

Abstract
Many inborn errors of metabolism can cause cardiomyopathy. Cardiomyopathy associated with glycogen storage includes PRKAG2-associated glycogen storage disease (GSD), Danon disease, infantile-onset Pompe disease (GSD II), GSD III, GSD IV, and phosphofructokinase deficiency (Tarui disease or GSD VII).
We present a 35-year-old female who presented with cardiomyopathy after a pregnancy complicated by primary hyperparathyroidism. She had enjoyed excellent health until her first pregnancy at age 33. One week postpartum, she developed dyspnea and an echocardiogram revealed left ventricular ejection fraction (LVEF) of 35%. A cardiac MRI was consistent with nonischemic cardiomyopathy with an infiltrative process. Endomyocardial biopsy showed striking sarcoplasmic vacuolization, excess glycogen by PAS staining, and frequent membrane-bound glycogen by electron microscopy, consistent with lysosomal GSD. Acid alpha-glucosidase (GAA) activity in skin fibroblasts was in the affected range for Pompe disease. Sequencing of the GAA gene revealed a paternally inherited pathogenic c.525delT (p.Glu176Argfs*45) and a de novo c.309C>G (p.Cys103Trp) with unknown pathogenicity. Testing of the familial mutations in her daughter indicated that the variants in the proband were in trans. 26-gene cardiomyopathy sequencing panel had normal results thereby excluding GSD III, Danon disease, Fabry disease, and PRKAG2-associated cardiomyopathy. Therefore, results strongly suggest a diagnosis of Pompe disease.
Pompe disease has a broad disease spectrum, including infantile-onset (IOPD) and late-onset (LOPD) forms. LOPD typically presents with proximal muscle weakness and respiratory insufficiency in childhood or late adulthood. Our case may represent a very unusual presentation of adult LOPD with isolated cardiomyopathy without skeletal muscle involvement or respiratory failure.
Keywords: Acid phosphatase, Cardiomyopathy, Electron microscopy, Hypertrophic, Membrane-bound, Pompe disease (glycogen storage disease type II), Vacuolation
Introduction
Many inborn errors of metabolism can cause cardiomyopathy. Several examples of these include mitochondrial disorders (Palecek et al. 2012), Fabry disease (Clarke 2007), glycogen storage diseases (GSDs), sphingolipid storage diseases, and mucopolysaccharidoses (Elliott et al. 2014). Cardiomyopathy associated with glycogen storage includes PRKAG2-associated glycogen storage (Arad et al. 2002), Danon disease (Nishino et al. 2000), infantile Pompe disease (GSD II) (Kishnani et al. 2006), GSD III (Kishnani et al. 2010), GSD IV (Bruno et al. 2004), and phosphofructokinase deficiency (Tarui disease or GSD VII) (Musumeci et al. 2012). We report an adult with late-onset Pompe disease (LOPD) presenting with cardiomyopathy during pregnancy complicated by primary hyperparathyroidism without evidence of skeletal or respiratory muscle involvement.
Case Report
The proband is a 35-year-old female who had enjoyed excellent health and competitive athletic activities until her first pregnancy at age 33. Her history was notable for a cardiac murmur at age 23 without clinical symptoms. Clinical evaluation showed normal cardiac findings. A murmur was again noted during a job application at age 33 with reassuring results on an electrocardiogram, echocardiogram, and treadmill stress test. She had no family history of cardiomyopathy or sudden death.
