

Abstract
Long non‐coding RNAs (lncRNAs) play diverse roles in physiological and pathological processes. Several lncRNAs have been suggested to modulate gene expression by guiding chromatin‐modifying complexes to specific sites in the genome. However, besides the example of Xist, clear‐cut evidence demonstrating this novel mode of regulation remains sparse. Here, we focus on HOTAIR, a lncRNA that is overexpressed in several tumor types and previously proposed to play a key role in gene silencing through direct recruitment of Polycomb Repressive Complex 2 (PRC2) to defined genomic loci. Using genetic tools and a novel RNA‐tethering system, we investigated the interplay between HOTAIR and PRC2 in gene silencing. Surprisingly, we observed that forced overexpression of HOTAIR in breast cancer cells leads to subtle transcriptomic changes that appear to be independent of PRC2. Mechanistically, we found that artificial tethering of HOTAIR to chromatin causes transcriptional repression, but that this effect does not require PRC2. Instead, PRC2 recruitment appears to be a consequence of gene silencing. We propose that PRC2 binding to RNA might serve functions other than chromatin targeting.
Keywords: chromatin, lincRNA, Polycomb, transcription
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology; Transcription
Introduction
Polycomb group proteins (PcG) are highly conserved factors that mainly act in the context of multi‐subunit nuclear complexes to maintain transcriptional repression. Their disruption interferes with various processes, ranging from genomic imprinting to cell identity and differentiation. The functions of PcG proteins rely on the regulation of chromatin structure, either through histone modifications or through chromatin compaction (Simon & Kingston, 2009). In Drosophila, four PcG complexes have been identified, while in mammals, only two complexes are well characterized so far: Polycomb Repressive Complex 2 (PRC2) and Polycomb Repressive Complex 1 (PRC1). The PRC2 is responsible for histone H3 lysine 27 (H3K27) di‐ and tri‐methylation (Margueron & Reinberg, 2011).
Although our understanding of how PRC2 contacts chromatin has improved, how it is specifically recruited to defined genomic loci is still only partially understood. The core PRC2 has no known sequence‐specific DNA‐binding domain. In Drosophila, DNA sequences known as Polycomb responsive elements (PREs) mediate PcG recruitment through a combination of specific transcription factors. Although similar mechanisms have been proposed in mammals (Arnold et al, 2013; Sing et al, 2009; Woo et al, 2010), they do not appear to be the general rule. Indeed, the specific transcription factors found to bind these putative mammalian PREs do not act consistently as PRC2 genomewide recruiters. Importantly, GC‐rich regions are frequently bound by PRC2 components (Ku et al, 2008) and they are, in some instances, sufficient to mediate PRC2 recruitment (Mendenhall et al, 2010; Jermann et al, 2014), although once again this cannot account for the specificity and dynamics of Polycomb recruitment in diverse developmental contexts.
It has been long known that RNAs carry out many functions independent of their protein‐coding potential (Cech & Steitz, 2014). Non‐coding RNAs are divided into various subclasses, one of which comprises the lncRNAs. LncRNAs are defined as RNA molecules longer than 200 nucleotides that are transcribed by RNA polymerase II, capped, spliced, and polyadenylated. Hence, with the exception of their lack of coding potential, lncRNAs fully resemble messenger RNAs. Many cellular functions have been ascribed to lncRNAs, although genetic inactivation has not always substantiated the initial observations (Rutenberg‐Schoenberg et al, 2016). Nonetheless, several lncRNAs are reported to influence transcription in the nucleus, in particular through the regulation of chromatin modifiers (Schmitz et al, 2016). The variety of lncRNAs and their tissue‐specific patterns of expression point toward potential functions in development.
Maybe not surprisingly, lncRNAs have been proposed to play an important role in the recruitment of PRC2 to specific chromatin regions, both in cis and in trans (Koziol & Rinn, 2010). The best‐studied example of lncRNA‐dependent cis‐targeting of chromatin modifiers is the localization of PRC2 to the inactive chromosome X (Xi), downstream of Xist RNA (Plath et al, 2003). A direct interaction between PRC2 and the conserved Xist A‐repeat region has been suggested to mediate this effect (Zhao et al, 2008). However, H3K27me3 deposition is still induced when Xist RNA is deleted for the A‐repeats (Kohlmaier et al, 2004; da Rocha et al, 2014), and recent studies aimed at characterizing the Xist interactome did not retrieve factors unambiguously linked to PRC2 (Chu et al, 2015; McHugh et al, 2015; Minajigi et al, 2015). While other domains of Xist could be involved in PRC2 targeting (da Rocha et al, 2014), direct physical interaction between Xist and PRC2 still remains to be proven. Importantly, PRC2 is not required for establishement of transcriptional silencing of the future inactive X; instead, it prevents aberant gene re‐activation in specific tissues (Kalantry et al, 2006).
The best‐known example of PRC2 targeting in trans by a lncRNA comes from the HOX antisense intergenic RNA HOTAIR. This is a 2,148‐nucleotide‐long RNA, originating from the HOXC locus, that has been reported to be necessary to target PRC2 in trans to the HOXD locus and additional genomic loci (Rinn et al, 2007; Chu et al, 2011). HOTAIR RNA adopts a defined secondary structure at its 5′ end, which is proposed to be critical for its interaction with the PRC2 in vitro (Tsai et al, 2010; Somarowthu et al, 2015). HOTAIR RNA also interacts with another repressive chromatin modifier, the LSD1/coREST/REST complex that catalyzes H3K4me2 demethylation. Hence, it has been proposed to act as a scaffold to coordinate recruitment of both the PRC2 and LSD1/coREST/REST complexes onto chromatin (Tsai et al, 2010). However, in mice, genetic deletion of the entire HOXC cluster (including HOTAIR) does not seem to impair H3K27me3 at the HOXD locus in any major way (Schorderet & Duboule, 2011). On the other hand, a more localized deletion of several kb including HOTAIR is reported to do so (Li et al, 2013). Deregulation of HOTAIR has also been observed in cancer cells (Gupta et al, 2010). Overexpression studies performed in a cell line model of triple‐negative breast cancer have linked elevated HOTAIR levels to a re‐targeting of PRC2 to several hundred genes, and it has been proposed that HOTAIR deregulation might contribute to tumor progression (Gupta et al, 2010).
Given the defined interaction suggested to occur between PRC2 and HOTAIR, it is surprising to note that in vitro and in vivo studies investigating the interplay between PRC2 and lncRNAs have reported a rather promiscuous binding of PRC2 and its cofactor JARID2 to RNA (Davidovich et al, 2013; Kaneko et al, 2013, 2014; Beltran et al, 2016). Altogether, the functional specificity of this interaction remains highly debated in the field (Brockdorff, 2013).
In the present study, we set out to further investigate the link between lncRNAs and PRC2 using HOTAIR as paradigm. To this end, we first evaluated the transcriptomic consequences of HOTAIR overexpression in breast cancer cells in the context of a functional or inactivated PRC2. The lack of substantial changes prompted us to study the role of HOTAIR at a local level in model cell lines enabling artificial tethering of HOTAIR at a reporter transgene. Our study provides evidence that HOTAIR RNA can indeed repress transcription in this context, but that this local effect is PRC2 independent.
Results
Overexpression of HOTAIR RNA leads to subtle, PRC2‐independent transcriptional changes in the MDA‐MB‐231 breast cancer cell line
To investigate the link between HOTAIR RNA and PRC2, we took advantage of an MDA‐MD‐231 breast cancer cell line in which we had knocked out EZH2 by genome editing (Wassef et al, 2015). HOTAIR RNA overexpression was previously reported to lead to the transcriptional repression of hundreds of genes in the same model, presumably in trans (Gupta et al, 2010). We overexpressed HOTAIR RNA in MDA‐MB‐231 EZH2+++ (wild‐type, original cell pool), MDA‐MB‐231 EZH2++− (clone with one EZH2 allele targeted, behaving as wild type), and MDA‐MB‐231 EZH2−−− breast cancer cells (subclone derived from the MDA‐MB‐231 EZH2++−, Fig 1A upper and lower panels). Transcript quantification by qRT–PCR revealed that HOTAIR RNA is expressed at similar levels in all three conditions (Fig 1A) and that its level of overexpression is comparable to a previous study (Gupta et al, 2010). As expected, overexpression of HOTAIR RNA has no effect on H3K27me3 global level (Fig 1A) and we did not detect any obvious change of cellular phenotypes such as cell proliferation (Fig EV1A). To get a global picture of HOTAIR‐mediated transcriptional regulation and of the contribution of PRC2 to this process, we performed RNA sequencing on MDA‐MB‐231 EZH2++− and MDA‐MB‐231 EZH2−−− cell types, both in the control condition and upon overexpression of HOTAIR RNA. We obtained good correlation between replicates as shown by the Pearson correlation value matrix (Appendix Table S1 and Fig EV1B). We subsequently focused on transcripts displaying the highest dispersion (higher interquartile) across the four conditions. Heatmap representing relative gene expression revealed that the two main clusters of differentially expressed genes are defined by EZH2 mutation and not by HOTAIR RNA overexpression (Fig 1A). In fact, we observed very similar correlation levels between duplicates and upon overexpression of HOTAIR (Fig EV1B). Nonetheless, we selected transcripts that were differentially expressed upon overexpression of HOTAIR RNA with an absolute expression fold change superior to 2 and a P‐value lower than 0.05. Within these criteria, very few transcripts were differentially expressed and most of them were upregulated regardless of whether PRC2 was functional or deficient (red dots on volcano plot, Figs 1C and EV1C). In addition, close examination of this set of genes revealed that they are characterized by a very low read count (red dots, Fig 1D). Of note, transcripts of genes that were previously reported to gain H3K27me3 upon overexpression of HOTAIR RNA (Gupta et al, 2010) or that are located within 100 kb of its binding sites identified by ChIRP (Chu et al, 2011) revealed a similar trend (Fig EV1D).
