

Abstract
Thiamine (vitamin B1) is essential to the health of all living organisms and deficiency has long been associated with diseases in animals such as fish, birds, alligators, and domesticated ruminant mammals. Thiamine is also implicated in several human diseases including Alzheimer's, diabetes, dementia, depression and, most notably, Wernicke–Korsakoff syndrome and Beriberi disease. Yet, highly sensitive and specific detection of thiamine remains an analytical challenge, as pM to nm levels of thiamine need to be detected in environmental and human samples, respectively, various phosphorylated variants need to be discriminated, and rapid on‐site detection would be highly desirable. Furthermore, appropriate sample preparation is mandatory, owing to the complexity of the relevant sample matrices including fish tissues, ocean water, and body fluids. This Review has two objectives. First, it provides a thorough overview of analytical techniques published for thiamine detection over the last 15 years. Second, it describes the principles of analytical approaches that are based on biorecognition and may open up new avenues for rapid and high‐throughput thiamine analysis. Most notably, periplasmic binding proteins, ribozymes, and aptamers are of particular interest, as they function as bioaffinity recognition elements that can fill an important assay technology gap, owing to the unavailability of thiamine‐specific commercial antibodies. Finally, the authors provide brief evaluations of key outcomes of the major assay concepts and suggest how innovative techniques could help develop sensitive and specific thiamine analytical test systems.
Keywords: biosensing, fluorescence spectroscopy, liquid chromatography, sample preparation, thiamine deficiency
1. Introduction
Thiamine (vitamin B1) is essential to the health of all living organisms.1 In its diphosphate form, thiamine serves as a cofactor for enzymes involved in carbohydrate metabolism, including transketolase, α‐ketoglutarate dehydrogenase, pyruvate dehydrogenase, and branched chain α‐keto acid dehydrogenase. These enzymes are involved in pathways that produce ATP, NADPH, and ribose‐5‐phosphate, which are critical for generating cellular energy and downstream production of amino acids, nucleic acids, and fatty acids. Deficiencies in thiamine have numerous deleterious effects observed in both people and the environment. Most notably, deficiencies are responsible for Wernicke–Korsakoff syndrome and Beriberi disease prevalent in alcoholics in developed nations and undernourished populations in developing nations.2 More recently, deficiencies have also been implicated in other conditions including diabetes,3 depression,4 Alzheimer's disease,5 and dementia.6 In an ecological context, thiamine deficiencies result in muscle weakness and reproductive failure of valuable fish, including lake trout and Atlantic salmon.7 Such deficiencies have also affected alligators,8 birds,9 as well as various species of livestock.10 Thiamine deficiencies may occur if conditions impair thiamine absorption;11 if dietary intake of thiamine‐containing foods is restricted;12 or if foods containing high levels of enzymes capable of thiamine cleavage (thiaminases) are concurrently consumed.13 As such, thiamine is an analyte of key importance in clinical, ecological, and veterinary studies, and interest in this analyte has prompted numerous analytical methodologies for its detection. Owing to its unusual properties, thiamine presents some unique analytical challenges, many of which were uncovered in early work beginning in the 1940′s. These caveats must be recognized in the development of new techniques, and readers are encouraged to look to previous excellent Review articles for thorough coverage of earlier developments.14, 15 By contrast, this Review focuses on analytical advances made in the last 15 years, but incorporates historic information, as needed, to ensure accurate and reproducible thiamine measurements.
2. Thiamine Properties
Thiamine is a highly water‐soluble vitamin with reduced solubility in alcohols and negligible solubility in less polar organic solvents.16 Commercially, it is available in the form of thiamine hydrochloride or thiamine mononitrate, with the latter markedly less water soluble and less hygroscopic.16, 17 Structurally, thiamine consists of 2‐methyl‐4‐aminopyrimidine attached via a methylene group to a thiazole ring, substituted with a methyl group in the 4 position and a hydroxyethyl group in the 5 position. Phosphorylated derivatives of the hydroxyl group include thiamine mono‐, di‐, and triphosphates (TMP, TDP, and TTP, respectively). Thiamine is stable under acidic conditions, but is labile in alkaline conditions with the opening of the thiazole ring to yield the thiol form. At low pH, thiamine is present with a positive charge on both the pyrimidine N1 nitrogen (pKa1≈4.8) and thiazole N3 nitrogen. At physiological pH, thiamine is a cation with a positive charge on the thiazole N3 nitrogen. With further increase in pH, its behavior is complex, passing through an uncharged pseudobase intermediate to yield its negatively charged thiol form (pKa2≈9.2) (Figure 1).18
Figure 1.
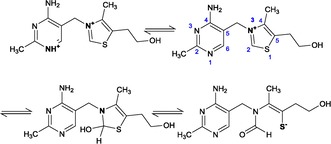
pH‐dependent forms of thiamine. Thiamine transitions upon increasing pH from cationic forms to the pseudobase and thiol forms.
Analytical methods based on the chemistry of thiamine take advantage of its cationic charge via ion exchange for isolation and separation techniques;19 detection relying on its activity in the UV range;20 ability to be oxidized and detected via fluorescence, chemiluminescence, and electrochemiluminescence,21, 22, 23 as well as polarity differences between thiamine, its phosphate esters, and other species (Figure 2).21, 24 Although intact thiamine is highly water soluble, chemical or biochemical cleavage at the methylene bridge allows subsequent extraction of the less polar 4‐methyl‐5‐(2‐hydroxyethyl)thiazole fragment into organic solvents prior to GC analysis25 or quantification by scintillation counting if the thiazole fragment is radiolabeled.26 Biological‐based methods take advantage of the role of thiamine as an essential vitamin for the growth of many microorganisms and, as noted above, in its diphosphate form, serving as a co‐factor for numerous enzymes.
Figure 2.
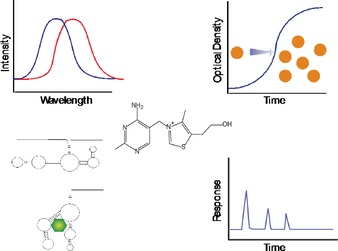
Approaches for thiamine analysis including fluorescence of the oxidized form of thiamine (thiochrome), thiamine requirement for microbiological growth, sensing based on biorecognition, and high performance liquid chromatography.
3. Sample Preparation
As thiamine is essential to the health of, and is present in, all organisms, sample matrices presented for analysis can range from environmental soils and water to a variety of human and animal foods as well as human urine and blood. Some generalities regarding best sample treatment practices are provided below.
Given the poor stability of thiamine under alkaline conditions, samples should be pH adjusted to the acidic range to avoid degradation. Environmental water samples should be filtered immediately upon sample collection to prevent possible uptake of thiamine by microorganisms and also possible release of thiamine from inorganic particulates.27 For tissue samples, Brown et al. noted the importance of storage conditions on thiamine forms.21 When stored at 4 °C, conversion of TMP to thiamine was observed in liver tissue samples from lake trout. Loss of TDP and formation of TMP was noted in muscle tissue stored at ambient temperature for 24 h, whereas in alewife muscle tissue, all thiamine species declined within an hour. Alewife contain high levels of thiaminases that break down thiamine and are likely responsible for the losses. In the majority of biological sample matrices, a mechanical homogenization step is required to ensure sample uniformity and free thiamine from its intracellular confines. This is commonly done in cold trichloroacetic acid (TCA) at a concentration of 2 % (v/v).21, 24 Proper implementation of the homogenization step is critical, with marked losses of thiamine and TMP using high‐speed tissue grinding or ultrasonic homogenization versus manual grinding of fish eggs.21 In human whole‐blood samples, overnight storage at −70 °C, but not −20 °C, is sufficient to ensure complete lysis of the red blood cells that contain the majority of thiamine (in the form of TDP) in the body.28 To ensure complete hemolysis, whole‐blood samples are typically frozen at −80 °C for at least 24 h, followed by TCA precipitation of proteins.29
Thiamine has exhibited non‐specific protein binding and TDP is associated with enzymes as a cofactor; therefore, protein precipitation and subsequent release of thiamine from these proteins is required for assaying biological sample matrices. Precipitation can be accomplished by using perchloric acid or TCA by boiling or autoclaving samples at 100 or 120 °C, respectively, for 10–30 minutes.21, 30, 31 For hemolyzed blood, centrifugation to remove cell debris, followed by the addition of cold 10 % TCA to the remaining supernatant has been considered sufficient to precipitate the proteins.28 Other means of acid extraction to release thiamine from sample matrices have included autoclaving homogenized samples at 120 °C in sulfuric acid or 10–100 mm HCl for 10–30 minutes.32, 33, 34 Some studies observed low recoveries with sulfuric acid, believed to be caused by either the presence of sulfite impurities, which can degrade thiamine by cleavage at the methylene bridge under acidic conditions, or adsorption of thiamine onto formed precipitates.14, 18 The latter has also been suggested with TCA.35 As recoveries with sulfuric acid tend to be low, HCl is preferred acid for the hydrolysis step. Some further caution may be warranted in the acid treatment of thiamine, as oxythiamine can be formed under strongly acidic conditions.18 This was confirmed recently, yielding low levels of oxythiamine in 100 mm acetic acid at elevated temperatures (100 °C for 1 h).36 As oxythiamine does not form a fluorescent product, the extent of this conversion cannot be monitored at the typical wavelengths used to quantify thiochrome, the fluorescent oxidation product of thiamine. Furthermore, extraction from protein matrices typically requires greater concentrations of TCA, a markedly stronger acid than acetic acid. If oxythiamine is formed to an appreciable degree under the conditions used to extract thiamine from protein matrices, the amount of thiamine present in a sample may be underestimated.