During the fifth month of her pregnancy, she developed sudden-onset dyspnea lasting for a week. One week after a normal delivery, dyspnea recurred and an echocardiogram revealed dilated cardiomyopathy with a left ventricular ejection fraction (LVEF) of 35% (normal 54–76%). A follow-up echocardiogram 7 months later showed moderate LV enlargement [LV volume of 81 ml (systolic) and 127 ml (diastolic)], mild hypertrophy [LV mass index (2D) of 141 g/m2; LV septum thickness of 12 mm (normal 7–10 mm); LV posterior wall thickness of 9 mm (normal 7–11 mm); and end-diastolic LV diameter of 55 mm (normal 39–53 mm)], and reduced systolic function [LVEF of 34% and LV longitudinal peak systolic strain of ~8% (normal < −18%)]. Grade 3/4 LV diastolic dysfunction consistent with severely elevated LV filling pressure as well as bi-atrial enlargement and mild right ventricular hypertrophy was also noted. The heterogeneous echo-bright appearance of myocardium was suggestive of an infiltrative cardiomyopathic process. A subsequent cardiac magnetic resonance imaging (MRI) confirmed LV enlargement (maximal end-diastolic wall thickness of 12 mm at mid septum) with pronounced wall motion abnormalities and moderately reduced systolic function with LVEF of 32% (normal 54–85%); LV volume/Body Surface Area (BSA) of 91 ml/m2 (systolic, normal 12–36 ml/m2) and 134 ml/m2 (diastolic, normal 49–90 ml/m2); and LV mass/BSA of 66 g/m2 (diastolic, normal 59–103 g/m2). Extensive biventricular delayed myocardial enhancement predominantly in the epicardial and transmural muscles was noted.
Pro-BNP level was elevated at 3,894 pg/ml. The patient was started on a loop diuretic, potassium sparing diuretic, digoxin, an angiotensin II receptor blocker, and a beta-blocker though the patient admitted to intermittent noncompliance. A placement of an implantable cardioverter-defibrillator was recommended but was declined by the patient. An endomyocardial biopsy was performed based on the cardiac MRI findings. Histologic analysis of formalin-fixed tissue revealed prominent myocyte vacuolization. The vacuoles contained glycogen by Periodic acid–Schiff (PAS) staining, with a minor component of diastase-resistant material on PAS diastase (PASD). Frequent membrane-bound glycogen was seen on electron microscopy; no abnormal glycogen structures were seen. The underlying cause of the myocardial glycogen accumulation was investigated. Acid alpha-glucosidase (GAA) activity was measured on dried blood spot in two different clinical labs. The first lab reported a level in the affected range, the second lab in the carrier range. GAA activity in skin fibroblasts (16.7% normal: 260±80 nmol/h/mg protein) and in heart biopsy tissue (0.07; normal: 0.89±0.42 μmol/min/g tissue) was clearly in the affected range and partially reduced in the muscle biopsy tissues (5.86; 38% of normal: 22.91±8.4 nmol/h/mg protein). However, both muscle and heart tissue glycogen content and structure tested in the low or normal ranges (muscle glycogen content 0.11 umol glucose/mg tissue; normal 0.39–1.5; muscle glycogen structure measured as glucose-1-phosphate/glucose ratio 72.22%; normal > 5%; heart glycogen content 0.14 umol glucose/mg tissue; normal > 0.047; and heart glycogen structure 23.91%; normal > 5%). Branching enzyme activity in the heart and muscle tissue was also normal.
Sequencing of the GAA gene and follow-up parental testing revealed a paternally inherited heterozygous c.525delT (p.Glu176Argfs*45) and a heterozygous de novo c.309C>G (p.Cys103Trp) variant. Mutations of cysteine 103 (p.Cys103Arg and p.Cys103Gly) have been reported in Pompe disease, while the pathogenicity of c.309C>G (p.Cys103Trp) is currently unknown. This mutation was not found in the proband’s mother. Polyphen, SIFT, and MutationTaster algorithms, that predict the effect of amino acid changes, predicted this change to be deleterious to protein function. The proband’s daughter was tested for the familial mutations in order to determine the phase of the variants in the proband, and was found to harbor the c.525delT but not c.309C>G. These results indicate that the GAA variants in the proband are in trans. A 26-gene cardiomyopathy sequencing panel was sent to exclude other causes of cardiomyopathy and yielded no pathogenic mutation in any of the tested genes that included AGL (GSD III), LAMP2 (Danon disease), GLA (Fabry disease), and PRKAG2. Urine glucose tetrasaccharides (Hex4) biomarker level was normal at 1.0 mmol/mol cr (<4). Creatine kinase (CK) activity levels were measured in the normal range.