Figure 1. Limited transcriptomic changes upon HOTAIR RNA overexpression in MDA‐MB‐231 breast cancer cells.
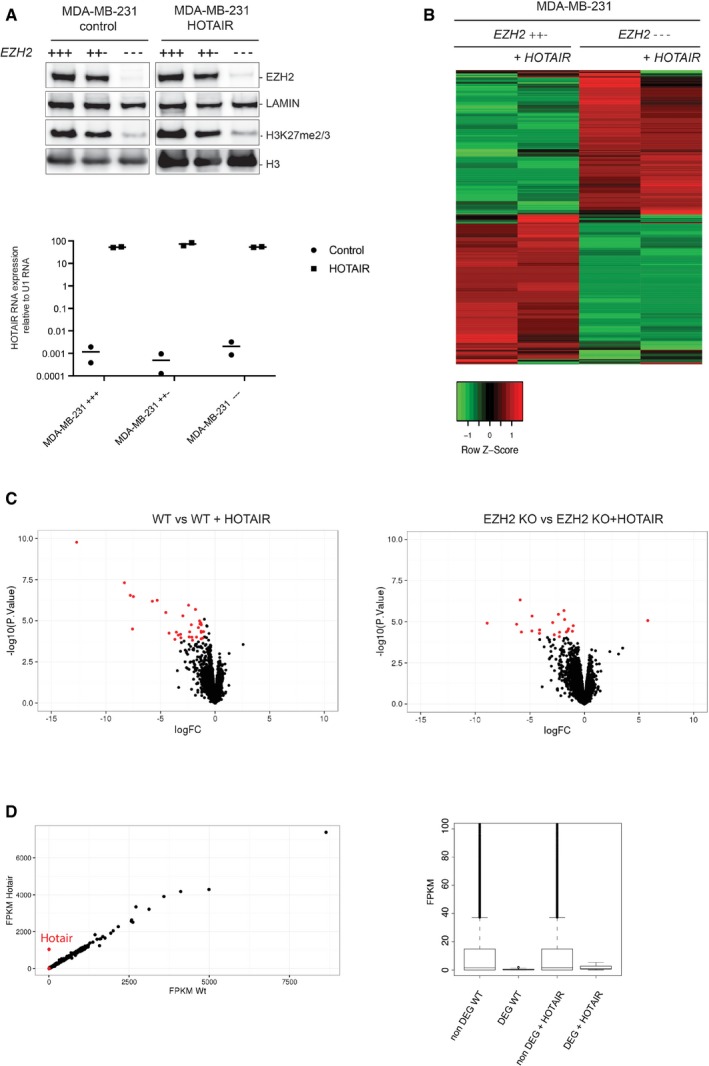
- Western blot analysis of nuclear extracts from indicated cell lines with antibodies for EZH2 and H3K27m2/3 mark. Lamin B1 and H3 are shown as loading controls (upper panel). qRT–PCR to test HOTAIR overexpression in the corresponding cell lines. Y‐axis represents HOTAIR expression relative to U1 RNA (individual experiments and mean, n = 2) (lower panel).
- Heatmap showing expression intensity of the 1,000 genes with the higher interquartile range. Genes up‐ or downregulated from MDA‐MB‐231 EZH2++− and MDA‐MB‐231 EZH2−−− with (+HOTAIR) or without HOTAIR samples are shown. Red indicates high expression, and green indicates low expression.
- Volcano plots representing gene expression change upon overexpression of HOTAIR in MDA‐MB‐231 EZH2++−, or MDA‐MB‐231 EZH2−−− (y‐axis: log10 P‐value, x‐axis: log2 fold change). Red dots represent genes whose expression changes by more than twofold with a P‐value < 0.05. P‐values: moderated t‐statistics.
- Left panel: Gene expression correlation between cells overexpressing HOTAIR or not. Expression is quantified as FPKM; red dots are differentially expressed genes (DEG). Right panel: Average FPKM for non‐DEG genes (> 1 FPKM in at least one of the four conditions) or DEG as defined in (C).
Source data are available online for this figure.
Figure EV1. HOTAIR overexpression has marginal phenotypic and transcriptomic consequences on MDA‐MB‐231.
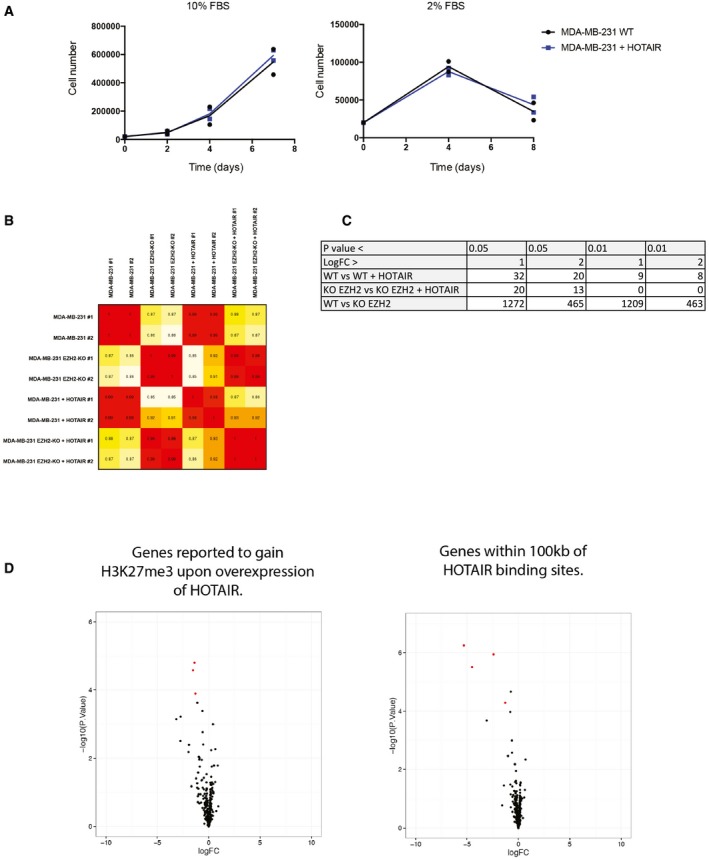
- Proliferation assays in two different cell culture conditions. Cells were counted with an automatic cell counter at the time indicated on the plot (individual experiments and mean, n = 2).
- Pearson correlation matrix between each single‐sequenced sample. Value varies between 1 and 0.85.
- Number of differentially expressed genes depending on cutoff parameter.
- Volcano plots representing gene expression change upon overexpression of HOTAIR in MDA‐MB‐231 EZH2++− and focusing on genes either previously reported to gain H3K27me3 upon overexpression of HOTAIR (left panel) or to be located with 100 kb of a binding sites for HOTAIR (right panel) (y‐axis: log10 P‐value, x‐axis: log2 fold change). Red dots represent genes whose expression changes by more than twofold with a P‐value < 0.05.
Source data are available online for this figure.
Altogether, these experiments suggest that HOTAIR RNA overexpression only marginally affects gene expression in the MDA‐MB‐231 breast cancer cell line and that this function does not critically require PRC2.
In vivo tethering of HOTAIR RNA induces gene silencing
The lack of a substantial effect of HOTAIR overexpression on gene expression profiles prompted us to develop a method to assess whether HOTAIR could have a more local impact on transcription. To this end, we set up an RNA‐tethering system to force the recruitment of HOTAIR at a reporter transgene.
This system exploits two well‐known heterologous tools: the bacteriophage MS2 coat protein (MS2BP), which binds to the MS2 stem loop RNA (MS2 loop), and the UAS/Gal4 tethering system. Both systems have been successfully used in eukaryotic cells, the former to tether MS2BP‐fused proteins to MS2 loop hybrid RNAs (Keryer‐Bibens et al, 2008) and the latter to target transcription factors or chromatin modifiers. The parental cell line (labeled 1) is T‐Rex HEK293 stably transfected with a luciferase reporter gene, the expression of which is controlled by the tk minimal promoter. UAS/Gal4‐binding sites enable the recruitment of a Gal4‐DNA binding domain fused to a protein of interest (Fig 2A). We derived a subclone constitutively expressing a Gal4‐DNA binding domain MS2 coat‐protein fusion protein (labeled 2). From this clone, we subsequently derived cells expressing either MS2 loop‐HOTAIR (labeled 3) or MS2 loop‐HOTAIR‐Rev (RNA antisense to HOTAIR, labeled 4) hybrid RNAs (Fig 2A). We checked the expression of the fused Gal4‐MS2BP protein by Western blot (Fig EV2A). Both MS2‐HOTAIR RNAs were expressed at similar levels (Fig EV2B). We confirmed the recruitment of Gal4‐MS2BP to the transgene by performing chromatin immunoprecipitation (Wang et al, 2005), using an antibody recognizing the Gal4‐binding domain (Fig 2B). RNA immunoprecipitation (RIP) with the same antibody further indicated that the fusion protein indeed interacts with the MS2‐HOTAIR or MS2‐HOTAIR‐Rev RNAs (Fig 2C).