Prior to analysis by HPLC, samples treated with TCA are washed with solvents such as water‐saturated MTBE or 3:2 ethyl acetate/hexane to remove the acid.21, 28 Such washed samples have been noted to retain their stability for at least 48 h.21, 28 Complete removal of TCA can be difficult to achieve and has led to increased fluorescence background in an HPLC method versus perchloric acid; hence, the latter was preferred.30 In food matrices, beyond acid–heat treatment, enzymatic digestion is employed with enzymes such as amylase, taka‐diastase, clara‐diastase, and papain, which break down starches and proteins to ensure release of thiamine.33 Thiamine itself is present as a food additive, but in naturally occurring matrices such as milk,25 meat,33 and whole grains,37 thiamine monophosphate, diphosphate, and triphosphate may also be present. If the total thiamine content is desired, enzymatic treatment with enzymes such as acid phosphatase can be used to convert thiamine phosphate esters into the parent molecule.33 This is followed by autoclaving to cease further enzyme activity.
Solid‐phase extraction (SPE) using reverse phase,27 reverse phase with ion‐pairing reagents,33 reverse phase alone after conversion to thiochrome,34 and weak cation exchange (WCX)20 have been employed as means for sample clean‐up or simplified analysis. A reverse‐phase SPE method for thiamine extraction has been reported, which takes advantage of the increased hydrophobic character of riboflavin versus thiamine for the quantification of both species in fish eggs. The respective concentrations are read with a fluorescence plate reader, eliminating the need for costly and less field practical analyses with HPLC.24 In another reverse‐phase method, filtered and pH‐adjusted seawater was first passed through a C18 resin, followed by a water wash, and then elution of thiamine using methanol.27 By subsequently evaporating the methanol and reconstituting with water, the authors suggested that this protocol minimized interferences by co‐eluting organic compounds when the samples were subsequently analyzed with HPLC. By using a C18 resin with ion‐pairing via sodium heptanesulfonate, thiamine and riboflavin could be isolated from meat extracts,33 whereas another method taking advantage of WCX has been used to provide partial purification of thiamine from sausage extracts prior to HPLC analysis.20
4. Analytical Methods Based on Chemical Properties
4.1. Colorimetric Approaches
The maximum absorbance of thiamine at 235 nm is shared with many other compounds, making direct spectrophotometric measurements useful only when coupled with a separation mechanism. Hence, thiamine concentrations have historically been determined through colorimetric assays that employ a variety of reagents that produce a visible change in color after reacting with thiamine. Most colorimetric assays developed in the 1930′s through 1950′s depended on the use of a diazotized amine—an aromatic amine that has been treated with nitrous acid—with coupling carried out under alkaline conditions. The two most commonly used were diazotized sulfanilic acid and diazotized p‐acetophenone, which react with thiamine to form pink‐ and purple–red‐colored compounds, respectively. The formation of colored products redshifts the maximum absorbance out of the UV range and away from many interferences while improving the molar extinction coefficient (Table S1.) Challenges with these reagents included reagent instability, lengthy extraction procedures, reaction with other interfering species, poor stability of the reaction products, and low intensity of the colored species. A timeline of the development of these historic methods is provided in the Supporting Information (Figure S1).
Interest remains in the development of modern colorimetric methods for the analysis of thiamine in pharmaceutical products, with numerous approaches reported within the last 20 years.38, 39, 40 Of these, two methods that provide a clear advantage over previous approaches are mentioned within. By taking advantage of the ability of thiamine to undergo oxidization, researchers utilized the reduction of potassium iodate (V) to yield iodide ions in the presence of thiamine. The iodide ions formed free iodine in the reaction mixture, which could subsequently oxidize leucocrystal violet dye to crystal violet, measurable at 589 nm.40 This approach lowered the detection limit approximately tenfold over that obtainable by diazonitized sulfanilic acid. In the second method, Liu et al. investigated the formation of complexes of thiamine with triphenylmethane dyes, including bromothymol blue and cresol red, in the presence of surfactants as an approach to spectrophotometrically determine thiamine concentrations.39 The phenolic group of these dyes ionically associates with thiamine through its thiazole nitrogen. By using these dyes and micellar solutions to ensure complex solubility, either the formation of a complex at a different wavelength or loss of the dye absorbance at its inherent wavelength could be monitored. This approach lowered the detection limit to the low ng mL−1 range and, thus, was a marked improvement over prior methods. However, one caveat to note with all reported colorimetric methods is the potential for interferences by other species. Even with relatively simple pharmaceutical tablet matrices, these modern methods were subject to interferences; therefore, it remains to be seen whether such methods can have broader applicability in more complex biological matrices. In addition, as most of the colorimetric methods yielded detection limits in the low μg mL−1 (ca. 3–30 μm) range, they have largely fallen out of favor in lieu of more sensitive fluorometric methods, especially when biological samples are considered.
4.2. Fluorescence Methods
Thiamine is most commonly quantified following oxidation to the blue fluorescent product thiochrome (Figure 3). This forms the basis for quantification in the widely employed AOAC and AACC methods.30, 41
Figure 3.
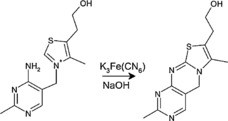
Oxidation of thiamine to thiochrome, shown here using potassium ferricyanide under alkaline conditions.
Oxidizing agents include mercury(II) chloride,42 cyanogen bromide,43, 44, 45 and potassium ferricyanide21 and, in all cases, the conversion forms both thiochrome and thiamine disulfide through competitive reactions.42 These reagents each have their benefits and limitations including improved dynamic range and lack of thiochrome fluorescence quenching,42 high fluorescence yield and minimization of redox interferences from biological matrices,43, 44, 45 as well as low cost, low toxicity, and ease of use, respectively, but suffer from high toxicity, short shelf‐life and reaction time dependence,43, 46 as well as reagent concentration dependence,45 respectively. Potassium ferricyanide is most commonly employed, owing to the aforementioned advantages and, hence, is the focus of this discussion, but a timeline of the development of other fluorometric reagents is provided in the Supporting Information (Figure S2). However, a limitation of this reagent is that, in excess, it can reduce formed thiochrome to non‐fluorescent products. Early experimenters of the thiochrome method attempted to measure thiochrome fluorescence directly in aqueous solutions,47 but later researchers incorporated an extraction of thiochrome into isobutanol to avoid possible quenching by ferricyanide, minimize the impact of other interferences present in the aqueous phase, and generate improved fluorescence intensity in the organic phase.48, 49, 50 An isobutanol extraction selectively isolates thiochrome formed from thiamine, but leaves behind that formed from the more polar phosphorylated esters. This step can thus be used to differentiate between free and esterified forms of thiamine while concomitantly eliminating interfering polar compounds that remain in the aqueous phase.50 Extraction of the formed thiochrome into isobutanol is independent of pH between 8 and 10, but decreases below pH 7.51 The organic extraction step could be eliminated in certain matrices by including a step that allows for the formation of a thiamine blank, as discussed below.
As the competing pathway for oxidation yields the formation of thiamine disulfide, the procedure and conditions for carrying out thiochrome formation is important. Formation of thiochrome occurs at pH values greater than 8.0 and its subsequent fluorescence intensity was found to be maximal at pH 12–13.52 Zajicek et al. reported that 5 N NaOH and 0.1 % potassium ferricyanide needed to be added simultaneously and directly to the thiamine sample, rather than allowing the fluids to enter more gradually along the tube walls.24 Mancinelli et al. noted unreliable results when an alkaline ferricyanide mixture was added, and instead added potassium ferricyanide followed by sodium hydroxide in their derivatization procedure.53 The addition of sodium sulfite, which stops the oxidation to thiochrome, has also been noted to improve reproducibility.54 Another avenue to increase the efficiency of thiochrome‐based detection involves including a miscible organic solvent such as ethanol or methanol in the aqueous alkaline media. This is believed to improve the yield of thiochrome versus thiamine disulfide, as well as increase the fluorescence intensity of formed thiochrome.55 Interferences with thiamine oxidation include ascorbic acid, polyphenols, and other substances with antioxidant capacity, which can consume ferricyanide,22, 56, 57 and polyphenols, which can additionally form complexes with thiamine.58 Polyvinylpyrrolidone (PVP), which has been used to precipitate polyphenolic species from wines,56 may be useful in overcoming some of these interferences.
As the predominant approaches for analysis involve thiochrome formation and fluorescence measurement, it is often desirable to develop a suitable blank to correct for endogenous fluorescence. Approaches have included omission of ferricyanide in the alkaline reagent during thiochrome formation,49 heating samples in alkaline solution at elevated temperatures,59 pH adjustment to 8.0–10.0 prior to isobutanol extraction,51 inclusion of benzenesulfonyl chloride that reacts with thiamine and prevents thiochrome formation,60 and enzymatic digestion of thiamine by using thiaminases.26 These efforts are less critical when HPLC is employed as a separation mechanism, but are necessary when fluorescence measurement alone is the downstream analytical approach.