The patient was also noted to have elevation of calcium (11.6 mg/dl, reference 8.9–10.1 mg/dl) and PTH (541 pg/ml, reference 15–65 pg/ml) and underwent partial parathyroidectomy due to a diagnosis of primary hyperparathyroidism; histologic analysis showed parathyroid hyperplasia. The onset of hyperparathyroidism could not be determined. Skeletal muscle biopsy performed at the same time as the parathyroidectomy showed only denervation atrophy; the specimen was not available for our review.
Several months after the surgery, she developed sudden decline in her cardiac function to an LVEF of 20–25% (normal 54–76%) with global hypokinesis and no evidence of LV dilation by echocardiography. CK-MB was 36 U/l (normal 7.0–25.0 U/l). An urgent placement of a centrifugal continuous-flow left ventricular assist device (LVAD) was required. Right ventricular myocardial tissue taken during the LVAD placement, 17 months after the first cardiac biopsy, showed progression in lysosomal glycogen storage with myocyte hypertrophy with striking sarcoplasmic vacuolization, excess glycogen by PAS staining, and diffusely increased lysosomal activity by acid phosphatase staining on frozen tissue (Fig. 1). PASD showed only minimal diastase-resistant material. Phosphofructokinase and myophosphorylase enzymatic activities were intact. There was widespread replacement fibrosis. Electron microscopy showed frequent membrane-bound glycogen, degenerative changes, and mildly increased mitochondrial numbers (Fig. 1); Rectus skeletal muscle obtained at the same time showed mild degenerative features and fiber atrophy. Extensive cautery artifact limited the interpretation, although PAS and PASD staining showed no evident glycogen deposition in the few well-preserved areas.
Fig. 1.
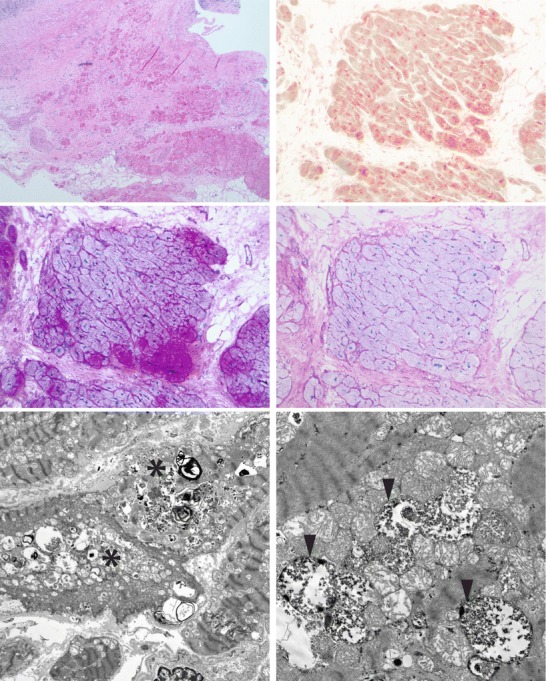
Histologic and ultrastructural features of myocardial tissue taken during LVAD placement, showing features of a lysosomal glycogen deposition disorder. Top row, left: paraffin-embedded H&E section showing extensive fibrosis and myocyte loss, myocyte hypertrophy, and vacuolization, with reactive, pericardial, mononuclear inflammation. Top row, right: frozen section stained for acid phosphatase activity showing diffusely increased lysosomal activity. Middle row, left: PAS staining of frozen section showing increased glycogen in myocytes, in an uneven pattern. Middle row, right: PASD (frozen) showing only minimal diastase-resistant material. Bottom row, left: electron microscopic image at 2200× showing vacuoles (asterisks) containing glycogen and lysosomal structures. Bottom row, right: electron microscopic image at 9300× showing that the glycogen is frequently membrane-bound (arrowheads)
The patient had multiple pulmonary function tests due to the diagnosis of LOPD with normal results with normal vital capacity in the upright and supine positions. Her neurological examination before and after the LVAD placement were both normal without muscle weakness. She had excellent muscle strength except slightly weak back extensors and normal grip and pinch strength. Her motor speech function, inspiratory/expiratory muscle strength, and lingual strength were essentially normal. Her 6-min walk test showed 76.9% predicted for her age and gender. Optimal cooperation from the patient was not possible for this test as she was fatigued from international traveling. She recovered to the point where she can practice Pilates and low weight resistance exercises after the LVAD placement. Enzyme replacement therapy (ERT) with alglucosidase alfa was discussed after the diagnosis of LOPD was made. It was felt that the therapy would not reverse the extensive fibrotic and irreversible damage evident in the cardiac muscle, nor alleviate cardiomyopathy as penetration of the enzyme through the fibrotic tissue was questionable. She was maintained on warfarin and aspirin but suffered an extensive thrombotic stroke that has precluded further ongoing evaluation for possible heart transplant.
