed] [Google Scholar]
- 10.Vogel BH, Bonagura V, Weinberg GA, et al. Newborn screening for SCID in New York State: experience from the first two years. J Clin Immunol. 2014;34(3):289–303. doi: 10.1007/s10875-014-0006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwan A, Church JA, Cowan MJ, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. 2013;132(1):140–50. doi: 10.1016/j.jaci.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwan A, Abraham RS, Currier A, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–38. doi: 10.1001/jama.2014.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffith LM, Cowan MJ, Notarangelo LD, et al. Primary Immune Deficiency Treatment Consortium (PIDTC) update. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2016.01.051. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dvorak CC, Cowan MJ, Logan BR, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. 2013;33(7):1156–64. doi: 10.1007/s10875-013-9917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwan A, Puck JM. History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol. 2015;39(3):194–205. doi: 10.1053/j.semperi.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grunebaum E, Cutz E, Roifman CM. Pulmonary alveolar proteinosis in patients with adenosine deaminase deficiency. J Allergy Clin Immunol. 2012;129(6):1588–93. doi: 10.1016/j.jaci.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Schuetz C, Neven B, Dvorak C, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood. 2014;123(2):281–289. doi: 10.1182/blood-2013-01-476432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cowan MJ, Gennery AR. Radiation-sensitive severe combined immunodeficiency: The arguments for and against conditioning before hematopoietic cell transplantation–what to do? J Allergy Clin Immunol. 2015;136(5):1178–85. doi: 10.1016/j.jaci.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mezzacappa ES, Endicott J. Parity mediates the association between infant feeding method and maternal depressive symptoms in the postpartum. Archives of Womens Mental Health. 2007;10(6):259–266. doi: 10.1007/s00737-007-0207-7. [DOI] [PubMed] [Google Scholar]
- 20.Ystrom E. Breastfeeding cessation and symptoms of anxiety and depression: a longitudinal cohort study. BMC pregnancy and childbirth. 2012;12(1):1. doi: 10.1186/1471-2393-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeLuca JM, Kearney MH, Norton SA. Parents' experiences of expanded newborn screening evaluations. Pediatrics. 2011;128(1):53–61. doi: 10.1542/peds.2010-3413. [DOI] [PubMed] [Google Scholar]
- 22.Heller K. Genetic counseling: DNA testing for the patient. Proc (Bayl Univ Med Cent) 2005;18(2):134–7. doi: 10.1080/08998280.2005.11928052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fanos JH, Puck JM. Family pictures: growing up with a brother with X-linked severe combined immunodeficiency. Am J Med Genet. 2001;98(1):57–63. [PubMed] [Google Scholar]
- 24.Kuo CY, Kohn DB. Gene therapy for the treatment of primary immune deficiencies. Current allergy and asthma reports. 2016 May 1;16(5):1–8. doi: 10.1007/s11882-016-0615-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohn D. Gene therapy outpaces haplo for X-SCID. Blood. 2015;25(23):3521–3522. doi: 10.1182/blood-2015-04-641720. [DOI] [PubMed] [Google Scholar]
- 26.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 27.Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hacein-Bey-Abina S, Pai S-Y, Gaspar HB, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371(15):1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassan A, Booth C, Brightwell A, et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase–deficient severe combined immunodeficiency. Blood. 2012;120(17):3615–24. doi: 10.1182/blood-2011-12-396879. [DOI] [PubMed] [Google Scholar]
- 30.https://clinicaltrials.gov/ct2/show/NCT01129544?term=SCID&rank=18
- 31.De Ravin SS, Wu X, Moir S, Anaya-O’Brien S, et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Science Translational Medicine. 2016;8(335):335–57. doi: 10.1126/scitranslmed.aad8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Punwani D, Zhang Y, Yu J, et al. Multisystem Anomalies in Severe Combined Immunodeficiency with Mutant BCL11B. New Engl J Med. 2016;375(22):2165–76. doi: 10.1056/NEJMoa1509164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wahlstrom J, Patel K, Eckhert E, Kong D, Horn B, Cowan MJ, Dvorak CC. Transplacental maternal engraftment and posttransplantation graft-versus-host disease in children with severe combined immunodeficiency. J of Allergy and Clin Immunol. 2016 Jun 16; doi: 10.1016/j.jaci.2016.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savic RM, Cowan MJ, Dvorak CC, et al. Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2013;19(11):1608–14. doi: 10.1016/j.bbmt.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dvorak CC, Hassan A, Slatter MA, et al. Comparison of outcomes of hematopoietic stem cell transplantation without chemotherapy conditioning by using matched sibling and unrelated donors for treatment of severe combined immunodeficiency. Journal of Allergy and Clinical Immunology. 2014;134(4):935–43. doi: 10.1016/j.jaci.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dvorak CC, Hung GY, Horn B, Dunn E, Oon CY, Cowan MJ. Megadose CD34+ cell grafts improve recovery of T cell engraftment but not B cell immunity in patients with severe combined immunodeficiency disease undergoing haplocompatible nonmyeloablative transplantation. Biology of Blood and Marrow Transplantation. 2008;14(10):1125–33. doi: 10.1016/j.bbmt.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Dimitrova D, Ong PY, O’Gorman MR, Church JA. Major histocompatibility complex class II deficiency complicated by Mycobacterium avium complex in a boy of mixed ethnicity. J Clin Immunol. 2014 Aug 1;34(6):677–80. doi: 10.1007/s10875-014-0048-x. [DOI] [PubMed] [Google Scholar]
- 38.Wu JT, Roan Y, Knight JA. Alpha-Fetoprotein and Congenital Disorders. New York, NY: Academic Press, Inc.; 1985. Serum AFP levels in normal infants: their clinical and physiological significance; pp. 111–123. [Google Scholar]
- 39.Patel NC, Hertel PM, Hanson IC, et al. Chronic rotavirus infection in an infant with severe combined immunodeficiency: successful treatment by hematopoietic stem cell transplantation. Clin Immunol. 2012;142(3):399–401. doi: 10.1016/j.clim.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]