4.3. Methods Based on Electrochemistry, Chemiluminescence and Electrochemiluminescence
Taking advantage of the ability of thiamine to be oxidized, numerous electrochemical methods have been developed (Table S2). The products of this oxidation vary depending on the conditions employed. By scanning the potential from 0 to +1.2 V by using a glassy carbon electrode under alkaline conditions, thiamine disulfide was initially formed with further oxidation, ultimately yielding the thiol form of thiamine in an irreversible oxidation process.61 Under alkaline conditions in the presence of high concentrations of methanol, thiochrome can be preferentially formed by using an applied potential of +0.4 V versus Ag/AgCl by using a glassy carbon electrode.62 By, instead, relying on an applied potential to oxidize thiamine to thiochrome, the inclusion of oxidizing reagents leading to the potential for interference can be eliminated. Kusube et al. combined the benefit of a chemical oxidant‐free process with the sensitivity afforded by fluorescence detection by using an in‐line electrochemical reactor in series with a fluorescence detector.62 This procedure permitted the specificity of fluorescence detection of thiochrome without the concomitant challenges inherent to chemical oxidation. Thiamine may also undergo reduction using electrochemical techniques.63 Unfortunately, while electrochemical methods offer comparatively low expense and simpler instrumentation than those relying on optics, they have traditionally not yielded the sensitivity that can be obtained using fluorescence detection. However, interest in their development may be reinvigorated with advances in materials. For example, a recent study used differential pulse adsorptive stripping voltammetry and a multi‐walled carbon nanotube paste electrode for the determination of thiamine. Such electrodes offer the benefits of improved conductivity, current sensitivity, and chemical inertness over traditional materials. This method yielded a linear range from 3 ng mL−1 to 0.88 μg mL−1 and reported a limit of detection of 1.1 ng mL−1 (ca. 3.3 nm).64
Chemiluminescence also occurs during thiamine oxidation with potassium ferricyanide enabling limits of detection of 9 μm.57 However, aside from the relatively high detection limit, absorption of the emitted radiation by riboflavin, if present, can result in an underestimation of thiamine content. At the same time, this can be used as an advantage with a well‐designed system. If a fluorophore, such as rhodamine B, is present and is capable of then emitting the absorbed light, an energy‐transfer mechanism can permit measurement instead at the fluorophore's intrinsic emission wavelength.23 An electrochemiluminescence method that takes advantage of this energy‐transfer emission of the fluorophore can be used to quantify thiamine at as little as 0.08 μg mL−1 (237 nm).23 Other methods utilizing chemiluminescence rely on the ability of thiamine to enhance or suppress the signal of traditional chemiluminescent reactions, leading to detection limits in the low nanometer range (Table S2.) Chemiluminescence offers the benefit of simpler instrumentation compared to fluorescence, as no light source is required, and the low non‐specific signal is useful in natural samples. However, aside from the few academic publications, these methods do not seem to have gained further traction within the analytical community.
4.4. Analysis by HPLC
Numerous methods for thiamine quantification by using HPLC have been reported, as covered in an excellent Review by Lynch and Young, and hence will only briefly be mentioned here.14 HPLC separations are the most practical approach if quantification of thiamine and possible phosphorylated thiamine species is desired or if thiamine is one of many analytes in a complex matrix that require quantification. Reverse phase is the most common separation mode by using C18 or PDVB columns, though ion pairing, hydrophilic interaction chromatography (HILIC), and amine columns have also been employed.20, 28, 31, 33, 34, 53, 56 Anionic ion‐pairing reagents include pentanesulfonate, hexanesulphonate, and heptanesulfonate, which interact with cationic thiamine to improve retention in reverse‐phase methods.33 In HILIC, thiamine is preferentially partitioned into the water layer adjacent to the hydrophilic stationary phase versus the mobile phase, which consists of a high organic content. Elution is accomplished by increasing the aqueous content of the mobile phase.20 Thiamine retention in HILIC is believed to be a function of not only its polarity, but also interactions via its positive charge with residual silanols on the stationary phase.65 Thiamine is poorly retained on reverse‐phase columns, but retention is efficient following its ring‐closure to thiochrome forming a more hydrophobic species. The order of elution of thiamine versus its phosphate esters is dependent on the separation conditions, with higher order phosphates eluting earlier in reverse phase methods and the opposite in normal phase methods. Although some methods utilize UV absorbance, allowing for detection down to 0.025 μg mL−1 (74 nm) at 232 nm,66 most methods use fluorescence detection following conversion of thiamine and its phosphate esters to their thiochrome derivatives. Oxidation to thiochrome is accomplished either by pre‐column or post‐column derivatization with reagents, as described above.28, 31 The advantage of the post‐column derivatization methods is that the separation does not need to be run at alkaline pH, which can adversely affect the useable life of silica‐based HPLC columns. However, this is a negligible concern when polymer or core–shell‐based HPLC columns are employed and the instrumentation complexity required for post‐column derivatization offsets any possible benefit.67 Methods based on fluorescence detection with modern instrumentation can achieve quantification limits of as little as 3 nm thiamine, with detection limits of as little as 1 nm routinely reported.28, 67 Methodologies relying on ultra‐performance liquid chromatography (UPLC) separations coupled with tandem mass spectrometry can achieve detection limits of 0.01 μg L−1 (30 pM) while simultaneously providing identity confirmation.68 HPLC methods have been employed for the quantification of thiamine and its phosphate esters in whole‐blood, plasma, erythrocytes, and breast milk.28, 29, 53, 68 For these samples, neither EDTA nor heparin used as an anticoagulant impacted the measurement of thiamine and its phosphate esters.53, 69 However, as EDTA likely inhibits phosphatase enzyme activity by complexing with the cofactor Mg2+,70 some researchers prefer its use as an anticoagulant to avoid the unintended cleavage of thiamine phosphate groups.29
4.5. Approaches Based on Optical Properties of Nanoparticles
Other approaches have relied on the modulation of nanoparticle (NP) aggregation or fluorescence via thiamine, as summarized in Table S3. Thiamine has been shown to cause aggregation of gold NPs, owing to its postulated interaction through its thiazole sulfur atom.71 The aggregation of the NPs can be monitored by a colorimetric change observed at 590 nm. This change did not occur with thiamine phosphates that allowed differentiation between free thiamine and TDP before and after treatment with alkaline phosphatase, which served to cleave phosphate groups. A limit of detection of 54 nm was reported with an assay range from 0.15 to 3.5 μm. Other researchers have attributed the aggregation of negatively charged AuNPs in the presence of thiamine to a neutralization of the NP surface charge.72 In a fluorescence‐based approach, captopril‐modified yttrium:europium NPs (Y2O3:Eu NPs) were found to exhibit an increase in fluorescence in the presence of increasing concentrations of thiamine.73 Captopril is a small‐molecule angiotensin‐converting inhibitor, which contains a free sulfhydryl and carboxylic acid group in its structure. The NPs were not prepared for oriented labeling of captopril or characterized, but it was assumed that thiamine interacted with the negatively charged functional groups of these particles through electrostatic interactions. A limit of detection of 144 nm and linear assay range to 44 μm was reported. In another approach, the fluorescence of ‘C‐dots’ (carbon‐coated core–shell silica particles) was reduced in the presence of Cu2+ via the formation of a charge‐transfer complex, but could be restored in a linear manner with increasing concentrations of thiamine.74 Thiamine was postulated to competitively form a complex with Cu2+ through its pyrimidine N1 nitrogen, which released the metal ion from the C‐dots and restored their fluorescence. In this work, the authors reported a calculated limit of detection of 280 nm, but it appears that this was an extrapolation of the linear relationship observed from 10–50 μm thiamine rather than measurements.
The novelty of these approaches merits their mention, but significant questions remain in their potential application. In the first example, although application of the method in food matrices was reported to be successful, aggregation of AuNPs is known to occur with various substances, including salt,75 which would place in question the specificity of this approach. In the second example with captopril‐modified NPs, the mechanism of fluorescence enhancement owing to thiamine was not investigated, nor was specificity towards other constituents assessed.73 In the third case with C‐dots, specificity was demonstrated with select components expected in physiological matrices,74 though the displacement mechanism would seem subject to competitive effects by other species capable of forming complexes with Cu2+ or preferential interaction with C‐dots in real samples.
5. Methods Based on Biorecognition
The majority of thiamine detection methods in use today use standard chemical techniques. However, as thiamine is an essential vitamin for many organisms and TDP is a cofactor for many enzymes in biological systems, sensors have been developed that take advantage of this natural requirement. Reported detection approaches have relied on whole cells, enzymes, and binding proteins. Thiamine assay development using such naturally derived molecules provides an alternative to immunological recognition as specific antibodies to essential nutrients are challenging, if not impossible, to develop. Although several commercial sources of thiamine antibodies are available, these antibodies are usually listed by the manufacturers to recognize only the thiamine‐carrier protein (bovine serum albumin or ovalbumin) conjugate and not the free vitamin. Furthermore, specificity information is sometimes not provided by manufacturers, which can yield misleading results. For example, in one instance, we unsuccessfully tried to develop an ELISA by using an antibody marketed to recognize thiamine, later finding upon further inquiry with the manufacturer that this antibody was suitable only for recognition of the conjugate. In another instance, we evaluated a commercially available ELISA for thiamine and found that it detected the provided proprietary standard, but not thiamine from a commercial vendor.76 Hence, assays relying on naturally derived binding elements are critical to thiamine analyses going forward.