Figure 2. MS2‐HOTAIR RNA causes repression when tethered to the luciferase transgene.
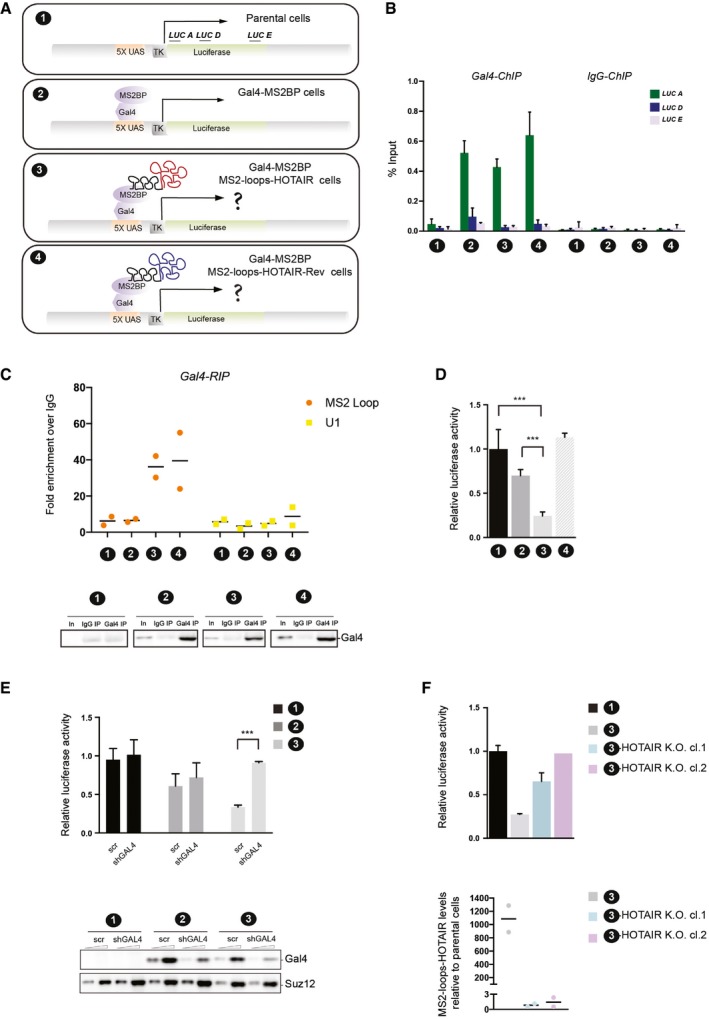
- Schematic representation of the RNA‐tethering system to chromatin. LUC A, LUC D and LUC E indicate primer sets along the luciferase transgene used for ChIP qPCR. Each cell model is labeled by a number which is used in all figure legends hereafter.
- ChIP experiments with Gal4 antibody in the cell lines indicated on the x‐axis. Y‐axis represents percent of input (mean ± SD, n = 3).
- RIP experiments with Gal4 antibody in the cell lines indicated on the x‐axis. MS2 loop and U1 primers were used in qRT–PCR. Y‐axis represents fold enrichment to IgG (individual experiments and mean, n = 2). Input (In) and IP were loaded and probed with Gal4 antibody (lower panel).
- Relative luciferase activity in the cell lines indicated on the x‐axis. Values represent the relative luciferase activity normalized to the amount of protein (mean ± SD, n = 4). Statistical analysis: unpaired t‐test, ***P < 0.001.
- Upper panel: Relative luciferase activity in the cell lines indicated in the right legend (mean ± SD, n = 4). Statistical analysis: unpaired t‐test, ***P < 0.001. Cells were infected either with scramble (scr) or shRNA targeting Gal4‐MS2BP (shGAL4). Lower panel: Western blot analysis with anti‐Gal4 antibody in the different cell models; SUZ12 was used as a loading control.
- Upper panel: Relative luciferase activity in the cell lines indicated in the right legend (mean ± SD, n ≥ 2). Lower panel: qRT–PCR to detect MS2‐HOTAIR RNA in the cell models indicated in the right legend. Y‐axis represents MS2 loop RNA levels normalized to actin and calculated over the parental cells (individual experiments and mean, n = 2).
Source data are available online for this figure.
Figure EV2. Expression of Gal4‐MS2CP fusion proteins and luciferase activity in clones overexpressing HOTAIR .
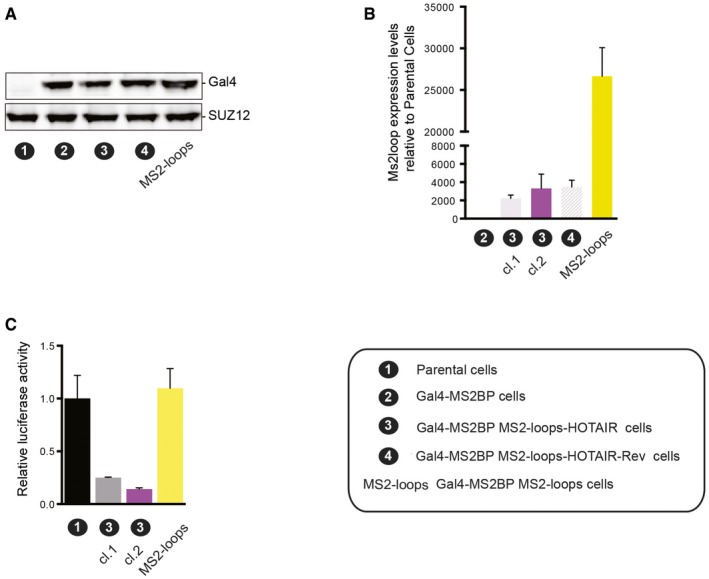
- Western blot analysis with Gal4 antibody on nuclear extracts from parental cell line (1) MS2BP, (2) MS2BP MS2‐HOTAIR (3) or MS2BP MS2‐HOTAIR‐Rev (4) and MS2 loop cell lines. SUZ12 was used as a loading control.
- qRT–PCR to detect MS2‐hybrid RNAs in the cell lines indicated on the x‐axis. Y‐axis represents MS2 loop RNA levels normalized to actin and calculated over the parental cells (mean ± SD, n = 2).
- Relative luciferase activity in the cell lines indicated on the x‐axis. Values represent the relative luciferase activity normalized to the amount of protein (mean ± SD, n = 3).
Having established the functionality of our system, we tested the transcriptional consequences of tethering HOTAIR RNA on the activity of the luciferase reporter. We observed a 75% reduction in luciferase activity in the presence of MS2‐HOTAIR RNA, a reduction that was not seen in the presence of MS2‐HOTAIR‐Rev RNA (Fig 2D) or in cells overexpressing the MS2 loops alone (Fig EV2B). To verify that this effect was not clone specific and that it required continuous tethering of MS2‐HOTAIR RNA, we used three different strategies. First, we confirmed the repression of the luciferase reporter in another clone expressing equal levels of MS2‐HOTAIR (Fig EV2B and C). Then, we checked that HOTAIR‐mediated transcriptional repression is relieved when preventing its recruitment by knocking down the Gal4‐MS2BP protein. Indeed, upon effective knockdown of the Gal4‐MS2BP protein by RNA interference (shGAL4) in the MS2BP and MS2BP MS2‐HOTAIR cell models (Fig 2E, lower panel), we observed a release of luciferase repression as compared to a scramble shRNA construct (Fig 2E, upper panel). Finally, we verified that abrogating HOTAIR expression by knocking out the MS2‐HOTAIR construct in the MS2BP MS2‐HOTAIR cell model also releases luciferase repression. In two clones knocked out for the MS2‐HOTAIR construct as shown by qRT–PCR (Fig 2F, lower panel, labeled 3‐HOTAIR K.O. cl.1 and 3‐HOTAIR K.O. cl.2), we could confirm a consistent increase in luciferase activity (Fig 2F, upper panel).
Altogether, these results extensively validate our approach to tether RNA to chromatin. More importantly, we demonstrate that forced recruitment of HOTAIR specifically leads to transcriptional repression.
Artificial tethering of HOTAIR RNA is associated with changes in chromatin structure
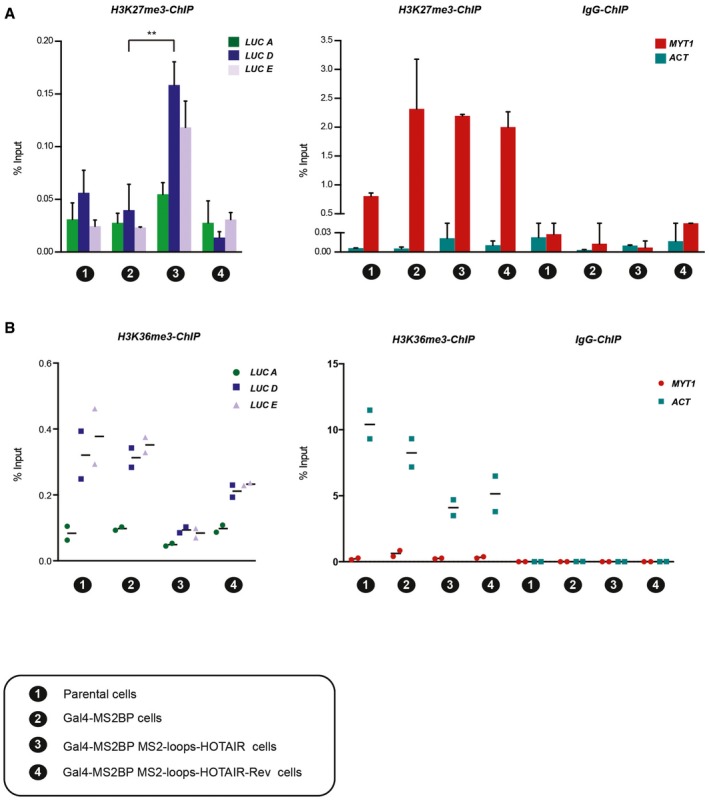
Given the observed gene silencing effect of HOTAIR RNA, we wished to explore its underlying mechanisms and, in particular, whether it involves specific chromatin regulatory activities. Therefore, we performed ChIP experiments in our MS2‐HOTAIR RNA‐tethered system to evaluate H3K27me3 enrichment upon recruitment of HOTAIR. This assay revealed increased enrichment of H3K27me3 downstream of the 5× UAS (LUC D and LUC E primers) specifically in cells expressing MS2‐HOTAIR RNA (Fig 3A). Importantly, not all repressive signatures were increased, since we did not observe any change when probing DNA methylation and H3K9me2 enrichment at the reporter transgene (Fig EV3B and C). Also, this effect required HOTAIR RNA recruitment as it was lost upon knockdown of the Gal4‐MS2BP protein (Fig EV3A). Of note, the gain of H3K27me3 in MS2‐HOTAIR cell line was relatively mild as compared to endogenous PRC2 target such as MYT1 (Figs 3A and EV3A). To determine whether other chromatin changes occur, we tested the enrichment of the H3K36me3 chromatin mark, which maps to the gene body of transcribed genes. In Gal4‐MS2BP and Gal4‐MS2BP MS2‐HOTAIR‐Rev cells, H3K36me3 levels in the gene body of the luciferase reporter (LUC D and LUC E primers) are 10 times lower than in the highly transcribed gene ACT (Fig 3B). Nonetheless, we could detect a reduction in H3K36me3 enrichment in MS2‐HOTAIR cells (Fig 3B). We observed similar trends when analyzing the enrichment for RNA polymerase II (Fig EV3B) but not for H3K27ac, which seems to have the same level of enrichment in all the model cell lines (Fig EV3B).
Figure 3. MS2‐HOTAIR RNA modulates chromatin structure.
-
A, BChIP experiments with H3K27me3 (A) or H3K36me3 (B) antibody in the cell models numbered on the x‐axis of each graph; corresponding legend is at the bottom of the figure. Enrichment for primers located along the luciferase reporter (left) and enrichment for control regions (MYT1 and ACT) (right). Y‐axis represents percent of input (mean ± SD, n = 3 in A; individual experiments and mean, n = 2 in B). Statistical analysis: unpaired t‐test, **P < 0.01.
Source data are available online for this figure.
Figure EV3. Chromatin regulation upon recruitment of HOTAIR .
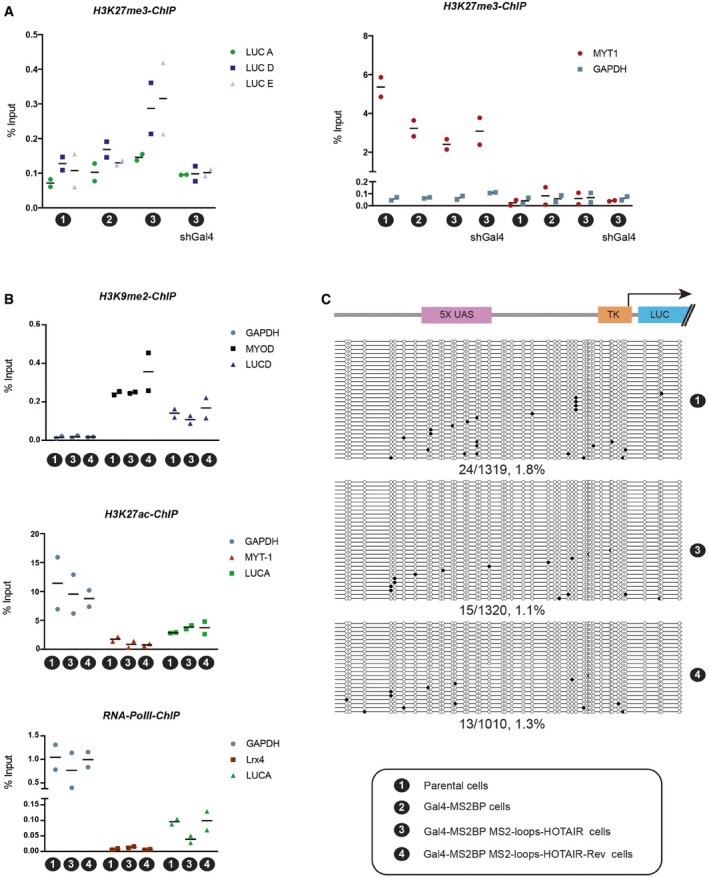
- ChIP experiments with H3K27me3 and primers located along the luciferase reporter (left) or control regions (MYT1 and ACT) (right) in the cell lines indicated on the x‐axis of each graph (individual experiments and mean, n = 2). A H3K27me3 antibody of different origin was used in this assay as compared to Fig 3.
- ChIP experiments with H3K9me2 (top), H3K27ac (middle), or RNA polymerase II (bottom) in the cell lines indicated on the x‐axis of each graph. Primers used are indicated in the color legend (individual experiments and mean, n = 2).
- Analysis of DNA methylation. Schematic representation of the portion of the reporter construct analyzed for DNA methylation is displayed on top. It comprises the Gal4‐binding sites (5× UAS), the TK minimal promoter and part of the luciferase gene. DNA methylation analysis by bisulfite cloning is shown below in three different cell lines as indicated on the right. Black circles indicate methylated CpGs, and white circles indicate unmethylated CpGs. The percentage of methylated CpG is indicated below each condition.
We conclude from these experiments that HOTAIR‐mediated transcriptional repression correlates with mild losses and gains of a subset of active and repressive chromatin marks, respectively.
Gain of H3K27me3 upon tethering of HOTAIR RNA does not reflect a specific interaction between HOTAIR and PRC2
The results described above could fit with the hypothesis that HOTAIR RNA recruits PRC2 to chromatin. However, recent studies lead to contrasting conclusions regarding the specificity of PRC2 binding to RNA (Davidovich et al, 2013; Kaneko et al, 2013, 2014; Beltran et al, 2016). These discrepancies might be due in part to the control used to determine whether an interaction is specific. Having established that HOTAIR‐Rev transcript does not lead to an increased enrichment of H3K27me3 when tethered at a transgene, but considering that it is identical in size to HOTAIR transcript and that it is also predicted to form secondary structures, we reasoned that it represents an ideal control for interaction assays. We probed PRC2‐HOTAIR RNA interaction through two methods: first by sucrose density gradient and then by electrophoretic mobility shift assays (EMSA). We used highly purified PRC2 (Fig EV4A) and in vitro transcribed full‐length HOTAIR or HOTAIR‐Rev (Fig EV4B) for these assays. Results from the two approaches were mutually consistent and showed that PRC2 binds RNA with high affinity and little specificity; PRC2 interacts equally well with HOTAIR and HOTAIR‐Rev but displays a slightly higher affinity for MS2 loop RNA in EMSA (Fig 4A and B). Next, we analyzed whether adding chromatin to the assay could impact the PRC2–HOTAIR interaction, as one might expect if HOTAIR acted as a bridge between PRC2 and chromatin. When we incubated chromatin with full‐length HOTAIR RNAs, the elution pattern of chromatin moved one fraction toward the RNA (Fig EV4C). This event is not specific, as both HOTAIR and HOTAIR‐Rev similarly affect the chromatin elution pattern. We then determined the effect of incubating all three partners together at an equimolar concentration: RNA, PRC2, and chromatin (Fig EV4D). We did not detect any obvious synergy between the three partners. Indeed, the elution pattern of PRC2 in the presence of HOTAIR and chromatin was similar to that observed with HOTAIR alone. Similarly, chromatin in the presence of HOTAIR and PRC2 shifts by one fraction as previously observed with HOTAIR alone. Of note, a recent report proposed that the interaction of PRC2 with RNA or chromatin is mutually exclusive, a conclusion which could be consistent with our observations (Beltran et al, 2016).
Figure EV4. PRC2 and HOTAIR in vitro production.

- Scheme for PRC2 production in Sf9 insect cells and purification over MiniQ column (left panel). Coomassie of eluted fractions. EZH2, SUZ12, EED‐FLAG and RbAp48 proteins are indicated (right panel).
- Agarose gel showing 2.2‐kb full‐length in vitro transcribed HOTAIR and HOTAIR‐Rev RNAs.
- Left: Scheme representing the interacting partners (chromatin and RNA) analyzed by sucrose gradient sedimentation on the right. Right: Native chromatin was incubated with or without biotinylated HOTAIR/HOTAIR‐Rev RNAs, and chromatin conformation was analyzed by density gradient centrifugation.
- Left: Scheme representing the interacting partners (chromatin, RNA and PRC2) analyzed by sucrose gradient on the right. Right: PRC2 and native chromatin were incubated with or without biotinylated HOTAIR/HOTAIR‐Rev RNAs and their profile analyzed by density gradient centrifugation.
Source data are available online for this figure.
Figure 4. PRC2 interacts with RNA with low specificity.
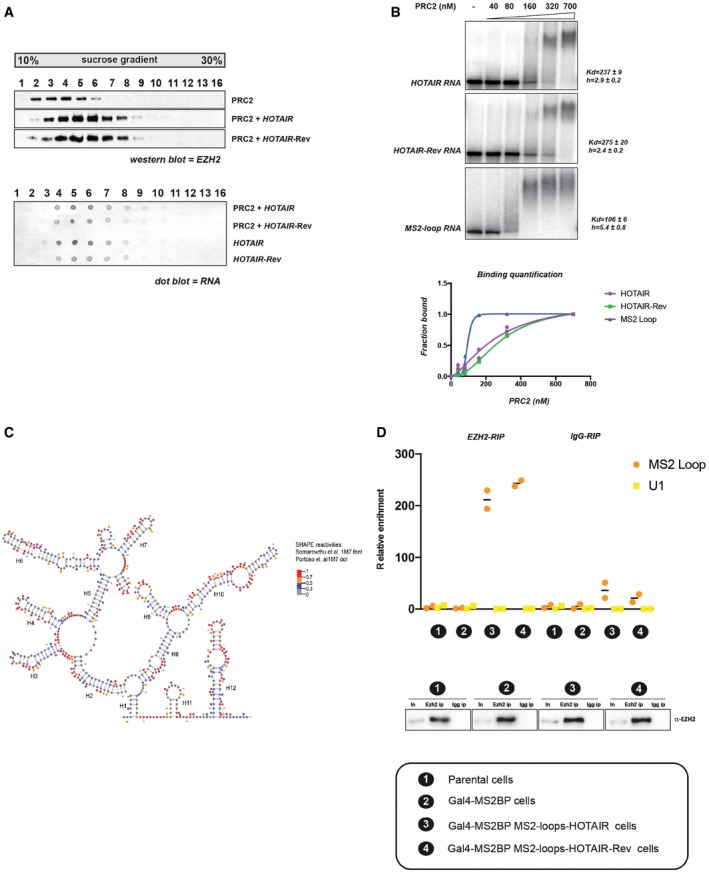
- PRC2 was incubated with or without biotinylated HOTAIR or HOTAIR reverse‐complement RNAs and analyzed by density gradient centrifugation on a linear sucrose gradient (10–30%). Individual fractions collected from sucrose gradient were probed by Western blot for EZH2 (upper panel) or by dot blot for biotinylated RNA (lower panel).
- Representative EMSA experiments showing binding of PRC2 to full‐length HOTAIR, HOTAIR‐Rev and MS2 loop RNA probes. Equilibrium dissociation constant (K d) values and Hill slope are calculated on biological replicates (n = 2). Corresponding binding curves of biological duplicate EMSA experiments (bottom panel).
- Predictive secondary structure for the first 530 bp of HOTAIR RNA from the RNAstructure software and VARNA visualization software. HOTAIR D1 domain as modeled by Somarowthu et al (2015) according to SHAPE‐CE probing is shown. SHAPE reactivities from Somarowthu et al (2015) are depicted by colored nucleotides; 1M7 SHAPE reactivity obtained in our experiment is represented by colored dots over the nucleotides. Highly reactive nucleotides are displayed in red and orange, and low reactive nucleotides are displayed in black or blue according to the values reported in the legend.
- EZH2 binds both MS2‐HOTAIR and MS2‐HOTAIR‐Rev RNAs in vivo. RIP experiments with EZH2 antibody in cell models indicated on the x‐axis. MS2 loop and U1 primers were used in qRT–PCR. Y‐axis represents relative enrichment (individual experiments and mean, n = 2). Input (In) and IP were loaded and probed with EZH2 antibody (lower panel). Correspondence between numbers and model cell lines is indicated at the bottom.
Source data are available online for this figure.
To exclude the possibility that the lack of specificity of PRC2 binding to RNA in vitro could be due to inappropriate folding of the RNA under our experimental settings, we probed HOTAIR RNA structure by SHAPE‐MaP (selective 2′‐hydroxyl acylation analyzed by primer extension) (Siegfried et al, 2014; Smola et al, 2015). Briefly, the RNA is incubated with small molecules that react with single‐stranded nucleotides, and high‐throughput sequencing is then used to identify the extent of mutations for each position. We used two different chemicals for this assay (NMIA and 1M7) and obtained SHAPE reactivity, which showed a good correlation between the two chemical probings as well as with the previously published data (see source data for Fig 4C). We then focused on results obtained with 1M7 (Figs 4C and EV5) for direct comparison with the previously published structure model of HOTAIR (Somarowthu et al, 2015). Our reactivity map was used as constrains to model HOTAIR secondary structure using the software RNAstructure (Deigan et al, 2009). The most stable secondary structure model predicted based on our 1M7 reactivities is slightly distinct from the previous report (Fig EV5); nonetheless, we observed a good overlap between the two structures as shown for the D1 domain (Fig 4C). In summary, the consistency with Somarowthu's thorough HOTAIR structure probing makes us confident that HOTAIR RNA is folded in a similar structure in both studies.
Figure EV5. HOTAIR domain 1 secondary structure.
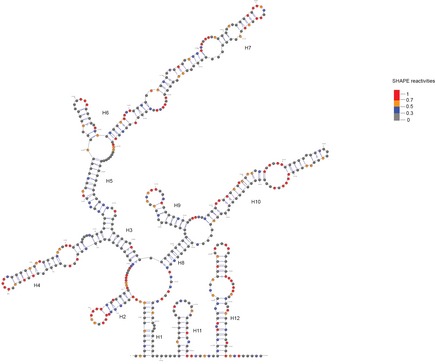
Secondary structure model for the first 530 bp of HOTAIR RNA obtained from the RNAstructure software using the 1M7 reactivity map obtained in this study and displayed with predictive secondary structure. HOTAIR D1 domain as mapped with 1M7 SHAPE probing from our experiment is shown. SHAPE reactivity is depicted by colored nucleotides. Highly reactive nucleotides are displayed in red and orange, and low reactive nucleotides are displayed in black or blue according to the values reported in the legend.Source data are available online for this figure.
Finally, we determined whether our in vitro results hold true in a cellular context. To address this question, we performed RIP pulling down RNAs interacting with EZH2 in our different cell models. As expected, we observed that EZH2 RIP is enriched for HOTAIR over the IgG control. However, we obtained a similar enrichment for HOTAIR‐Rev, thus corroborating the in vitro findings (Fig 4D). Although we cannot exclude the possibility that the MS2 loops interfere with HOTAIR structure in the artificial tethering assay, the similarity between our in vitro interaction experiments (HOTAIR without MS2 loops) and the RIP experiment in a cellular context (HOTAIR with MS2 loops) suggests that it is not the case.
Altogether, our experiments confirm the lack of specificity in the interaction of PRC2 with RNAs both in vitro and in cultured cells.
HOTAIR‐mediated transcriptional repression does not require PRC2
A previous study reported that simply inhibiting transcription is sufficient to trigger the recruitment of PRC2 to many loci across the genome (Riising et al, 2014). In light of those findings and the results of our interaction assays, we considered the possibility that the observed increased H3K27me3 enrichment subsequent to HOTAIR tethering might not be caused by direct HOTAIR‐mediated recruitment of PRC2, but might rather occur as a consequence of reduced transcription. To clarify this point, we employed CRISPR/Cas9 to knock out two essential PRC2 components (EED and SUZ12, Fig 5A). Deleting either EED or SUZ12 led to a complete loss of H3K27me3 both at the global level (Fig 5A, top panel) and at the local level (Fig 5B, lower panel). Yet, in two different MS2‐HOTAIR‐expressing subclones deleted for EED (cl.1 and cl.2) or SUZ12 (cl.1 and cl.2) proteins, we observed the same transcriptional repression as in the MS2‐HOTAIR parental cell line with wild‐type PRC2 components (Fig 5C).
Figure 5. PRC2 is dispensable for HOTAIR‐mediated transcriptional repression.
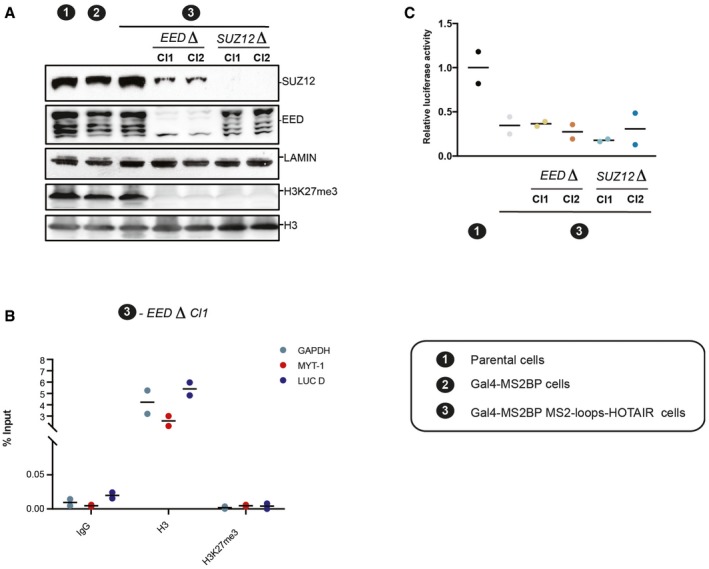
- Western blot analysis of nuclear extract from indicated cell lines with antibodies for SUZ12, EED and H3K27m2/3 mark. Lamin and H3 are shown as loading controls.
- ChIP experiments with IgG, histone H3, or H3K27me3 antibodies in the cell model indicated on top. Enrichment for the primers indicated on the right (individual experiments and mean, n = 2)
- Relative luciferase activity in the cell lines indicated on the x‐axis. Values represent the relative luciferase activity normalized to the amount of protein (individual experiments and mean, n = 2).
Altogether, our results demonstrate that the silencing of the luciferase reporter requires the continuous presence of MS2‐HOTAIR RNA but not H3K27me3 deposition. It suggests therefore that the MS2‐HOTAIR transcript modulates transcription independently of PRC2.
Discussion
While an unexpected proportion of eukaryotic genomes is transcribed, many of the resulting transcripts are non‐coding RNAs. Among them, the subclass of lncRNAs has been implicated in the regulation of a variety of cellular functions. In particular, nuclear lncRNAs have been found to modulate transcription through the targeting of chromatin modifiers to specific genomic regions. One such example is HOTAIR, a lncRNA which was reported to promote breast cancers through the aberrant targeting of PRC2 and consequently inappropriate gene silencing (Gupta et al, 2010). However, when we overexpressed HOTAIR RNA in MDA‐MB‐231 breast cancer cells, either in the presence or absence of PRC2, we detected few transcriptomic changes. While the reasons for the discrepancy with the previous report remain unclear, it underscores the need for caution when considering the potential contribution of HOTAIR transcript to tumorigenesis.
Considering the lack of broad trans effects of overexpressing HOTAIR RNA in MDA‐MB‐231 cell line, we sought a more direct way to gauge whether and how HOTAIR RNA regulates transcription. To address this question, we established cell models enabling to artificially tether HOTAIR RNA at a stably integrated reporter transgene. This approach revealed that, at least in this specific context and assuming that the MS2 loops do not interfere with HOTAIR structure, HOTAIR RNA can repress transcription. Several mechanisms could mediate this repressive activity. The most trivial model would be that HOTAIR RNA recruitment directly interferes with the RNA polymerase machinery, that is, by steric hindrance. Although we cannot formally exclude this hypothesis, it is undermined by the fact that the recruitment of the transcript antisense to HOTAIR (an RNA of identical size) did no impact on reporter activity. An alternative hypothesis is that HOTAIR recruits chromatin modifiers, which in turn modulate transcription. Accordingly, we observed that HOTAIR artificial recruitment is paralleled by mild changes in chromatin structure (histone methylation). However, when we deleted essential components of PRC2 that abrogate its methyltransferase activity, this did not affect the repressive activity of HOTAIR RNA. This shows that at least some of the major changes in chromatin composition upon recruitment of HOTAIR RNA are a secondary consequence of changes in transcription in our model. Last, HOTAIR may interfere with transcription by interacting with yet unknown factors. Unfortunately, our attempt to use yeast three‐hybrid system to identify such factors was unsuccessful (data not shown). Further investigation will therefore be required to address this point.
The interaction between PRC2 and RNA has retained a great deal of attention; however, different studies have reached contrasting conclusions (Davidovich et al, 2013; Cifuentes‐Rojas et al, 2014; Beltran et al, 2016). Our results strongly support the weak specificity but strong affinity of PRC2 for RNAs (Davidovich et al, 2013; Cifuentes‐Rojas et al, 2014; Beltran et al, 2016). The authors of the latest study proposed that chromatin and RNA might compete for binding to PRC2. In agreement with this idea, we did not find evidence for a complex between RNA, chromatin, and PRC2. We also observed that an excess of RNA could reduce PRC2 enzymatic activity on chromatin but not on another substrate, JARID2 (data not shown). This result is consistent with a specific competition between chromatin and RNA for interaction with PRC2. It is proposed that this antagonism could explain why active transcription prevents PRC2 recruitment (Beltran et al, 2016). Intriguingly, the inhibitory activity of RNA on chromatin‐modifying enzymes is not exclusive to the PRC2, but appears to be a rather common property, since it has also been observed for SET9 (Kaneko et al, 2014), G9A (data not shown), BRG1‐BAF (Cajigas et al, 2015), and DNMT1 (Di Ruscio et al, 2013), even though these enzymes have very distinct functions in transcriptional regulation. It is possible that the affinity of chromatin modifiers for RNA is important to compete with and therefore prevent low affinity and random binding to chromatin. Further studies will be required to test this hypothesis.
Materials and Methods
Recombinant proteins and PRC2 purification
hPRC2 production in SF9 insect cells was performed upon co‐infection with EZH2‐His, SUZ12‐His, RBBP4‐Strep‐TAG, and EED‐Flag‐tagged baculoviruses. Cells were lysed in BC300 buffer (300 mM KCl, 10% glycerol, 25 mM Tris–HCl pH 8, 1 mM EDTA), sonicated, and clarified by centrifugation before incubation with Flag beads (M2 beads SIGMA). PRC2 was eluted with Flag peptide and further purified on a MiniQ column to assure homogeneity and complete removal of nucleic acid contaminants. Fraction content was verified on Coomassie.
In vitro RNA transcription with biotinylated or radiolabeled UTP
One microgram of linearized pBluescript plasmid expressing HOTAIR reverse‐complement or MS2 loop RNA was in vitro transcribed for 3 h at 37°C using the MEGAscript T7 transcription kit (AM1334). After DNase treatment, when biotinylated, samples were purified over the MEGAclear™ transcription clean‐up kit (AM1908) and checked for full length on agarose gel. When radiolabeled, samples were cleaned with acid phenol/chloroform and precipitated at −20°C with 2.5 vol EtOH and 1/10 3 M NaAcet pH 5.3, 70% EtOH‐washed, and resuspended in DEPC H2O. RNAs were successively quantified by UV absorbance at 260 nm. Purity and integrity of all RNA batches were examined on a 0.8% agarose gel.
Refolding of in vitro transcribed RNA
In vitro transcribed RNA was heated at 95°C for 3 min, then immediately placed on ice for 2 min, added 2× refolding buffer (20 mM Tris pH 7; 200 mM KCl; 20 mM MgCl2), and refolded at RT for 20 min.
Nucleosome reconstitution
Nucleosomes were assembled from 5S 12 repeat DNA (Dorigo et al, 2004) and purified HeLa cell histone octamers by salt dialysis through a linear gradient (2.2 M NaCl to 0.4 M NaCl) for 20 h, followed by a step dialysis against TE.
Sucrose gradient sedimentation analysis
Equimolar PRC2, in vitro reconstituted chromatin, and in vitro transcribed and refolded biotinylated RNAs were incubated together prior to sucrose gradients in HEB buffer (25 mM Hepes, 40 mM KCl, 0.2 mM EDTA, 1 mM DTT) for 1 h at RT.
Sucrose gradients were prepared using a gradient maker (Biocomp) according to the manufacturer's instruction and centrifuged for 16 h at 55,000 g in a Beckmann 60Ti rotor. Fractions were collected manually (250 μl each fraction), and 30 μl of each sample was loaded on NuPAGE Novex 4–12% Bis–Tris protein for Western blot analysis; 20 μl of each sample added with 0.5% SDS was loaded on 0.8% agarose gel, and 1 μl for each sample was spotted on positively charged nylon membrane nylon for dot blot.
Dot blot
One micoliter from each sucrose gradient centrifugated fraction was spotted on positively charged nylon membrane, let dry for 30 min, and UV‐cross‐linked. RNA was revealed using the biotin chromogenic detection kit (KO661 Thermo Fischer Scientific) following the manual instructions.
Electrophoretic mobility shift assay
Five nanomolar refolded RNA was incubated with increasing concentration of PRC2 in binding buffer (50 mM Tris–HCl, pH 7.5 at 25°C, 100 mM KCl, 5 mM MgCl2, 0.5 mM ZnCl2, 0.1 mM CaCl2, 2 mM 2‐mercaptoethanol, 0.1 mg/ml BSA, 0.1 mg/ml fragmented yeast tRNA, 5% v/v glycerol, 0.025% w/v bromophenol blue, and 0.025% w/v xylene cyanol) at 30°C for 30 min. Samples were cooled to 4°C for 10 min and loaded on a 0.7% agarose gel in 1× TBE buffer at 4°C. Gels were vacuum‐dried for 45 min at 80°C on a nylon membrane and two sheets of Whatman 3‐mm chromatography paper. Dried gels were exposed to phosphorimaging plates, and signal acquisition was performed with a Typhoon Trio phosphorimager (GE Healthcare). Densitometry was carried out with ImageJ software and data fitted to a sigmoidal binding curve with Prism Software. Data ranges for both dissociation constants and Hill coefficients were calculated on the basis of two replicates.
Cloning
Construction of templates for in vitro transcription: HOTAIR cDNAs from LZRS‐HOTAIR (purchased from Addgene, plasmid #26110, deposited by Howard Chang) were cloned into pBluescript plasmid digested with BamHI. pCR4‐24XMS2SL‐stable (Addgene plasmid #31865, deposited by Robert Singer) was used to produce in vitro MS2 loop RNA. Fusion protein vector: pFLAG_NLS_MS2‐MS2 plasmid was a kind gift from Richard Breathnach (Gesnel et al, 2009). Gal4‐DBD was cloned upstream MS2‐MS2 dimer coat protein with EcoRV/XbaI sites. MS2 loop‐RNA hybrid constructs: The MS2 loop repeat fragment was digested BamHI/BglII from pCR4‐24XMS2SL‐stable and inserted in the modified mammalian expression vector pCDNA4/TO linearized with BamHI restriction enzyme. To this plasmid, HOTAIR cDNA, digested BamHI from LZRS‐HOTAIR was ligated 5′ to the MS2 loop repeats. Both orientations were checked by restriction enzyme digestion and sequencing. Cloning the shGal4 sequence into the pLKO.1 hygro vector (Addgene plasmid #24150, deposited by Bob Weinberg) was performed according to the pKLO‐TRC cloning vector procedure at http://www.addgene.org/tools/protocols/plko/. The target sequence for Gal4 was ATCGAACAAGCATGCGATATT.
The deletion cassette for HOTAIR was built by compatible restriction enzyme digestion and ligation and verified both by restriction enzyme digestion and sequencing at all steps. Briefly, 500 bp was amplified from the pCDNA4/TO HOTAIR‐MS2 loop plasmid comprising the promoter region and ligated to the hygromycin B resistance cassette followed by 640 bp of HOTAIR cDNA fragment, located 300 bp downstream the J. Rinn HOTAIR start site in a pBluescript plasmid. The gRNA target site, designed using the http://crispor.tefor.net/ Web site, GAGAGCACCTCCGGGATATT was comprised within the first 300 bp of HOTAIR cDNA and cloned into the gRNA vector (Addgene plasmid #41824, deposited by George Church) according to the Addgene procedure.
The deletion cassette for hEED was built cloning hygromycin B resistance cassette between left and right region homologs to EED exon 2. The gRNA target site GCACCTGGAAGGAAAAGTTG was cloned into the gRNA vector according to the Addgene procedure. The deletion cassette for hSUZ12 was done as for EED with left and right arm homologs to SUZ12 exon 10. The gRNA target site GAGACTCTCTGAATTTCTAG was cloned into the gRNA vector according to the Addgene procedure.
Cell culture and transfections
T‐Rex 293 cells (Invitrogen) were grown according to the manufacturer's instructions. MDA‐MB‐231‐derived cell lines were previously described (Wassef et al, 2015). All cell lines were tested for the absence of mycoplasma on a monthly basis. All transfections were performed using PEI (polyethylenimine) at 3:1 ratio to DNA.
First, 5XGal4RE‐tk‐Luc‐Neo plasmid was stably integrated into the cells and selected for G418 resistance (0.5 μg/ml). One highly expressing luciferase clone was stably transfected and selected for pFLAG_Gal4DBD_NLS_MS2‐MS2 bearing puromycin resistance (10 μg/ml). Subsequently a single clone verified by Western blot for the expression of the fused Gal4DBD_NLS_MS2‐MS2 protein was transfected with each MS2 loop‐RNA hybrid plasmid bearing Zeocin resistance. Resistant clones selected for Zeocin (0.4 μg/ml) were tested for expression of the different MS2 loop‐RNA hybrid constructs by strand‐specific RT–PCR and qRT–PCR.
Co‐transfection with gRNA targeting HOTAIR, hCas9, and the targeted HOTAIR construct was performed with PEI. Hygromycin B selection was performed at 0.3 μg/ml.
Co‐transfections with gRNAs targeting EED or SUZ12, hCas9, and each of the EED and SUZ12 targeted constructs were performed with PEI. Hygromycin B selection was performed at 0.3 μg/ml.
Retroviral vector production and transduction
Production of shGal4 lentiviral vector was performed in 293T cells. Transduction and selection of target cells were performed according to the online Addgene procedure. Hygromycin B was added at 0.3 μg/ml. Production of overexpressing HOTAIR retroviral vectors was performed in 293T cells. Transduction of target cells was performed as for the lentiviral vector.
Quantification of mRNA levels by qRT–PCR
Total RNA was isolated following TRIzol reagent (Invitrogen) extraction instructions. cDNA was synthetized using SuperScript III reverse transcriptase kit (18080044 Invitrogen), and quantitative PCR was performed with technical triplicate using SYBR green reagent (Roche) on a ViiA7 equipment (Applied Biosystems). At least three biological independent experiments were performed for each assay.
Luciferase assay
Luciferase reporter activities were measured in whole‐cell lysates using the Luciferase Assay System (Promega, #E15020) and Fluostar Optima BMG Labtech luminometer. All experiments were done in triplicate and normalized for protein concentration (Bradford).
Chromatin immunoprecipitation
ChIPs were performed as previously described (Sanulli et al, 2015). 1.2 × 107 cells were plated in 15‐cm plates 2 days before cross‐linking. Quantification was done as previously described for the qRT–PCR. Primers sequences and antibodies used are provided in Appendix Tables S2 and S3.
DNA methylation analysis
Genomic DNA was treated with bisulfite using EpiTect Bisulfite Kit (Qiagen) and PCR‐amplified using a nested PCR strategy. The PCR products were cloned using New England Biolab PCR cloning kit, and individual clones were analyzed by Sanger sequencing. DNA methylation analysis was performed using Quma (Kumaki et al, 2008) (http://quma.cdb.riken.jp/).
RNA immunoprecipitation
RNA immunoprecipitation experiments were performed as previously described (Rinn et al, 2007) with the following modifications. Two 15‐cm plates with 1.5 × 107 cells each were plated 2 days before the experiment, a pre‐clearing step for 1–2 h at 4°C before the IP was performed with ON blocked beads (BSA 10 mg/ml as 100× and salmon sperm 10 mg/ml as 10×) in PBS or RIP buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP‐40 added with protease inhibitors, PMSF, and RNase inhibitors). One‐fourth of the immunoprecipitated material was tested in Western blot analysis, and the rest was resuspended in TRIzol. Co‐precipitated RNAs were isolated, and qRT–PCR for MS2 loop RNA and U1 RNA was performed as described above. Primer sequences and antibodies used are provided in Appendix Tables S2 and S3.
RNA‐seq analysis
Total RNA from MDA‐MB‐231 cell lines was isolated by TRIzol extraction and quality‐verified by Bionalyzer. Isolated RNA was used to prepare cDNA libraries and amplified with primers containing sequences required for the Illumina platform. PCR products were cleaned and subjected to 100‐bp paired‐end sequencing on an Illumina Hi‐seq 2500. Sequenced reads from duplicate samples were assembled on the human genome hg19, using tophat_2.0.6 (Kim et al, 2013).
The Htseq software (v0.6.0.) was used to define the number of reads associated with each gene. TMM normalization from the edgeR package v3.6.2 (Robinson & Oshlack, 2010) was first applied. As described in the guideline of limma R package v3.20.4, normalized counts were processed by the voom method (Law et al, 2014) to convert them into log2 counts per million with associated precision weights. The differential expression was estimated with the limma package. The P‐values were adjusted for multiple testing using the Benjamini–Hochberg procedure. Finally, differentially expressed genes with a log fold change > 1, FPKM > 1, and adjusted P‐value < 0.05 were used for downstream analysis. Genes' FPKM was estimated using the Cuffquant and Cuffnorm tools of the Cufflinks suite (v2.2.1).
The hierarchical clustering was performed using a Pearson correlation distance and a Ward linkage (R v3.2.0, hclust function).
Baculoviruses production
RBBP4‐Strep‐TAG baculovirus was produced according to the Bac‐to‐Bac Baculovirus Expression Systems (Invitrogen) starting from pFASTbac vectors.
SHAPE‐MaP
SHAPE‐MaP structure probing was performed as described by Smola et al (2015). Refolded RNA was incubated with 10 mM 1M7(+) (1‐methyl‐7‐nitroisatoic anhydride), 10 mM NMIA (+) (N‐methylisatoic anhydride) or an equal amount of pure DMSO as a control (−) for 3 min or 22 min at 37°C, respectively, due to the different half‐lives of the SHAPE reagents. The samples were then purified by G50 columns and subsequently fragmented to obtain 300‐bp RNA fragments. Reverse transcription was then performed in the presence of Mn2+ using SuperScript III reverse transcriptase kit (Invitrogen); finally, samples (+) and (−) were purified using G50 columns. In parallel, an RNA‐denatured sample was treated following the same steps as the (+) samples as a second negative control (−). SHAPE reactions (+) and (−) were then sequenced using an Ion Torrent sequencing platform, and sequencing data were taken into a bioinformatics pipeline to obtain SHAPE reactivities for 1M7 or NMIA for each RNA nucleotide and normalized for DMSO negative control and RNA‐denatured negative control. The bioinformatics script provided by the Weeks laboratory was adapted for Ion Torrent output files by A. Saadi and Y. Ponty (manuscript in preparation).
To generate the HOTAIR secondary structure maps using the software RNAstructure, SHAPE 1M7 reactivity was used to provide pseudo‐energy constraints, while VARNA software was used to visualize the predicted structure (Darty et al, 2009). Resulting structures were manually evaluated for match with NMIA probing data. SHAPE reactivities are listed in the source data for Fig 4C.
Nuclear extract
For nuclear extract preparation, cells were incubated with buffer A (10 mM Hepes pH 7.9, 2.5 mM MgCl2, 0.25 M sucrose, 0.1% NP‐40, 0.5 mM DTT, 1 mM PSMF) for 10 min on ice, centrifuged at 7,000 g for 10 min, resuspended in buffer B (25 mM Hepes pH 7.9, 1.5 mM MgCl2, 700 mM NaCl, 0.5 mM DTT, 0.1 mM EDTA, 20% glycerol), sonicated, and centrifuged at 21,000 g for 15 min.
Data access
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO series accession number GSE72524 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72524).
Author contributions
MP, ZB, RR, AMo, AMi, and MW performed experiments. MP, IV, NS, BS, and RM analyzed data. MP and RM wrote the manuscript. All authors edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Dr. Breathnach Richard for providing the pFLAG_NLS_MS2‐MS2 plasmid, Margaux Charruel for technical help, Armelle Luscan for providing EED and SUZ12 targeting vectors, Anne‐Catherine Dock‐Bregeon for advices on RNA SHAPE, Afaf Saadi and Yann Ponty for help with processing the raw SHAPE‐MaP Data, and Delphine Allouche for advices on the SHAPE‐MaP experiments. We also thank Drs. Edith Heard, Antoine Graindorge, Serena Sanulli, Daniel Holoch, and Mythili Ganapathi for critical reading of the manuscript. Work in RM laboratory is funded by an ERC‐Stg (REPODDID) grant and the Institut National du Cancer (INCa, grant 2012‐1‐PLBIO). High‐throughput sequencing has been performed by the NGS platform of the Institut Curie and supported by the grants ANR‐10‐EQPX‐03 and ANR10‐INBS‐09‐08 from the Agence Nationale de le Recherche (investissements d'avenir) and by the Canceropôle Ile‐de‐France.
The EMBO Journal (2017) 36: 981–994
See also: MR Blanco & M Guttman (April 2017)
References
- Arnold P, Scholer A, Pachkov M, Balwierz PJ, Jorgensen H, Stadler MB, van Nimwegen E, Schubeler D (2013) Modeling of epigenome dynamics identifies transcription factors that mediate Polycomb targeting. Genome Res 23: 60–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran M, Yates CM, Skalska L, Dawson M, Reis FP, Viiri K, Fisher CL, Sibley CR, Foster BM, Bartke T, Ule J, Jenner RG (2016) The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res 26: 896–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N (2013) Noncoding RNA and Polycomb recruitment. RNA 19: 429–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajigas I, Leib DE, Cochrane J, Luo H, Swyter KR, Chen S, Clark BS, Thompson J, Yates JR III, Kingston RE, Kohtz JD (2015) Evf2 lncRNA/BRG1/DLX1 interactions reveal RNA‐dependent inhibition of chromatin remodeling. Development 142: 2641–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, Steitz JA (2014) The noncoding RNA revolution‐trashing old rules to forge new ones. Cell 157: 77–94 [DOI] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA‐chromatin interactions. Mol Cell 44: 667–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY (2015) Systematic discovery of Xist RNA binding proteins. Cell 161: 404–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes‐Rojas C, Hernandez AJ, Sarma K, Lee JT (2014) Regulatory interactions between RNA and Polycomb repressive complex 2. Mol Cell 55: 171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K, Denise A, Ponty Y (2009) VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25: 1974–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Zheng L, Goodrich KJ, Cech TR (2013) Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol 20: 1250–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, Weeks KM (2009) Accurate SHAPE‐directed RNA structure determination. Proc Natl Acad Sci USA 106: 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, Figueroa ME, De Figueiredo Pontes LL, Alberich‐Jorda M, Zhang P, Wu M, D'Alo F, Melnick A, Leone G, Ebralidze KK, Pradhan S, Rinn JL, Tenen DG (2013) DNMT1‐interacting RNAs block gene‐specific DNA methylation. Nature 503: 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ (2004) Nucleosome arrays reveal the two‐start organization of the chromatin fiber. Science 306: 1571–1573 [DOI] [PubMed] [Google Scholar]
- Gesnel MC, Del Gatto‐Konczak F, Breathnach R (2009) Combined use of MS2 and PP7 coat fusions shows that TIA‐1 dominates hnRNP A1 for K‐SAM exon splicing control. J Biomed Biotechnol 2009: 104853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non‐coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464: 1071–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermann P, Hoerner L, Burger L, Schubeler D (2014) Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci USA 111: E3415–E3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantry S, Mills KC, Yee D, Otte AP, Panning B, Magnuson T (2006) The Polycomb group protein EED protects the inactive X‐chromosome from differentiation‐induced reactivation. Nat Cell Biol 8: 195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Son J, Shen SS, Reinberg D, Bonasio R (2013) PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol 20: 1258–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldana‐Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D (2014) Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell 53: 290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keryer‐Bibens C, Barreau C, Osborne HB (2008) Tethering of proteins to RNAs by bacteriophage proteins. Biol Cell 100: 125–138 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A (2004) A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2: E171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziol MJ, Rinn JL (2010) RNA traffic control of chromatin complexes. Curr Opin Genet Dev 20: 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaki Y, Oda M, Okano M (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36: W170–W175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK (2014) Voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol 15: R29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu B, Wapinski OL, Tsai MC, Qu K, Zhang J, Carlson JC, Lin M, Fang F, Gupta RA, Helms JA, Chang HY (2013) Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep 5: 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D (2011) The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya‐Jones A, Blanco M, Burghard C, Moradian A, Sweredoski MJ, Shishkin AA, Su J, Lander ES, Hess S, Plath K, Guttman M (2015) The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE (2010) GC‐rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 6: e1001244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, Lee JT (2015) Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA‐directed chromosome conformation. Science 349: aab2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk‐Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300: 131–135 [DOI] [PubMed] [Google Scholar]
- Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K (2014) Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55: 347–360 [DOI] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol 11: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha ST, Boeva V, Escamilla‐Del‐Arenal M, Ancelin K, Granier C, Matias NR, Sanulli S, Chow J, Schulz E, Picard C, Kaneko S, Helin K, Reinberg D, Stewart AF, Wutz A, Margueron R, Heard E (2014) Jarid2 is implicated in the initial Xist‐induced targeting of PRC2 to the inactive X chromosome. Mol Cell 53: 301–316 [DOI] [PubMed] [Google Scholar]
- Rutenberg‐Schoenberg M, Sexton AN, Simon MD (2016) The Properties of long noncoding RNAs that regulate chromatin. Annu Rev Genomics Hum Genet 17: 69–94 [DOI] [PubMed] [Google Scholar]
- Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M, Michaud A, Lombard B, da Rocha ST, Offer J, Loew D, Servant N, Wassef M, Burlina F, Gamblin SJ, Heard E, Margueron R (2015) Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol Cell 57: 769–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz SU, Grote P, Herrmann BG (2016) Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci 73: 2491–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorderet P, Duboule D (2011) Structural and functional differences in the long non‐coding RNA hotair in mouse and human. PLoS Genet 7: e1002071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried NA, Busan S, Rice GM, Nelson JA, Weeks KM (2014) RNA motif discovery by SHAPE and mutational profiling (SHAPE‐MaP). Nat Methods 11: 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Kingston RE (2009) Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10: 697–708 [DOI] [PubMed] [Google Scholar]
- Sing A, Pannell D, Karaiskakis A, Sturgeon K, Djabali M, Ellis J, Lipshitz HD, Cordes SP (2009) A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell 138: 885–897 [DOI] [PubMed] [Google Scholar]
- Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM (2015) Selective 2′‐hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE‐MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc 10: 1643–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somarowthu S, Legiewicz M, Chillon I, Marcia M, Liu F, Pyle AM (2015) HOTAIR forms an intricate and modular secondary structure. Mol Cell 58: 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Cowan CA, Chipperfield H, Powers RD (2005) Gene expression in the preimplantation embryo: in‐vitro developmental changes. Reprod Biomed Online 10: 607–616 [DOI] [PubMed] [Google Scholar]
- Wassef M, Rodilla V, Teissandier A, Zeitouni B, Gruel N, Sadacca B, Irondelle M, Charruel M, Ducos B, Michaud A, Caron M, Marangoni E, Chavrier P, Le Tourneau C, Kamal M, Pasmant E, Vidaud M, Servant N, Reyal F, Meseure D et al (2015) Impaired PRC2 activity promotes transcriptional instability and favors breast tumorigenesis. Genes Dev 29: 2547–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CJ, Kharchenko PV, Daheron L, Park PJ, Kingston RE (2010) A region of the human HOXD cluster that confers polycomb‐group responsiveness. Cell 140: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5