Discussion
Based on myocardial histology, low enzyme activity in fibroblast tissue and cardiac muscle, and the identification of two GAA changes in trans, LOPD is the favored diagnosis in this case.
Pompe disease has a broad disease spectrum, including infantile-onset (IOPD) and late-onset (LOPD) forms. LOPD typically presents with proximal muscle weakness and respiratory insufficiency in childhood or late adulthood (Kishnani et al. 2006). Cardiac involvement in LOPD include aortic aneurysm (El-Gharbawy et al. 2011), arrhythmias, ventricular dysfunction, but cardiomyopathy is rare and, if found, is not the primary presentation (Soliman et al. 2008; Morris et al. 2015). Lee et al. reported six children with Pompe disease who developed cardiomyopathy at ages ranging from 1 month to 3 years (Lee et al. 2014). All of the patients also had muscular involvement with hypotonia or developmental delay at the time of presentation. Ben-Ami et al. reported a 45-year-old man with dilated cardiomyopathy and marked myocardial hypertrophy, in whom endomyocardial biopsy and staining indicated abnormal glycogen storage. Enzyme and genetic tests and other clinical history were not described in this case (Ben-Ami et al. 2001). In the current case, a diagnosis of LOPD is most likely based on histology, ultrastructure, enzymology, and GAA genotype. Two myocardial biopsies showed membrane-bound glycogen associated with lysosomal structures, suggesting a lysosomal GSD. The presence of glycogen granules with a normal structure argues against a polyglucosan disorder. PRKAG2-associated cardiomyopathy, Danon disease, Fabry disease, GSD IV, and GSD III were ruled out by sequencing and/or enzymology. Normal staining of phosphofructokinase and myophosphorylase activity by immunohistochemistry ruled out GSD VII and GSD V. Some mitochondrial disorders can cause isolated cardiomyopathy but would not cause glycogen storage in histology. GAA sequencing revealed a pathogenic mutation c.525delT and a missense variant c.309C>G in trans. Notably, de novo variants in GAA are rare and the de novo variant in our case may represent a post-zygotic or germline event. GAA activity in skin fibroblasts, the current gold standard for laboratory diagnosis (Kishnani et al. 2006), was clearly in the affected range. Urine-Hex4 and CK can be normal in Pompe disease. In our extensive experience, skeletal muscle biopsy can often be normal in affected in patients with LOPD if the biopsy is not taken from an affected tissue. It is possible that at the current age our proband has yet to manifest skeletal myopathy or respiratory failure and other LOPD symptoms, whereas pregnancy and hyperparathyroidism triggered exacerbation of cardiomyopathy (Andersson et al. 2004).
Conclusion
We described a rare presentation of adult LOPD that presented with isolated cardiomyopathy after a pregnancy, complicated by hyperparathyroidism. Histology, enzymology, and molecular analysis of GAA support the diagnosis. LOPD can present with cardiomyopathy even in an absence of skeletal muscle involvement. Cardiac muscle pathology with specific staining supported by genetic and enzyme testing can lead to the diagnosis.
Synopsis
LOPD can present with an isolated cardiomyopathy and endomyocardial biopsy can be diagnostic.
Compliance with Ethics Guidelines
Conflict of Interest
Mari Mori, Lauren A. Bailey, Catherine W. Rehder, Januario Estrada, Deeksha S. Bali, Anne F. Buckley, Jennifer S. Li, Joseph G. Rogers, and Priya S. Kishnani declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Contributions of Individual Authors
Mari Mori and Priya S. Kishnani: Patient evaluation, literature search, drafting and revisions of the manuscript.
Lauren A. Bailey: Collection of clinical data.
Catherine W. Rehder: Molecular analysis of the proband and daughter, and critical revision of the manuscript.
Januario Estrada: Preparation and interpretation of skeletal and cardiac muscle tissues.
Deeksha S. Bali: Enzyme activity analysis of fibroblast, and skeletal/cardiac muscle tissues, and critical revisions of the manuscript.
Jennifer S. Li and Joseph G. Rogers: Patient evaluation and critical revisions of the manuscript.
Anne F. Buckley: Interpretation of skeletal and cardiac muscle tissues and critical revisions of the manuscript.
Contributor Information
Mari Mori, Email: mari.mori@duke.edu.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Andersson P, Rydberg E, Willenheimer R. Primary hyperparathyroidism and heart disease—a review. Eur Heart J. 2004;25:1776–1787. doi: 10.1016/j.ehj.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Arad M, Benson DW, Perez-Atayde AR, et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–362. doi: 10.1172/JCI0214571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ami R, Puglisi J, Haider T, Mehta D. The Mount Sinai Hospital clinicalpathological conference: a 45-year-old man with Pompe’s disease and dilated cardiomyopathy. Mt Sinai J Med. 2001;68:205–212. [PubMed] [Google Scholar]
- Bruno C, van Diggelen OP, Cassandrini D, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV) Neurology. 2004;63:1053–1058. doi: 10.1212/01.WNL.0000138429.11433.0D. [DOI] [PubMed] [Google Scholar]
- Clarke JTR. Narrative review: Fabry disease. Ann Intern Med. 2007;146:425–433. doi: 10.7326/0003-4819-146-6-200703200-00007. [DOI] [PubMed] [Google Scholar]
- El-Gharbawy AH, Bhat G, Murillo JE, et al. Expanding the clinical spectrum of late-onset Pompe disease: dilated arteriopathy involving the thoracic aorta, a novel vascular phenotype uncovered. Mol Genet Metab. 2011;103:362–366. doi: 10.1016/j.ymgme.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 2014;35:2733–2779. doi: 10.1093/eurheartj/ehu199. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–463. doi: 10.1097/GIM.0b013e3181e655b6. [DOI] [PubMed] [Google Scholar]
- Lee DH, Qiu WJ, Lee J, et al. Hypertrophic cardiomyopathy in Pompe disease is not limited to the classic infantile-onset phenotype. JIMD Rep. 2014;17:71–75. doi: 10.1007/8904_2014_339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DA, Blaschke D, Krebs A, et al. Structural and functional cardiac analyses using modern and sensitive myocardial techniques in adult Pompe disease. Int J Cardiovasc Imaging. 2015;31:947–956. doi: 10.1007/s10554-015-0629-7. [DOI] [PubMed] [Google Scholar]
- Musumeci O, Bruno C, Mongini T, et al. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII) Neuromuscul Disord. 2012;22:325–330. doi: 10.1016/j.nmd.2011.10.022. [DOI] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- Palecek T, Tesarova M, Kuchynka P, et al. Hypertrophic cardiomyopathy due to the mitochondrial DNA mutation m.3303C>T diagnosed in an adult male. Int Heart J. 2012;53:383–387. doi: 10.1536/ihj.53.383. [DOI] [PubMed] [Google Scholar]
- Soliman OI, Van Der Beek NA, Van Doorn PA, et al. Cardiac involvement in adults with Pompe disease. J Intern Med. 2008;264:333–339. doi: 10.1111/j.1365-2796.2008.01966.x. [DOI] [PubMed] [Google Scholar]