5.1. Microbiological Assays
Early work monitored the growth of microorganisms including bacteria, yeasts, and algae to detect thiamine levels. Some of these bioassays could achieve exceptionally low limits of detection—for example, as little as 2 ng L−1 (6 pM) thiamine could be detected by monitoring the growth of the marine algae Monochrysis lutheri.77 However, complications of these assays included the prolonged times for growth, accessibility of the given organisms, the ability of many of these organisms to respond to thiamine precursors and fragments, and growth or inhibition thereof by other constituents of the sample matrix, as outlined in Table S4. Such microbiological‐based assays are commercially available, relying on turbidity changes with the growth of Lactobacillus fermentum coated in microtiter plate wells. Although this assay can detect as little as 3 ng L−1 thiamine, it requires 48 h of incubation prior to collection of results and may not yield reliable results in all sample matrices. For example, in our hands, the assay was not successful for the analysis of thiamine in fish egg extracts (unpublished results).
Recent advances have been made, utilizing metabolic measurements or modified organisms that specifically recognize thiamine to develop new sensors without the prolonged assay times and specificity challenges of the former. In one assay, whole cells of the yeast Saccharomyces cerevisiae were immobilized onto a Teflon membrane‐covered dissolved oxygen probe.78 When thiamine was introduced, the metabolic activity of the yeast increased, resulting in a decrease in available oxygen. As Teflon is permeable to oxygen, the decrease in the amount of dissolved oxygen could be monitored amperometrically as an indirect measure of thiamine concentration. This assay yielded a linear response to thiamine concentration in the range of 5–100 nm. However, this sensor also responded (at a lower level) to vitamin B2, B6, and nicotinic acid; therefore, it is not specific to thiamine.
The nmt1, an abbreviation for “no message in thiamine”, promoter of the fission yeast Schizosaccharomyces pombe is tightly regulated by thiamine.79 The expression levels of the mRNA leading to a 39 kDa protein involved in thiamine biosynthesis are inversely proportional to the thiamine concentration, with no expression at thiamine concentrations greater than 0.5 μm. In a novel approach for thiamine detection, S. pombe was engineered to express MFα1, a peptide pheromone involved in cell‐to‐cell communication.80 Expression of MFα1 was regulated by the nmt1 promoter, which was inhibited in a concentration‐dependent manner by thiamine (Figure 4). This whole‐cell biosensor was coupled with a unique surface composed of immobilized hydrophobins, which are small, amphipathic proteins. These hydrophobins were expressed as a fusion protein with the yeast α‐factor, which allowed an anti‐α‐factor antibody to be bound to the surface of a microtiter plate. To measure the concentration of MFα1 in a sample of yeast culture supernatants, the sample was incubated with an anti‐α‐factor antibody prior to introduction to the plate where the remaining unbound antibody could be captured by the immobilized α‐hydrophobins. Thus, in this competitive format, the lower the concentration of MFα1 in the sample, the more antibody would be bound at the surface, which could then be detected by an appropriate secondary antibody with colorimetric signal amplification. As the expression of MFα1 in the yeast cells was regulated by thiamine, the quantification of the latter could indirectly be made by detection of the former. A reduction in MFα1 pheromone secretion by S. pombe could be observed following a 4 h incubation period with thiamine, and as little as 10 nm thiamine could be detected when coupled with the above described assay for MFα1. The sensitivity of this assay could be attributed not only to the enzymatic amplification of the ELISA, but also to the production of four MFα1 molecules for every precursor protein expressed upon promotion by nmt1.
Figure 4.
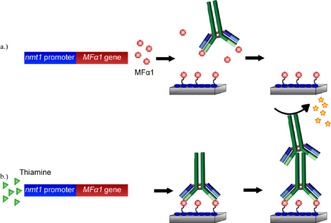
a) In the absence of thiamine, MFα1 is synthesized at high levels by Schizosaccharomyces pombe. In the subsequent ELISA, the MFα1 present in the culture supernatant competes with surface immobilized α‐conjugate for the available anti‐α‐factor antibody. Upon washing the plate, no anti‐α‐factor antibody remains bound and, hence, is not detected by the secondary antibody/enzyme conjugate. b) In the presence of thiamine, the nmt1 promoter is repressed and synthesis of MFα1 is prevented. The anti‐α‐factor antibody can bind maximally and subsequently be detected by a secondary antibody/enzyme conjugate and visible substrate. Drawn from concepts described in Ref. 80.
5.2. Biorecognition by Enzymes
As an assessment of thiamine deficiency in clinical samples, measurement of the activity of enzymes requiring TDP as a cofactor is commonly carried out. One such measure is the erythrocyte transketolase activity (ETKA) assay that measures the production of sedoheptulose‐7‐phosphate per minute per liter of blood.81 In cases of thiamine deficiency, the activity of transketolase increases when exogenous TDP is introduced. The percentage increase in enzyme activity is subsequently measured and is known as the thiamine pyrophosphate effect (TPPE). In patients without thiamine deficiency, the TPPE is 0–15 %; with mild deficiency 16–25 %; and with severe deficiency >25 %.82 Despite the widespread use of the transketolase assay, many documented drawbacks to this assay have been revealed to date.83 The weakest aspect of this assay is that it does not yield thiamine concentration information directly, but rather an indirect measure via the transketolase activity. This introduces the possibility of factors other than exogenous TDP that may influence ETKA and the TPPE, such as magnesium, which is also a cofactor for transketolase. Additionally, expression of the apoenzyme may be suppressed, the variant of the apoenzyme produced in certain individuals may have lower affinity for TDP, or the conversion of thiamine to TDP may be impaired; all of these would falsely mimic a thiamine deficiency by the transketolase assay.81, 83 Other issues with the transketolase assay include poor inter‐batch reproducibility and sample stability, owing to fast inactivation of transketolase84 and difficulty in standardization, resulting in upper limit reference values ranging from 15.5 to 40 %.85
Pyruvate and lactate assays have also been able to provide information on thiamine status, owing to TDP′s role as a cofactor for pyruvate dehydrogenase, which participates in the conversion of pyruvate to acetyl‐CoA, a starting substrate for the Krebs cycle.86 Without the presence of TDP, pyruvate remains extremely stable to decarboxylation and may reversibly convert to lactate by lactate dehydrogenase (LDH). To test the effects of thiamine supplementation, LDH is added to whole blood to convert pyruvate to lactate and pyruvate is subsequently measured by the stoichiometric decrease in NADH absorbance at 340 nm. Likewise, lactate is measured by the increase in NADH absorbance at 340 nm after addition of LDH converts lactate to pyruvate. As TDP allows pyruvate to undergo decarboxylation rather than becoming available for conversion to lactate, the extent of the decrease in pyruvate and lactate following thiamine supplementation serves as an indicator of thiamine status.
Aside from clinical samples, there has been little work using TDP‐dependent enzymes for biosensor development. However, one biosensor approach utilizing pyruvate oxidase (POX) has been reported.87 POX uses pyruvate, oxygen, and phosphate to yield acetylphosphate, carbon dioxide, and hydrogen peroxide in the POX‐mediated formation of acetylphosphate [Eq. (1)]:
| (1) |
In an approach based on a similar premise to that described above using S. cerevisiae, a dissolved oxygen probe was covered with a Teflon membrane onto which POX was subsequently immobilized. In the presence of Flavin adenine dinucleotide (FAD) and Mg2+, with controlled addition of thiamine, the POX activity reportedly increased, yielding a greater consumption of dissolved oxygen. This assay yielded a linear response to thiamine concentration in the range of 0.025 to 0.5 μm. Although thiamine diphosphate is the typical cofactor for pyruvate oxidase, this sensor apparently responded to the non‐phosphorylated molecule. The differences in enzyme activity resulting from the phosphorylated or parent forms of thiamine would be of specificity interest. Also, a baseline level of enzyme activity in the absence of exogenous thiamine was observed. Although not suggested within, this system could instead be coupled with horseradish peroxidase and appropriate substrates, which would allow a directly proportional signal as a function of the hydrogen peroxide produced through colorimetric or fluorescence measurements.
5.3. Biorecognition by Other Entities
As thiamine is essential for all life forms, many organisms have mechanisms in place to allow uptake of thiamine from their environment, to sequester thiamine from intake, or, in organisms capable of its synthesis, to control the endogenous synthesis of thiamine from its precursors. Riboswitches are RNA molecules that control gene expression upon binding by small‐molecule targets. These molecules consist of an aptamer component that provides specific recognition of the target and a regulatory component, which, upon target binding, can up‐ or down‐regulate gene expression. In the presence of thiamine pyrophosphate (TPP), the TPP riboswitch serves to turn off genes, resulting in decreased expression of proteins involved in TPP biosynthesis. The kinetics of this are concentration dependent with high levels of TPP rapidly shutting down biosynthetic protein production and lower levels doing so less immediately by tailoring the protein expression to the available TPP.88 Its function in plants, bacteria, and fungi is to regulate genes responsible for uptake or synthesis of thiamine.89 The aptamer component of the riboswitch is an 84‐nucleotide sequence (Figure 5 a) with a reported K D for TPP of 850 pM.88, 90 This structure does not recognize the thiazole portion, but recognizes the 4‐amino‐5‐hydroxymethyl‐2‐methylpyrimidine (HMP) and the pyrophosphate portions of TPP in separate binding pockets.89 By using the aptamer component of the TPP riboswitch, Lau et al. designed a sensor that resulted in an increase in fluorescence upon TPP binding by the displacement of a quencher conjugated to a complementary DNA strand.88 The aptamer was modified on the 5′ terminus with a quencher DNA (QDNA) binding site, a short spacer sequence, and fluorophore DNA (FDNA) binding site (Figure 5 b). In the absence of TPP, a 16‐nucleotide QDNA sequence and 20‐nucleotide FDNA sequence could hybridize to their respective sites. In the presence of TPP, the aptamer portion of the sequence would recognize its target with high affinity, while concomitantly displacing the quencher sequence (Figure 5 c). The release of the quencher sequence removed the close proximity previously held by the fluorophore and the quencher and resulted in an increase in fluorescence. The range of this assay was linear between 1 μm and 1 mm TPP, with a signal enhancement being four times the background at the maximum concentration. Owing to the concentration dependence of the riboswitch in nature, a time dependence is associated with attaining the maximum signal in the sensor, for example, requiring 5 min at 1 mm, but 120 min for 10 μm TPP. A limit of detection of 10 nm was noted and, though not specified, was presumably attainable if the assay time was extended. This assay yielded high specificity towards TPP versus TMP, thiamine, and oxythiamine, demonstrating the excellent binding fidelity of naturally derived aptamers. One challenge in the broader utility of this approach, however, is the 1:1 relationship between the aptamer binding event and the fluorophore signal, which limits its potential sensitivity.
Figure 5.
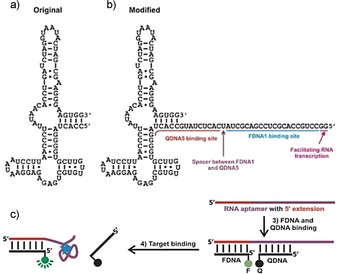
a) The TPP‐binding aptamer portion of the TPP‐dependent ribozyme, b) TPP‐binding aptamer modified with fluorophore‐modified DNA (FDNA) and quencher‐modified DNA (QDNA) binding sites at the 5′ terminus, and c) QDNA and FDNA hybridize to their respective binding sites within the aptamer sequence. In the absence of TPP, the fluorophore and quencher are in close proximity and a low fluorescence signal results. In the presence of TPP, the QDNA is displaced and the fluorescence signal of the FDNA is restored. Reprinted from Ref. 88 with permission. Copyright (2010) Wiley‐VCH.
Artificial constructs have been able to take advantage of nature's target selectivity combined with synthetic elements to mediate gene regulation. So‐called ‘aptazymes’ have been developed, which consist of an aptamer binding domain to provide specific recognition and a ribozyme domain that undergoes self‐cleavage upon target binding. One such artificial ribozyme has been developed by using TPP recognition by the aptamer domain of its natural riboswitch.91 By utilizing this artificial construct in conjunction with DNAse I and graphene oxide (GO), Li et al. developed a novel approach to the detection of TPP.92 DNAse I cleaves phosphodiester bonds of single‐ and double‐stranded DNA. The TPP hammerhead aptazyme cleaves itself in the absence of TPP, but remains intact in its presence. GO has the unique property of being selective for adsorbing ssDNA, whereas dsDNA or DNA–RNA hybrids are not retained. GO additionally has a fluorescence quenching effect on fluorophores attached to adsorbed ssDNA along with a protective effect on the enzymatic cleavage of ssDNA. These properties have made GO attractive for sensing applications in which the fluorescence from the fluorophore linked to the adsorbed probe is restored upon target hybridization forming dsDNA.93 In this assay for TDP (Figure 6), a ssDNA probe labeled with a fluorophore was adsorbed onto GO, where the fluorescence signal undergoes complete quenching.92
Figure 6.
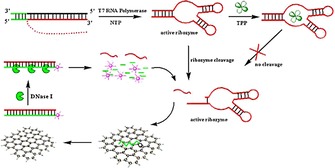
The TPP‐dependent ribozyme undergoes self‐cleavage releasing a small segment of RNA. This RNA can hybridize to fluorophore‐labeled complementary DNA adsorbed onto graphene oxide. The hybridized DNA–RNA complex is then released from the GO, where the DNA portion is degraded by DNAse I. The released RNA can then participate in another cycle of hybridization, DNA degradation, and fluorophore release yielding an amplification event. In the presence of TPP, its binding to the ribozyme inactivates the ribozyme's self‐cleavage and, thus, a reduction in signal results. Reprinted from Ref. 92 with permission from. Copyright (2013) Elsevier.
The aptazyme cleaves a segment of the RNA from the TPP riboswitch in the absence of TPP, whereas this cleavage is inhibited in the presence of TPP in a concentration‐dependent manner. The released segment of RNA can then hybridize to the fluorophore‐labeled complementary ssDNA probe, which causes its release from the graphene oxide and subsequent rise of the fluorescence signal. Upon release from the GO, the DNA component of the DNA–RNA hybrid would be degraded by DNAse I, yielding the release of the fluorophore and the original RNA segment. This original RNA segment could then participate in another cycle of hybridization with GO‐adsorbed ssDNA, leading to signal enhancement. Through the inclusion of DNAse I, this re‐hybridization of RNA and release of ssDNA allows for amplification of the original TPP–aptazyme 1:1 binding event. The range of this assay was reported to be between 0.5 and 100 μm TDP, with little cross‐reactivity towards thiamine or TMP. The challenge with either of these sensors is the RNA‐based platform. RNA is readily subject to degradation by RNA nucleases in the environment, thus limiting the utility for thiamine detection in biological sample matrices. Substituting DNA bases for RNA nucleotides in synthetic sequences has not yielded functional aptamers (unpublished results); thus, the development of a nuclease‐resistant thiamine aptamer is one of our current interests.
Thiamine binding proteins are found in various life forms, including plants, bacteria, and yeasts, which allow storage and subsequent utilization of the vitamin by an organism. Taking advantage of this natural requirement, a competitive radioassay for thiamine by using Sepharose‐immobilized thiamine binding proteins from Buckwheat seeds was developed.59 In this assay, thiazole‐2–14C thiamine was used as a competitor for thiamine in food samples for the immobilized protein. An assay range of 1–10 μm was achieved and could be used for the analysis of thiamine alone as thiamine phosphates are not recognized by plant binding proteins.
Periplasmic binding proteins are produced in high quantities by gram‐negative bacteria such as Escherichia coli and Salmonella typhimurium with mediate uptake of small molecules including sugars, amino acids, inorganic ions, and vitamins.94 These proteins undergo a significant conformational shift upon target binding, which opens up the possibility for homogeneous sensor platforms. Hanes et al. developed a sensor that took advantage of the conformational change of the E. coli thiamine periplasmic binding protein (TBP) to change the local environment of an environmentally sensitive fluorophore upon target binding.95 In this sensor, the site‐specific modification of the protein via a S62C mutation allowed thiol‐modification with a coumarin or pyrene derivative (Figure 7).
Figure 7.
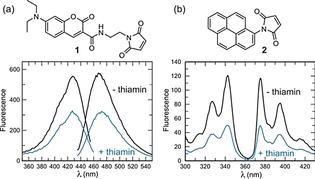
Measurement of the fluorescence change with MDCC (1) and pyrene (2) labeled TbpA. a) Fluorescence excitation and emission scans of 200 nm MDCC‐TbpA. Upon addition of a saturating concentration of thiamin (2 μm), the fluorescence is reduced 47 % at 467 nm. b) Fluorescence excitation and emission scans of 200 nm pyrene‐TbpA. Upon addition of thiamin, the fluorescence is reduced 57 % at 375 nm. Reproduced from Ref. 95 with permission. Copyright (2011) Royal Society of Chemistry.
In the presence of thiamine, the fluorescence from either derivative was decreased. A similar approach that took advantage of the conformational change of E. coli TBP, but relying on fluorescence resonance energy transfer (FRET) between fluorescent proteins eCFP and Venus, has been reported.96 FRET changes of up to 25 % could be observed upon incubation with 100 μm thiamine in vitro and the system could also be utilized as an in vitro measurement in E. coli in vivo.
Rather than focusing on the conformational shift, we recently developed a high‐throughput assay by using the E. coli TBP for specific recognition of thiamine, much in the same way an antibody would be used in an ELISA (Figure 8).76
Figure 8.
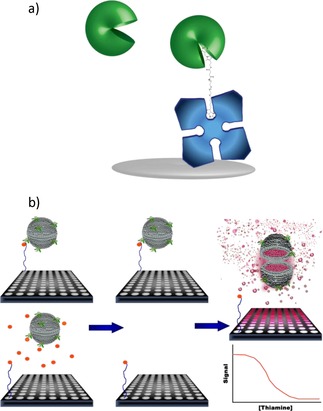
Competitive assay for thiamine using fluorescent‐dye‐encapsulating liposomes for signal amplification and the thiamine binding protein for specific biorecognition. a) A thiamine derivative was designed with a long PEG spacer between thiamine monosuccinate and biotin to accommodate the binding sites of both TBP used for thiamine recognition and tetrameric streptavidin used for immobilization. The TBP undergoes a significant conformational shift upon binding. b) Competitive assay with thiamine monosuccinate‐PEG‐biotin immobilized through streptavidin in microtiter plates and detected via TBP conjugated to the lipid bilayer of sulforhodamine B encapsulating liposomes (left). In the absence of free thiamine in the sample (top), TBP liposomes bind maximally to the immobilized thiamine derivative; in the presence of free thiamine in the sample (bottom), owing to competition, fewer TBP‐liposomes can bind to the immobilized thiamine derivative. After incubation with the sample, unbound materials are removed (middle) and liposomes remaining bound are lysed to release dye yielding a signal inversely proportional to thiamine concentration (right). Reprinted with permission from Ref. 76. Copyright (2016) American Chemical Society.
Here, the TBP served in lieu of an antibody and fluorescent dye‐encapsulating liposomes provided signal amplification. In this competitive assay format, a PEGylated thiamine derivative was immobilized onto the surface of a microtiter plate (Figure 8 a) and served as a competitor to thiamine potentially in a sample for the available TBP‐conjugated liposomes (Figure 8 b). The liposomes entrap hundreds of thousands to millions of fluorophore molecules, hence providing a significant amplification of any binding event. A signal inversely proportional to the thiamine concentration resulted, with a limit of detection of 0.5 nm. This method demonstrated excellent specificity towards thiamine and its phosphorylated derivatives, with no cross‐reactivity towards constitutive fragments of thiamine or most thiamine analogues. Only thiamine impurity E, which has a thione group between the sulfur and nitrogen atoms of the thiazole ring, yielded a response, but at concentrations 1000‐fold higher than thiamine. This compound is an impurity formed during thiamine manufacturing and would not be found at such high concentrations in nature. The liposome‐based platform provided the needed amplification, whereas the TBP provided excellent specificity in this assay.
6. Utility of Thiamine in the Detection of Other Species
Although not assays for the thiamine molecule, thiamine has been recently used as a probe itself for assaying other analytes. Examples include the detection of prion proteins,97 uric acid,98 hypochlorite,99 and thiaminase activity.100 It has been used as a fluorimetric substrate in conjunction with co‐immobilized uricase and horseradish peroxidase in an assay for uric acid98 and, in an assay for hypochlorite, alkaline ferrocyanide could be reduced by the analyte to ferricyanide, which could serve to oxidize thiamine.99 Thiamine radiolabeled on its thiazole ring with 13C is also used in an assay for thiaminase activity in biological samples.100 This assay relies on the increased hydrophobicity of the enzymatically cleaved 13C‐4‐methyl‐5‐(2‐hydroxyethyl)thiazole and ability to partition this product into an organic solvent.
7. Summary and Outlook
The profound importance of thiamine to the health of all organisms and the shortcomings of previous thiamine analysis methods have together fueled an ongoing interest in improving methodologies for monitoring thiamine in a variety of matrices. Numerous colorimetric, fluorescence, electrochemical, and biological‐based approaches have been developed, each with their own inherent benefits and caveats (Table 1). Although colorimetric approaches were once widely used, owing to the inherently simple detection principle, they have largely fallen out of favor in lieu of more sensitive fluorometric methods, owing to their high limits of detection (μm) and risk of interferences, especially when biological samples are considered. As a result, the most commonly employed methodologies have relied on fluorescence detection following chromatographic separation. These established methodologies offer the benefit of sensitive quantification of thiamine in multi‐component mixtures and speciation of thiamine phosphates. Advances in HPLC separations and improvements in detection capabilities offer the ability to reach impressive pM levels of detection suited for well‐equipped laboratory environments.
Table 1.
Summary of advantages and disadvantages of each strategy using methods with the lowest reported limit of detection.
| Technique | Methodology | Limit of Detection | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| Colorimetric | Ionic association of thiamine with micellar triphenylmethane dyes. | 66 nm | ‐ Spectrophotometric measurements are easy to carry out with widely available equipment. | ‐ Interferences with common constituents, including vitamin B2, vitamin B6, and vitamin C. | 39 |
| UV | HPLC using a C18 column and gradient elution. | 74 nm | ‐ HPLC with UV detectors are widely available. ‐ No derivatization procedure needed. ‐ Ability to simultaneously quantify other compounds. | ‐ Level of expertise required. ‐ Expense and maintenance of instrumentation. | 66 |
| Fluorescence | HPLC using a C18 column and gradient elution. | 1 nm | ‐ Quantification of different thiamine phosphate forms. | ‐ Derivatization to thiochrome required. ‐ HPLC with fluorescence detectors less commonly available. | 67 |
| Chemiluminescence | Flow‐injection technique with detection based on suppression of luminol/KIO4 chemiluminescence by thiamine. | 1 nm | ‐ Rapid analyses. | ‐ Mechanism of chemiluminescence suppression not elucidated; thus, specificity for solely thiamine not proven. ‐ No further development of this technique since 2002. | 101 |
| Electrochemical | Fast Fourier transform square‐wave voltammetry. | 0.56 nm | ‐ Low limit of detection. | ‐ Specificity not assessed. ‐ Reported limit of detection markedly lower than similar techniques and further validation would be beneficial. | 102 |
| Mass spectrometry | UPLC‐MS/MS using a C18 column and gradient elution. | 30 pM | ‐ Limit of detection applicable to environmental water samples. ‐ Identity confirmation. | ‐ Expertise required. ‐ Expense and maintenance of instrumentation. | 68 |
| NP‐based approaches | Fluorescence enhancement of 4‐amino‐6‐hydroxy‐2‐mercaptopyrimidine gold NPs. | 6 fM | ‐ Reported limit of detection markedly lower than all other methods. ‐ Reported specificity towards thiamine demonstrated versus 10,000‐fold higher concentrations of possible interferences including riboflavin, vitamin B12, and ascorbic acid. | ‐ Mechanism of association with nanoparticles (for example, via electrostatic or coordination complexes) is not specific to only thiamine. ‐ Mechanism of fluorescence enhancement not investigated. ‐ Reported limit of detection markedly lower than similar techniques and further validation would be beneficial. | 72 |
| Whole‐cell biosensors | Growth of the marine algae Monochrysis lutheri. | 6 pM | ‐ Limit of detection applicable to environmental water samples. | ‐ Non‐specific growth may result from thiamine fragments or other molecules in biological matrices. ‐ Prolonged time periods until results. ‐ Need to maintain viable microorganisms. | 77 |
| Enzymes | Increase in enzymatic activity of pyruvate oxidase in the presence of thiamine yielding consumption of oxygen. | 25 nm | ‐ Accessibility of equipment for dissolved oxygen measurements. | ‐ Maintenance of immobilized enzyme activity/long‐term stability. ‐ Basal level of enzyme activity in the absence of exogenous thiamine. | 87 |
| Riboswitches | FRET relying on conformational change of RNA aptamer portion upon binding to TPP. | 10 nm | ‐ High specificity towards TPP versus thiamine, TMP, and oxythiamine. | ‐ Strong potential for RNA degradation limiting utility with real sample matrices. | 88 |
| Periplasmic binding proteins | Competitive, heterogeneous assay using fluorescent‐dye encapsulating liposomes conjugated to the thiamine periplasmic binding protein from E. coli. | 0.5 nm | ‐ High‐throughput microtiter plate‐based platform. ‐ Exquisite specificity towards thiamine versus fragments and analogues. | ‐ Components not commercially available. ‐ Validation in environmental matrices required. | 76 |
Looking ahead, simpler high‐performing methodologies now seem attainable in the near future. We consider most promising the biorecognition‐based approaches that have been developed in recent years. Although RNA‐based aptamer approaches as an affinity recognition system may be a dead‐end strategy, owing to the rapid degradation of RNA when in contact with environmental or biological samples, such approaches may be viable with modified RNA or DNA nucleotides. We are currently investigating the possibility of DNA‐based aptamers for thiamine, which would open up a multitude of field‐appropriate biosensors while affording custom specificity through counter‐SELEX measures. The described catalytic enzyme‐based approaches could be further coupled with more sensitive and commonly used enzymatic amplification strategies such as horseradish peroxidase and result in simple colorimetric or fluorescence‐based high‐throughput strategies. Most interesting and promising is a periplasmic binding approach, as it can be used similar to antibodies, which shows immense sensitivity and specificity toward thiamine, and has also shown potential in real‐world samples. Still, as decades of analytical work with thiamine has demonstrated that, independent of any detection strategy, significant emphasis must still be placed on sample preparation. The reproducible release of thiamine from sample matrices, as well as its stability, requires careful consideration. This release is usually carried out under harsh chemical conditions that must be mediated prior to downstream analysis by using biological entities. Methods going forward must encompass not only a sensitive and specific approach to thiamine detection, but one that can withstand the challenges of sample analysis by using complex environmental and biological matrices.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Katie Edwards is a Scientist and Lecturer in biosensors and bioanalytical techniques at Cornell University (USA). She earned her M.S. and Ph.D. in toxicology from Cornell University and B.S. in Chemistry from Clarkson University (USA). Her work has focused on the advancement of liposomes as analytical tools, utilizing her background in organic synthesis, lipid formulation, and novel biorecognition strategies to develop high‐throughput laboratory‐based methods and point‐of‐care diagnostics for analytes of environmental, national security, and clinical interest.

Biographical Information
Cliff Kraft is a Professor of Fisheries and Aquatic Sciences in the Department of Natural Resources at Cornell University (USA). He studies ecosystem interactions that influence human use and impacts upon organisms living in lakes and rivers, including a focus on understanding how ecological competition for thiamine (vitamin B1) leads to mortality in highly valued fisheries and other organisms.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funding during the preparation of this manuscript was provided in part by the Great Lakes Fishery Trust project #2013.1299 and Multistate Federal Formula Grant project #2012‐13‐132.
K. A. Edwards, N. Tu-Maung, K. Cheng, B. Wang, A. J. Baeumner, C. E. Kraft, ChemistryOpen 2017, 6, 178.
Contributor Information
Dr. Katie A. Edwards, Email: kae24@cornell.edu.
Dr. Clifford E. Kraft, Email: cek7@cornell.edu
References
- 1. Kraft C. E., Angert E., Quarterly Review of Biology, 2017. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Feinberg J. F., Am. Fam. Physician 1980, 22, 129–133; [PubMed] [Google Scholar]
- 2b. Davis R. E., Icke G. C., Adv. Clin. Chem. 1983, 23, 93–140. [DOI] [PubMed] [Google Scholar]
- 3. Page G. L., Laight D., Cummings M. H., Int. J. Clin. Pract. 2011, 65, 684–690. [DOI] [PubMed] [Google Scholar]
- 4. Zhang G., Ding H., Chen H., Ye X., Li H., Lin X., Ke Z., J. Nutr. 2013, 143, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu′o′ng K. V., Nguyen L. T., Am. J. Alzheimers Dis. Other Demen. 2011, 26, 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gibson G. E., Hirsch J. A., Fonzetti P., Jordan B. D., Cirio R. T., Elder J., Ann. NY Acad. Sci. 2016, 1367, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carvalho P. S., Tillitt D. E., Zajicek J. L., Claunch R. A., Honeyfield D. C., Fitzsimons J. D., Brown S. B., J. Aquat. Anim. Health 2009, 21, 315–325; [DOI] [PubMed] [Google Scholar]; Keinanen M., Uddstrom A., Mikkonen J., Casini M., Ponni J., Myllyla T., Aro E., Vuorinen P. J., ICES J. Mar. Sci. 2012, 69, 516–528; [Google Scholar]; Ketola H. G., Johnson J. H., Rinchard J., Verdoliva F. J., Penney M. E., Greulich A. W., Lloyd R. C., N Am J Fish Manage 2009, 29, 895–902. [Google Scholar]
- 8. Sepulveda M. S., Wiebe J. J., Honeyfield D. C., Rauschenberger H. R., Hinterkopf J. P., Johnson W. E., Gross T. S., J. Wildl. Dis. 2004, 40, 782–786. [DOI] [PubMed] [Google Scholar]
- 9. Balk L., Hagerroth P. A., Akerman G., Hanson M., Tjarnlund U., Hansson T., Hallgrimsson G. T., Zebuhr Y., Broman D., Morner T., Sundberg H., Proc. Natl. Acad. Sci. USA 2009, 106, 12001–12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.M. Levy in Overview of Polioencephalomalacia, Vol.
- 11. Tomasulo P. A., Kater R. M., Iber F. L., Am. J. Clin. Nutr. 1968, 21, 1341–1344. [DOI] [PubMed] [Google Scholar]
- 12. Ahoua L., Etienne W., Fermon F., Godain G., Brown V., Kadjo K., Bouaffou K., Legros D., Guerin P. J., Food Nutr. Bull. Suppl. 2007, 28, 283–290. [DOI] [PubMed] [Google Scholar]
- 13. Vimokesant S. L., Hilker D. M., Nakornchai S., Rungruangsak K., Dhanamitta S., Am. J. Clin. Nutr. 1975, 28, 1458–1463. [DOI] [PubMed] [Google Scholar]
- 14. Lynch P. L., Young I. S., J. Chromatogr. A 2000, 881, 267–284. [DOI] [PubMed] [Google Scholar]
- 15. Voigt M. N., Eitenmiller R. R., J. Food Prot. 1978, 41, 730–738. [DOI] [PubMed] [Google Scholar]
- 16. Aquilina G., Bories G., Chesson A., Cocconcelli P., Knecht J., Dierick N., Gralak M., Gropp J., EFSA J. 2011, 9, 2413 [17 pp.]. [Google Scholar]
- 17. Macek T. J., Feller B. A., Hanus E. J., J. Am. Pharm. Assoc. 1950, 39, 365–369. [DOI] [PubMed] [Google Scholar]
- 18. Perez-Caballero G., Perez-Arevalo J. F., Morales-Hipolito E. A., Carbajal-Arenas M. E., Rojas-Hernandez A., J. Mex. Chem. Soc. 2011, 55, 126–131. [Google Scholar]
- 19. Herr D. S., Ind. Eng. Chem. 1945, 37, 631–634. [Google Scholar]
- 20. Gratacós-Cubarsi M., Sarraga C., Clariana M., Regueiro J. A., Castellari M., Meat Sci. 2011, 87, 234–238. [DOI] [PubMed] [Google Scholar]
- 21. Brown S. B., Honeyfield D. C., Vandenbyllaardt L., Am. Fish. Soc. Symp. 1998, 21, 73–81. [Google Scholar]
- 22. Grekas N., Calokerinos A. C., Talanta 1990, 37, 1043–1048. [DOI] [PubMed] [Google Scholar]
- 23. Zhang C., Zhou G., Zhang Z., Aizawa M., Anal. Chim. Acta 1999, 394, 165–170. [Google Scholar]
- 24. Zajicek J. L., Tillitt D. E., Brown S. B., Brown L. R., Honeyfield D. C., Fitzsimons J. D., J. Aquat. Anim. Health 2005, 17, 95–105. [Google Scholar]
- 25. Echols R. E., Miller R. H., Foster W., J. Dairy Sci. 1986, 69, 1246–1249. [DOI] [PubMed] [Google Scholar]
- 26. Kennedy C. A., McCleary B. V., Analyst 1981, 106, 344–351. [DOI] [PubMed] [Google Scholar]
- 27. Okbamichael M., Sañudo-Wilhelmy S. A., Limnol. Oceanogr. 2005, 3, 241–246. [Google Scholar]
- 28. Lu J., Frank E. L., Clin. Chem. 2008, 54, 901–906. [DOI] [PubMed] [Google Scholar]
- 29. Körner R. W., Vierzig A., Roth B., Muller C., J. Chromatogr. B 2009, 877, 1882–1886. [DOI] [PubMed] [Google Scholar]
- 30. Batifoulier F., Verny M. A., Besson C., Demigne C., Remesy C., J. Chromatogr. B 2005, 816, 67–72. [DOI] [PubMed] [Google Scholar]
- 31. Lalić J., Denić M., Sunarić S., Kocić G., Trutić N., Mitić S., Jovanović T., CyTA–Journal of Food 2014, 12, 203–209. [Google Scholar]
- 32. Ang C. Y. W., Moseley F. A., J. Ag. Food Chem. 1980, 28, 483–486. [DOI] [PubMed] [Google Scholar]
- 33. Barna É., Dworschák E., J. Chromatogr. A 1994, 668, 359–363. [DOI] [PubMed] [Google Scholar]
- 34. Koski P., Pakarinen M., Nakari T., Soivio A., Hartikainen K., Dis. Aquat. Organ. 1999, 37, 209–220. [DOI] [PubMed] [Google Scholar]
- 35. Udenfriend S. in Vitamins coenzymes and their metabolites, Vol. 2 (Ed. S. Udenfriend), Academic Press, New York, NY, 1969, pp. 291–334. [Google Scholar]
- 36. Zhang F., Masania J., Anwar A., Xue M., Zehnder D., Kanji H., Rabbani N., Thornalley P. J., Kidney Int. 2016, 90, 396–403. [DOI] [PubMed] [Google Scholar]
- 37. Buchholz M., Drotleff A. M., Ternes W., J. Cereal Sci. 2012, 56, 109–114. [Google Scholar]
- 38. Al-Ahmary K. M., Eur. J. Chem. 2014, 5, 81–84. [Google Scholar]
- 39. Liu S., Zhang Z., Liu Q., Luo H., Zheng W., J. Pharm. Biomed. Anal. 2002, 30, 685–694. [DOI] [PubMed] [Google Scholar]
- 40. Szpikowska-Sroka B., J. Anal. Chem. 2013, 68, 218. [Google Scholar]
- 41. Haller A., Altman R. B., Souliere M. F., Blanchard S. C., Micura R., Proc. Natl. Acad. Sci. USA 2013, 110, 4188–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martinez-Lozano C., Perez-Ruiz T., Tomas V., Abellan C., Analyst 1990, 115, 217–220. [Google Scholar]
- 43. Ratanaubolchai K., Jareonsanti J., Panijpan B., Clin. Chem. 1981, 27, 1777–1778. [PubMed] [Google Scholar]
- 44. Ratanaubolchai K., Panijpan B., Clin. Chem. 1979, 25, 1670–1671. [PubMed] [Google Scholar]
- 45. Wyatt D. T., Lee M., Hillman R. E., Clin. Chem. 1989, 35, 2173–2178. [PubMed] [Google Scholar]
- 46. Wyatt D. T., Hillman R. E. in [11] Cyanogen bromide-based assay of thiamin, Vol. Volume 279 Academic Press, 1997, pp. 91–97. [DOI] [PubMed] [Google Scholar]
- 47.
- 47a. Bessey O. A., Lowry O., Davis E., J. Biol. Chem. 1952, 195, 453–458; [PubMed] [Google Scholar]
- 47b. Burch H. B., Bessey O. A., Love R. H., Lowry O. H., J. Biol. Chem. 1952, 198, 477–490; [PubMed] [Google Scholar]
- 47c. Roth H., Biochem. Ztschr. 1938, 897, 52–55; [Google Scholar]
- 47d. Westenbrink H., Goudsmit J., Nature 1938, 142, 150–151; [Google Scholar]
- 47e. Widenbauer F., Heckler F., Z. Kinderheilkunde 1939, 60, 683–690. [Google Scholar]
- 48. Hennessy D. J., Ind. Eng. Chem. 1941, 13, 216–218. [Google Scholar]
- 49. Hennessy D. J., Cerecedo L. R., J. Am. Chem. Soc. 1939, 61, 179–183. [Google Scholar]
- 50. Westenbrink H. G. K., Goudsmit J., Nature 1938, 142, 150–151. [Google Scholar]
- 51. Mickelsen O., Condiff H., Keys A., J. Biol. Chem. 1945, 160, 361–370. [Google Scholar]
- 52. Pérez-Ruiz T., Martinez-Lozano C., Sanz A., Guillen A., J. Pharm. Biomed. Anal. 2004, 34, 551–557. [DOI] [PubMed] [Google Scholar]
- 53. Mancinelli R., Ceccanti M., Guiducci M. S., Sasso G. F., Sebastiani G., Attilia M. L., Allen J. P., J. Chromatogr. B 2003, 789, 355–363. [DOI] [PubMed] [Google Scholar]
- 54. Gauch R., Leuenberger U., Muller U., Z. Lebensm. Unters. Forsch. 1992, 195, 312–315. [DOI] [PubMed] [Google Scholar]
- 55. Bubeshko N. N., Stsiapura V. I., Stepuro I. I., J. Appl. Spectrosc. 2011, 78, 337–343; [Google Scholar]; Penttinen H. K., Acta Chem. Scand. 1976, 30, 659–663. [Google Scholar]
- 56. Liddicoat C., Hucker B., Liang H., Vriesekoop F., Food Chem. 2015, 177, 325–329. [DOI] [PubMed] [Google Scholar]
- 57. Panijpan B., Vilartsakdanon P., J. Sci. Soc. Thailand 1977, 3, 131–134. [Google Scholar]
- 58. Ratanaubolchai K., Panijpan B., Int. J. Vitam. Nutr. Res. 1980, 50, 3–9. [PubMed] [Google Scholar]
- 59. Kozik A., J Biochem Biophys Methods 1994, 28, 147–154. [DOI] [PubMed] [Google Scholar]
- 60.
- 60a. Soliman A. G., J. Assoc. Off Anal. Chem. 1981, 64, 616–622; [PubMed] [Google Scholar]
- 60b. Urban F., Goldman M. L., J. Biol. Chem. 1944, 152, 329–337. [Google Scholar]
- 61. Hart J. P., Norman M. D., Tsang S., Analyst 1995, 120, 1059–1064. [Google Scholar]
- 62. Kusube K., Abe K., Hiroshima O., Ishiguro Y., Ishikawa S., Hoshida H., Chem. Pharm. Bull. 1983, 31, 3589–3594. [DOI] [PubMed] [Google Scholar]
- 63. Ciszewski A., Studnickova M., Fischer O., Bioelectrochem. Bioenerg. 1984, 13, 25–38. [Google Scholar]
- 64. Brahman P. K., Dar R. A., Pitre K. S., Sens. Actuators B Chem. 2013, 177, 807–812. [Google Scholar]
- 65. Karatapanis A. E., Fiamegos Y. C., Stalikas C. D., Chromatographia 2010, 71, 751–759. [Google Scholar]
- 66.S. Joseph in Analysis of water-soluble vitamins from multivitamin tablets for nutrition labeling, Vol. 2016 Agilent Technologies, Inc., 2011.
- 67. Basiri B., Sutton J. M., Hanberry B. S., Zastre J. A., Bartlett M. G., Biomed. Chromatogr. 2016, 30, 35–41. [DOI] [PubMed] [Google Scholar]
- 68. Hampel D., York E. R., Allen L. H., J. Chromatogr. B 2012, 903, 7–13. [DOI] [PubMed] [Google Scholar]
- 69. Ihara H., Matsumoto T., Shino Y., Hashizume N., J. Clin. Lab. Anal. 2005, 19, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bettendorff L., Michel-Cahay C., Grandfils C., De Rycker C., Schoffeniels E., J. Neurochem. 1987, 49, 495–502. [DOI] [PubMed] [Google Scholar]
- 71. Molina-Delgado M. Á., Aguilar-Caballos M. P., Gómez-Hens A., Microchim. Acta 2016, 183, 1385–1390. [Google Scholar]
- 72. Shankar S., John S. A., RSC Adv. 2015, 5, 49920–49925. [Google Scholar]
- 73. Bayandori Moghaddam A., Gudarzy F., Ganjkhanlou Y., J. Fluoresc. 2014, 24, 1025–1030. [DOI] [PubMed] [Google Scholar]
- 74. Purbia R., Paria S., Biosens. Bioelectron. 2016, 79, 467–475. [DOI] [PubMed] [Google Scholar]
- 75. Sun L., Zhang Z., Wang S., Zhang J., Li H., Ren L., Weng J., Zhang Q., Nanoscale Res. Lett. 2009, 4, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Edwards K. A., Seog W. J., Han L., Feder S., Kraft C. E., Baeumner A. J., Anal. Chem. 2016, 88, 8248–8256. [DOI] [PubMed] [Google Scholar]
- 77. Carlucci A. F., Silbernagel S. B., Can. J. Microbiol. 1966, 12, 1079–1089. [DOI] [PubMed] [Google Scholar]
- 78. Akyilmaz E., Yaşa İ., Dinçkaya E., Anal. Biochem. 2006, 354, 78–84. [DOI] [PubMed] [Google Scholar]
- 79. Maundrell K., J. Biol. Chem. 1990, 265, 10857–10864. [PubMed] [Google Scholar]
- 80. Hennig S., Rodel G., Ostermann K., Sensors 2016, 16, 602 [19 pp.]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kjosen B., Seim S. H., Am. J. Clin. Nutr. 1977, 30, 1591–1596. [DOI] [PubMed] [Google Scholar]
- 82. Ahmed M., Azizi-Namini P., Yan A. T., Keith M., Heart Failure Rev. 2015, 20, 1–11. [DOI] [PubMed] [Google Scholar]
- 83. Baines M., Davies G., Ann. Clin. Biochem. 1988, 25, 698–705. [DOI] [PubMed] [Google Scholar]
- 84. Puxty J. A., Haskew A. E., Ratcliffe J. G., McMurray J., Ann. Clin. Biochem. 1985, 22, 423–427. [DOI] [PubMed] [Google Scholar]
- 85. Camilo M. E., Morgan M. Y., Sherlock S., Scand. J. Gastroenterol. 1981, 16, 273–279. [DOI] [PubMed] [Google Scholar]
- 86. Falder S., Silla R., Phillips M., Rea S., Gurfinkel R., Baur E., Bartley A., Wood F. M., Fear M. W., Burns 2010, 36, 261–269. [DOI] [PubMed] [Google Scholar]
- 87. Akyilmaz E., Yorganci E., Biosens. Bioelectron. 2008, 23, 1874–1877. [DOI] [PubMed] [Google Scholar]
- 88. Lau P. S., Coombes B. K., Li Y., Angew. Chem. Int. Ed. 2010, 49, 7938–7942; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8110–8114. [Google Scholar]
- 89. Serganov A., Polonskaia A., Phan A. T., Breaker R. R., Patel D. J., Nature 2006, 441, 1167–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Welz R., Breaker R. R., RNA 2007, 13, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wieland M., Benz A., Klauser B., Hartig J. S., Angew. Chem. Int. Ed. 2009, 48, 2715–2718; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2753–2756. [Google Scholar]
- 92. Li X., Song J., Wang Y., Cheng T., Anal. Chim. Acta 2013, 797, 95–101. [DOI] [PubMed] [Google Scholar]
- 93. Lu C. H., Yang H. H., Zhu C. L., Chen X., Chen G. N., Angew. Chem. Int. Ed. 2009, 48, 4785–4787; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4879–4881. [Google Scholar]
- 94. Dwyer M. A., Hellinga H. W., Curr. Opin. Struct. Biol. 2004, 14, 495–504. [DOI] [PubMed] [Google Scholar]
- 95. Hanes J. W., Chatterjee D., Soriano E. V., Ealick S. E., Begley T. P., Chem. Commun. 2011, 47, 2273–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Peroza E. A., Boumezbeur A.-H., Zamboni N., Analyst 2015, 140, 4540–4548. [DOI] [PubMed] [Google Scholar]
- 97. Wustoni S., Hideshima S., Kuroiwa S., Nakanishi T., Hashimoto M., Mori Y., Osaka T., Biosens. Bioelectron. 2015, 67, 256–262. [DOI] [PubMed] [Google Scholar]
- 98. Gong Z., Zhang Z., Anal. Lett. 1996, 29, 695–709. [Google Scholar]
- 99. Zhu J., Liu S., Liu Z., Li Y., Qiao M., Hu X., RSC Adv. 2014, 4, 5990–5994. [Google Scholar]
- 100. Zajicek J. L., Tillitt D. E., Honeyfield D. C., Brown S. B., Fitzsimons J. D., J. Aquat. Anim. Health 2005, 17, 82–94. [Google Scholar]
- 101. Song Z., Hou S., J. Pharm. Biomed. Anal. 2002, 28, 683–691. [DOI] [PubMed] [Google Scholar]
- 102. Norouzi P., Garakani T., Rashedi H., Zamani H., Ganjali M., Int. J. Electrochem. Sci. 2010, 10, 639–652. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary