

Abstract
The IL-1β induced activation of the p38MAPK/MAPK-activated protein kinase 2 (MK2) pathway in hepatocytes is important for control of the acute phase response and regulation of liver regeneration. Many aspects of the regulatory relevance of this pathway have been investigated in immune cells in the context of inflammation. However, very little is known about concentration-dependent activation kinetics and signal propagation in hepatocytes and the role of MK2. We established a mathematical model for IL-1β-induced activation of the p38MAPK/MK2 pathway in hepatocytes that was calibrated to quantitative data on time- and IL-1β concentration-dependent phosphorylation of p38MAPK and MK2 in primary mouse hepatocytes. This analysis showed that, in hepatocytes, signal transduction from IL-1β via p38MAPK to MK2 is characterized by strong signal amplification. Quantification of p38MAPK and MK2 revealed that, in hepatocytes, at maximum, 11.3% of p38MAPK molecules and 36.5% of MK2 molecules are activated in response to IL-1β. The mathematical model was experimentally validated by employing phosphatase inhibitors and the p38MAPK inhibitor SB203580. Model simulations predicted an IC50 of 1–1.2 μm for SB203580 in hepatocytes. In silico analyses and experimental validation demonstrated that the kinase activity of p38MAPK determines signal amplitude, whereas phosphatase activity affects both signal amplitude and duration. p38MAPK and MK2 concentrations and responsiveness toward IL-1β were quantitatively compared between hepatocytes and macrophages. In macrophages, the absolute p38MAPK and MK2 concentration was significantly higher. Finally, in line with experimental observations, the mathematical model predicted a significantly higher half-maximal effective concentration for IL-1β-induced pathway activation in macrophages compared with hepatocytes, underscoring the importance of cell type-specific differences in pathway regulation.
Keywords: cell signaling, computational biology, hepatocyte, liver, macrophage, mathematical modeling, MAPK, p38 MAPK, systems biology, interleukin 1β
Introduction
MAPK cascades are integral components of signaling networks that are crucial for translating extracellular stimuli into a wide range of cellular responses (1). In particular, p38MAPK is activated in response to environmental stress, pathogen-associated molecular patterns, DNA damage, and inflammatory cytokines such as IL-1β via phosphorylation of threonine and tyrosine residues located at positions 180 and 182, respectively (2). p38MAPK, in turn, triggers a variety of different downstream targets, including the two members of the MAPK-activated protein kinase (MAPKAP or MK) MK24 and MK3 (3). p38MAPK-driven processes play a critical role in the physiology of several organs (4) and in the pathogenesis of acute and chronic diseases (5–8). Because of its crucial role in the regulation of inflammatory cytokine expression, in particular signaling via the p38MAPK/MK2 pathway module, it is increasingly recognized as a therapeutic target for the treatment of inflammatory diseases (9–11).
So far, the p38MAPK pathway has been primarily studied in the context of immune cells, but because this pathway is literally expressed in all organs of mammals, it remains to be addressed whether context-specific differences in the extent of pathway activation exist. In the liver, the p38MAPK pathway is involved in the regulation of the acute phase response (12) and plays a role in the coordinated progression of other processes that are largely controlled via inflammatory reactions. Additionally, it has been proposed that activation of p38MAPK contributes to termination of liver regeneration (13) by inhibiting cell cycle progression, DNA replication, and hepatocyte proliferation (13–15). Although the role of the p38MAPK/MK2 pathway in regulating the functions of macrophages has been studied in much detail, information regarding its specific role in regulating responses of hepatocytes during acute phase reaction and liver regeneration is rather limited. In particular, knowledge of the function of MK2 in hepatocytes is sparse. Accordingly, there is almost no information regarding the stimuli that trigger MK2 activation in hepatocytes, nor there is knowledge of the kinetics of MK2 activation and of target genes that are MK2-dependently controlled in hepatocytes. The data available so far indicate that activation of the p38MAPK/MK2 pathway in hepatocytes is involved in the regulation of acute phase protein synthesis in response to IL-6 (16) and is critical for the cross-regulation of IL-6-type cytokine signaling in hepatocytes by IL-1β (17). In particular, activation of MK2 mediates the inhibitory effects of IL-1β on surface expression of the signal-transducing subunit of the IL-6 receptor complex gp130 (18), which is central for IL-6-type cytokine signal transduction and regulation of the expression of acute phase proteins (12). Apart from this, it was reported recently that oncostatin M (OSM) activates MK2 in hepatocytes and that MK2 is involved in regulation of the expression of the oncostatin M receptor transcript in hepatocytes (19).
Based on a systems biology approach, this work provides an extensive quantitative analysis of the IL-1β-dependent activation kinetics of the p38MAPK/MK2 pathway in hepatocytes and reports the establishment of a mathematical model of the p38MAPK/MK2 pathway in hepatocytes for in silico analysis. The mathematical model is validated by time-resolved quantitative data on concentration-dependent activation of the p38MAPK/MK2 signaling module by IL-1β in primary mouse hepatocytes. Of note, comparative analysis of the cellular concentrations of p38MAPK and MK2 between hepatocytes and macrophages suggests that there are substantial differences between these two cell types with respect to the concentrations of p38MAPK and MK2 and the cell type-specific responsiveness toward IL-1β.
Results
Determination of cell volume and quantification of intracellular p38MAPK and MK2 in primary mouse hepatocytes
To establish a procedure to estimate the intracellular concentration of p38MAPK and MK2 molecules in hepatocytes, first the cellular volume of primary mouse hepatocytes was determined. Live-cell fluorescence microscopy was used to obtain optical slices that were analyzed in 40 independent measurements (supplemental Fig. S1A). Based on this, 15.475 ± 4.044 pl (S.D.) was determined as the total volume of primary mouse hepatocytes. The applied approach was validated by measurements on fluorescent microspheres (FocalCheck, Molecular Probes). The measured diameter of the microspheres was 15.25 ± 0.36 μm (S.D.), which closely agrees with the specifications of the manufacturer (15.4 μm). Accordingly, the calculated volume of the microspheres (V = πd3 = 1.912 pl) was in agreement with the measured volume of the microspheres of 1.903 ± 0.0554 pl (S.D.), thereby validating our approach to determine the volume of hepatocytes.
The intracellular concentrations of total p38MAPK, total MK2, and the phosphorylated variants, respectively, were assessed for wild-type hepatocytes as well as for MK2-deficient hepatocytes by quantitative immunoblotting using a co-separated internal standard with known quantities of MK2 and p38MAPK and a known total degree of phosphorylation of the activation motif of these molecules (supplemental Fig. 1, B–F). The internal standard was calibrated based on recombinantly expressed and in vitro phosphorylated GST-tagged p38MAPK and MK2, respectively. The global phosphorylation degree of the activation motif in both GST fusion proteins was determined by mass spectrometry. Fig. 1 exemplarily represents the procedure we applied to quantify phosphorylated p38MAPK molecules. In total, 18 independent experiments (biological replicates) in multiple technical replicates were performed, and 1676 data points on the time-resolved and concentration-dependent activation of the p38MAPK/MK2 signaling module in primary mouse hepatocytes in response to IL-1β were generated to calibrate and validate our mathematical model for the p38MAPK/MK2 pathway in hepatocytes.
Figure 1.

Quantification of p38MAPK phosphorylation in primary mouse hepatocytes. A, immunoblotting of whole cell lysates in co-separation with an internal standard affords the quantification of intracellular phosphorylated p38MAPK molecule concentration. Primary mouse hepatocytes were treated with 0, 0.1, 0.5, and 1 ng·ml−1 IL-1β, respectively, for up to 60 min. The cells lysed in Triton lysis buffer and specimen were applied randomly to SDS-PAGE and subsequent Western blotting in co-separation with a dilution series (1, 5, 10, 20, and 40 μl) of an internal standard (int). After incubation with specific antibodies, the nitrocellulose membrane was cut in half to fit on the documentation screen, and chemiluminescence was captured at a 16-bit charge-coupled device. B and C, quantitative determination of IL-1β-induced p38MAPK phosphorylation kinetics. The intensity of the chemiluminescent signals was quantified, and, in consideration of cell volume and the internal standard, the intracellular concentration of p38MAPK phosphorylation as a function of IL-1β and time was quantified. a. u.: arbitrary units.
ODE model formulation and calculation of time- and dose-dependent IL-1β induced p38MAPK and MK2 activation
Based on the law of mass action and custom kinetic rate laws, a set of nonlinear ordinary differential equations (ODE) was formulated, describing the IL-1β-dependent p38MAPK and MK2 phosphorylation and its negative feedback in mouse hepatocytes (Fig. 2 and supplemental Information S2), consisting of six reaction rates that constitute an ODE system with five equations. The complexity of the model was chosen so that only the essential features of the pathway that are important for this study are included while maintaining an adequate description of the experimental data. Some simplifications were possible to make the system computationally more tractable and to increase the number of identifiable parameters.
Figure 2.
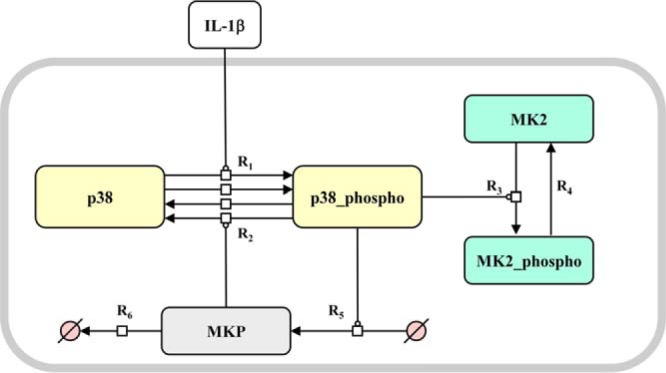
ODE model of IL-1β-dependent p38MAPK/MK2 activation. A, mathematical model of IL-1β-dependent p38MAPK/MK2 activation. Shown is a graphical representation of the ODE model of IL-1β-dependent p38MAPK and MK2 signal transduction. R1, basal and IL-1β-induced activation (Thr-180/Tyr-182 phosphorylation) of p38MAPK; R2, dephosphorylation of p38MAPK by MKP; R3, activation (Thr-222 phosphorylation) of MK2 by activated p38MAPK; R4, dephosphorylation of MK2; R5, activated p38MAPK induces the expression of MKP; R6, degradation of MKP. The reaction rates of the system are forming a five-dimensional ODE.
On that account, the IL1R complex comprising TAK1, Myd88, interleukin-1 receptor-associated kinase (IRAK), TAB1 and 2, and MKK6 were summarized as IL-1β-mediated p38MAPK activation (phosphorylation) (Fig. 2, R1). To describe basal activation, we introduced the parameter p38_act_basal (R1), which is linked to the basal activation (phosphorylation) of p38MAPK independent of IL-1β. Furthermore, because of the lack of specificity of phosphatases, the contributing phosphatases in the model were summarized. The system is down-regulated by MAPK phosphatases (MKP) whose transcription is activated by phosphorylated p38MAPK (R5). MKP deactivates (dephosphorylates) the activated p38MAPK (R2). This leads to a negative feedback. MKP is degraded with a linear rate (R6). The MAPKAP kinase MK2 is activated (phosphorylated) by activated p38MAPK (R3). Because of basal phosphatase activity, p38MAPK and MK2 are dephosphorylated with a linear rate (R4 and R2).
To calculate the absolute concentration of the key systems components, the unknown calibration parameter values of the internal standard were simultaneously estimated with the mathematical model. Using this approach, the total intracellular concentration of p38MAPK (359 nm, i.e. 3.35 × 106 molecules/cell) and MK2 (4637 nm, i.e. 4.32 × 107 molecules/cell) in primary mouse hepatocytes was determined. Under basal conditions (0 ng·ml−1 IL-1β), only 0.36% (1.3 nm) of all p38MAPK molecules was activated, as indicated by the determination of the activating phosphorylation of p38MAPK at Thr-180/Tyr-182, and 1.9% (85.6 nm) of the MK2 molecules was phosphorylated on Thr-222 under the respective control conditions. Treatment of mouse hepatocytes with IL-1β resulted in a dose-dependent transient activation of p38MAPK and MK2 with a peak at 12–20 min, and saturation of the activation was achieved at IL-1β concentrations above 20 ng·ml−1 (Figs. 3 and 4). IL-1β induced activation of p38MAPK, and MK2-mediated signal transduction was accompanied by significant signal amplification because 57 fm IL-1β results in an increase in p38MAPK phosphorylation at Thr-180/Tyr-182 of 20,960 fm, and this subsequently leads to an increase in MK2 phosphorylation at Thr-222 of 1,063,000 fm (Fig. 4).
Figure 3.

Quantitative determination of IL-1β-induced p38MAPK and MK2 phosphorylation kinetics. Primary mouse hepatocytes were treated with 0–40 ng·ml−1 IL-1β for up to 60 min. Whole cell lysates were applied to Western blotting analysis with a serial dilution of an internal standard, and phosphorylation of p38MAPK at Thr-180/Tyr-182 as well as phosphorylation of MK2 at Thr-222 was detected by respective phosphorylation-specific antibodies. Signal intensities were determined and converted to intracellular concentrations. The solid lines indicate the fit to the mathematical model based on ordinary differential equations, whereas symbols indicate the respective experimental data. In total, the experimental data used for model fitting are from 18 independent experiments (biological replicates). Each experiment was done in up to three technical replicates to account for measuring inaccuracy.
Figure 4.

Simulation of the p38MAPK and MK2 phosphorylation kinetics as a function of IL-1β concentration and time. A and B, simulations based on the fitted model illustrate the time- and dose-dependent kinetics of IL-1β-dependent p38MAPK (A) and MK2 (B) activation, where the intrinsic signal amplification is reflected. Signal amplification is roughly IL-1β concentration (conc)-independent, and signal transduction occurs under minimal deformation.
Parameter estimation and identifiability
In total, 169 parameter values were estimated from 1676 data points (see supplemental Information S2 for details regarding parameter estimation, the estimated parameter values, and the data used for the parameter estimation). To prevent convergence to possible local optima, 1000 parameter estimation runs with different initial parameter guesses were performed (20). More than 650 runs directed to the same optimum, which is a strong indication that the global optimum was identified. In the remaining 350 runs, several local minima were identified at likelihood values that are significantly worse than the global optimum. Therefore, they were neglected for further investigations.
To reliably study the systems behavior in silico, it is important to establish a structurally and practically identifiable set of parameter values (21). To investigate the identifiability and confidence intervals of the parameters, the likelihood profile was calculated (Fig. 5). The analysis revealed that, except for the parameter p38_dea_const, all parameters are identifiable. The parameter profile of the parameter p38_dea_const suggests that the constant dephosphorylation rate of p38MAPK is negligible compared with the MKP-induced dephosphorylation. For model simulations predicting the differential contributions of signal-inducing kinases and signal-terminating phosphatases (Fig. 6, A and C), a parameter set was used that was obtained by excluding experiments in which inhibitors were applied.
Figure 5.

Parameter likelihood profiles. Likelihood profiles of the dynamic model parameters (dePhospho_inh, il1b_sat, init_MK2, init_p38, mk2_act_p38, mk2_dea, mk2_inh_sb, mkp_prod_pp38, p38_act_basal, p38_act_il1b, p38_dea_const, and p38_dea_mkp). Black lines represent the profile likelihood. The parameter set of the optimum is indicated with a green dot. Red lines indicate the 95% confidence level. Blue lines indicate the global minimum of the parameter estimation process. The analysis revealed that all model parameters are structurally identifiable. The parameter p38_dea_const is practically non-identifiable. The parameter profile suggests that the parameter is compatible with zero; i.e. the constant dephosphorylation rate of p38MAPK is not relevant for our system.
Figure 6.

In silico analysis (predictions) and experimental validation. A and C, contribution of kinases and phosphatases to signal amplitude and duration. Simulations of parameter perturbations that are reflecting changes in kinase and phosphatase activity are showing different effects on MK2 phosphorylation. The perturbation of the kinases was simulated by changing the parameter mk2_act_p38 by a factor indicated in the figure. This has mainly an effect on the signal amplitude of MK2 phosphorylation. The perturbation of the phosphatases was modeled by changing the parameter p38_dea_MKP. This affects both signal amplitude and duration of MK2 phosphorylation. B, SB203580 inhibited IL-1β induced MK2 phosphorylation. Primary mouse hepatocytes were treated with the p38MAPK inhibitor SB203580 for 30 min prior to treatment with 1 ng·ml−1 IL-1β. Whole cell lysates were applied to Western blotting analysis with a serial dilution of an internal phosphostandard, and phosphorylation of MK2 at Thr-222 was detected by specific antibodies. Signal intensities were determined and converted to intracellular concentrations. The data were fitted to the mathematical model (solid lines). D, inhibition of p38MAPK and MK2 dephosphorylation. Primary mouse hepatocytes were treated with 200 μm sodium orthovanadate and 200 μm β-glycerophosphate for inhibition of phosphatases prior to treatment with 1 ng·ml−1 IL-1β. Whole cell lysates were applied to Western blotting analysis, phosphorylation of MK2 at Thr-222 (B) was detected by specific antibodies, and the signal intensities were determined. The data were fitted to the mathematical model (solid lines).
Signal amplification across the p38MAPK/MK2 cascade with minimal deformation
In silico simulations based on the complete dataset revealed the time- and dose-dependent kinetics of IL-1β-dependent p38MAPK and MK2 phosphorylation. This analysis showed that an intrinsic signal amplification of up to 60-fold occurs (Fig. 4, compare A and B). Signal amplification is to a large extent IL-1β concentration-independent, and signal transduction occurs under minimal deformation from p38MAPK to MK2 in terms of time and concentration dependence.
In silico studies suggest a distinct contribution of signal-inducing kinases and signal-terminating phosphatases to signal amplitude and duration
Transient p38MAPK and MK2 signal activation are overall effects of both kinase and phosphatase activity. To characterize their distinct contribution to signal propagation, model simulations for different perturbation conditions were performed using the parameter set obtained by excluding perturbation experiments from the complete dataset as described above. Specifically, the parameter mk2_act_p38 was varied to characterize the contribution of kinases to signal progression. This analysis suggested that kinases mainly affect signal amplitude and only to a lesser extent the signal duration of IL-1β-induced activation of p38MAPK and MK2 (Fig. 6A), respectively. On the other hand, modulation of the parameter p38_dea_mkp was used to assess the contribution of phosphatases to signal progression. The resulting model simulations revealed that phosphatases modulate the activation kinetics. The maxima are reached with a significant delay upon both signal amplitude and duration of IL-1β-induced p38MAPK activation and MK2 activation (Fig. 6C), respectively.
Experimental validation shows that p38MAPK controls signal amplitude rather than signal duration, whereas phosphatases control both signal amplitude and signal duration of p38MAPK-mediated MK2 activation
To validate the in silico analyses, primary mouse hepatocytes were treated with the p38MAPK-specific inhibitor SB203580 prior to IL-1β treatment. Perturbation of signal transduction through inhibition of p38MAPK activity by SB203580 resulted in a concentration-dependent down-regulation of IL-1β-induced MK2 phosphorylation. Therefore, the signal duration remained unaffected (Fig. 6B). For primary mouse hepatocytes, the model predicted an IC50 of 1.127 μm for SB203580 (Fig. 7). Treatment of primary mouse hepatocytes with a combination of the phosphatase inhibitors sodium orthovanadate and β-glycerophosphate prior to IL-1β treatment particularly resulted in sustained phosphorylation of p38MAPK and MK2, which was enhanced and prolonged (Fig. 6D). These results confirm the predictions from the in silico analyses, indicating that kinase activity has an impact on signal amplitude rather than signal duration and that the phosphatase activity controls signal duration and amplitude.
Figure 7.
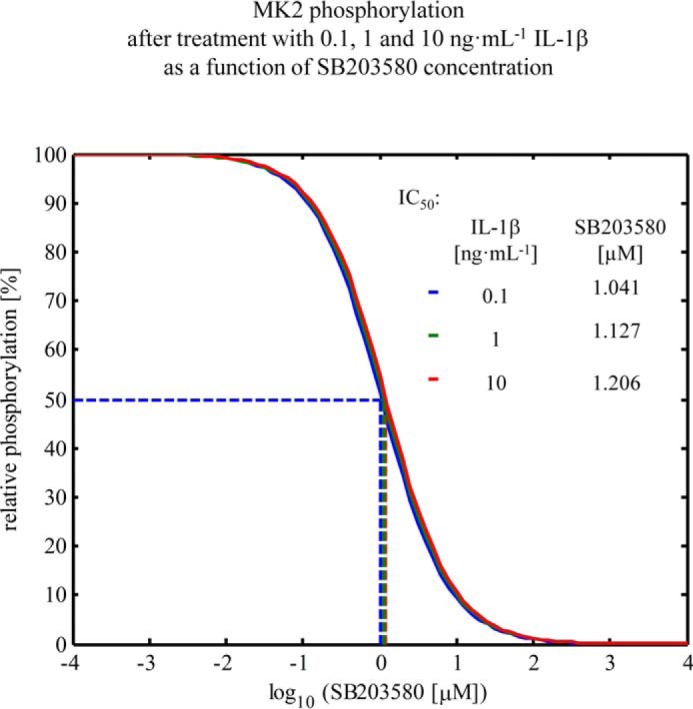
Inhibition of MK2 phosphorylation by SB203580. Down-regulation of IL-1β-induced MK2 phosphorylation by applying the inhibitor SB203580, as simulated with the model for the time point 20, and treatment with different concentrations of IL-1β. At 1.127 μm SB203580, the amount of phosphorylated MK2 is around 50% of the original amount of phosphorylated MK2 without SB203580. This does not depend on the used IL-1β concentration.
The concentration of p38MAPK and MK2 as well as the responsiveness toward IL-1β significantly differs between hepatocytes and macrophages
As mentioned above, the regulatory relevance of the p38MAPK/MK2 pathway, and in particular of MK2, has been investigated mainly in immune cells. Likewise, systems biology-based analyses of this pathway only exist for immune cells such as macrophages but not for hepatocytes. To elucidate potential differences between macrophages and hepatocytes, comparative analyses of the concentration of p38MAPK and MK2 were performed in primary mouse hepatocytes and bone marrow-derived macrophages. As depicted in Fig. 9A, the p38MAPK concentration in macrophages (1.4 μm) is significantly higher compared with hepatocytes (0.36 μm). The concentration of MK2 in macrophages (13.2 μm) is around three times higher than in hepatocytes (4.6 μm). Although, in hepatocytes, a stimulus with 1 ng·ml−1 IL-1β resulted in more than 5-fold increased activation of the p38MAPK/MK2 pathway compared with the basal phosphorylation level (Fig. 9A, right panels), the analysis of the phosphorylated p38MAPK and MK2 in macrophages showed that a strong pathway induction only occurs in response to higher doses of IL-1β. To delineate potential cell type-specific differences in the kinetics of signal transmission via the p38MAPK/MK2 pathway, time course experiments for both cell types with different IL-1β inputs were performed (Fig. 9B). The quantitative data obtained for hepatocyte could be described by the mathematical model established for activation of the p38MAPK/MK2 pathway in hepatocytes and thereby further validated the model. On the other hand, the determinations in macrophages were used to recalibrate the mathematical model for quantitative characterization of the activation kinetics of the pathway observed in macrophages. With the two calibrated models, it was possible to calculate the cell type-specific responsiveness of p38MAPK and MK2 to different IL-1β doses (Fig. 9C). For macrophages, the half-maximal effective concentration of IL-1β for p38MAPK and MK2 phosphorylation, respectively, is significantly higher than in hepatocytes, indicating that there are substantial cell type-specific differences in the responsiveness of hepatocytes and macrophages toward IL-1β.
Figure 9.

Comparative analyses between hepatocytes and macrophages. A, absolute concentrations. The concentrations of total and phosphorylated p38MAPK and MK2 were measured with the internal standard for different durations and concentrations of IL-1β treatment of macrophages (red) and hepatocytes (gray), respectively. The total concentrations are significantly higher in macrophages in comparison with hepatocytes. The criteria for significance were a two-way analysis of variance and post hoc pairwise comparisons of each IL-1β treatment condition of hepatocytes and macrophages, respectively, based on multiple two-tailed t tests. The p values were adjusted according to Bonferroni to account for the multiple comparisons problem. ****, p < 0.0001. B, kinetics. The kinetics of phosphorylated MK2 and p38MAPK were measured in macrophages and hepatocytes for different IL-1β inputs (points). For hepatocytes, the solid lines indicate the prediction of the calibrated model. For macrophages, the mathematical model was fitted to the measured macrophage data (solid line). C, model prediction of dose responses. Shown is relative phosphorylation dependent on the IL-1β input dose as predicted with the calibrated models. The bands show the prediction uncertainty calculated by integrating all parameter sets along the corresponding profile likelihoods.
Discussion
The p38MAPK pathway is considered to be important for the pathogenesis of a variety of diseases. Therefore, it plays a role as a potential therapeutic target, with implications in inflammation, acute phase response, liver regeneration, cell differentiation, cell growth, cell death, senescence, and tumorigenesis. In the context of liver regeneration, the p38MAPK pathway contributes to coordination of the termination of liver regeneration. Over 20 different p38MAPK inhibitors have entered clinical trials (22, 23). However, little is known about the intracellular abundance and activation kinetics of p38MAPK and its downstream target MK2 in hepatocytes.
The aim of our work was to elucidate the intracellular concentration as well as the activation kinetics of p38MAPK and its downstream target MK2 induced by IL-1β in hepatocytes and to exemplify existing concepts of signal amplification (24), signal deformation (25), and signal amplitude and duration (26, 27) to provide a mathematical model of the p38MAPK/MK2 pathway in the context of acute phase response and liver regeneration. The experimental data and the model analysis presented indicate that primary mouse hepatocytes have a cell volume of 15.475 ± 4.044 pl (S.D.) and that they comprise about 12.9 times higher concentrations of MK2 than p38MAPK (4637 versus 359 nm) (Fig. 8). The data indicate that the relationship of molecules that are already activated under basal conditions differs 65.8-fold between p38MAPK and MK2 (1.3 versus 85.6 nm). This indicates that, in primary mouse hepatocytes under basal conditions, a relative small proportion of activated p38MAPK molecules (1.32 nm) maintains a considerable higher activation state of MK2 (85.5 nm). Importantly, the data and the mathematical model provide evidence that, in primary hepatocytes, at maximum only 11.3% of all p38MAPK but 36.5% of all MK2 molecules contribute to intracellular signal transduction induced by IL-1β.
Figure 8.

Schematic of the signal amplification within the IL-1β-induced p38MAPK and MK2 pathway in hepatocytes. Under basal conditions, only a small fraction of p38MAPK and MK2 is activated, i.e. 1.3 nm of 359 nm and 85.6 nm of 4637 nm, respectively. After treatment with up to 2.3 nm IL-1β, under saturating conditions, the concentration of phosphorylated p38MAPK and MK2 rises transiently to 40.6 and 1693 nm. Under subsaturated conditions, e.g. 57 pm, IL-1β induces an activation of approximately 21 nm p38MAPK via the IL1 receptor complex. This subsequently induces an activation of 1063 nm MK2.
To our knowledge, this is the first report providing quantitative and time-resolved data on p38MAPK and MK2 and their activating phosphorylation in primary hepatocytes. Likewise, an applied quantitative mathematical model describing signal transduction of IL-1β via the p38MAPK/MK2 pathway in primary hepatocytes has so far not been published. In the context of LPS-mediated p38MAPK signal transduction and TNFα production in monocytes in the context of rheumatoid arthritis, quantitative data on the intracellular concentrations of p38MAPK and MK2 have been provided (28–30). In these reports, the concentration of p38MAPK was ∼4 μm in the monocytic cell line U937, >1 μm in Thp1 cells, and >0.3 μm in primary peripheral blood mononuclear cells, whereas the concentrations of MK2 were >6 μm, >1 μm, and >1 μm in U937, Thp1, and peripheral blood mononuclear cells, respectively. In our study, we observed a p38MAPK concentration of 1.4 μm and a MK2 concentration of 13.2 μm in primary murine bone marrow-derived macrophages. These data suggest a similar ratio of MK2/p38MAPK between the different monocytic cell types investigated in the abovementioned reports and of the MK2/p38MAPK ratio described in our study for primary bone marrow-derived macrophages. However, the observation that there are significant differences in the concentrations of p38MAPK and MK2 between monocyte and primary hepatocytes, as demonstrated in this study, clearly indicates that there are substantial cell type-specific differences with respect to the concentrations of the different molecules, underlining the need for a more differentiated view on cell type and context. This consideration is further supported by the results of our model-based analyses indicating that the half-maximal effective concentration for the IL-1β-inducible activation of MK2 in hepatocytes is at least three times lower than in macrophages (Fig. 9C). In the future, it will be of interest to elucidate the contribution of distinct intracellular concentrations of p38MAPK and MK2 to functional differences between hepatocytes and macrophages. In addition, and because it is known that p38MAPK and MK2 shuttle between different subcellular compartments, such as the nucleus and cytoplasm, during their activation process, it would be interesting to introduce cellular compartmentation in the recent model. However, so far little is known about the subcellular specificity of the phosphorylation and dephosphorylation reactions and the parameters of the molecular fluxes between the different compartments, leaving these considerations beyond the scope of this study.
By our mathematical modeling approach, we identified signal amplification, robustness against signal deformation and determinants for signal amplitude and duration as key properties of the IL-1β-induced activation of the p38MAPK/MK2 pathway in primary hepatocytes. Koshland et al. (24) combined existing concepts of signal amplification in biological systems to two forms, i.e. magnitude amplification and sensitivity amplification, respectively, to counter redundancy and ambiguity in the diversity of definitions. Magnitude amplification occurs whenever the output molecules are produced in far greater numbers than the stimulus molecules. Sensitivity amplification deals with the relative change in a response compared with the stimulus (24). The IL-1β-induced and p38MAPK-mediated MK2 phosphorylation in hepatocytes mainly accounts for magnitude amplification. Magnitude amplification could be demonstrated by data-driven in silico studies based on quantitative immunoblotting measurements. 57 fm IL-1β results in the phosphorylation (activation) of 20,960 pm p38MAPK and, subsequently, in the phosphorylation of 1,063,000 pm MK2. Signal transmission from p38MAPK to MK2 results in a 40- to 60-fold amplification of the signal, which is roughly independent from the IL-1β concentration applied and closely fits the experimental data. Signal amplification also persists upon inhibition of phosphatase and kinase activity. Grubelnik et al. (25) pointed out that biological amplification cascades show a high similarity to electrical engineered amplification assemblies and play a crucial role in signal transduction systems with high amplification rates and fast input-to-output signals, which preserve the form of input signals without or with minimal deformation. This is in line with this work. Signal amplification across the p38MAPK/MK2 cascade in hepatocytes is to a large extent IL-1β concentration-independent, and signal transduction occurs under minimal deformation from p38MAPK to MK2 in terms of time and concentration dependence. Heinrich et al. (27) found by mathematical theory and Hornberg et al. (26) found by experiments on ERK phosphorylation in normal rat kidney fibroblasts, respectively, that phosphatases have a more pronounced effect than kinases on the rate and duration of signaling, whereas signal amplitude is controlled primarily by kinases. In our study, we acknowledge these findings by experimental data and mathematical modeling for signal transduction across the p38MAPK/MK2 cascade in hepatocytes and provide ascertained parameter values that can be linked to other mathematical models in the respective context of acute phase reaction and liver regeneration.
The induction, dynamics, and termination of a signal or signal transduction pathway are the result of complex processing and integration of synergistic and/or antagonistic regulatory patterns provided by pathways that act in parallel or in series (31). Therefore, the kinetic features of its activation, the amplitude and the duration of a signal, and not only the bare events of activation or deactivation determine its biological impact. Factors that further characterize a signaling pathway and determine the possibility to influence its activation kinetics are the concentration and the availability of signaling molecules involved and the question whether signal amplification occurs within a defined signaling module. To sufficiently address these points, signaling pathways should be regarded as quantitative dynamic systems. This requires knowledge of the concentration and the availability of signaling molecules within the respective cell type.
Experimental procedures
Materials
Primary antibodies were obtained from Cell Signaling Technology (p38 MAPK, phospho-p38 MAPK (Thr-180/Tyr-182), MAPKAPK-2, and phospho-MAPKAPK-2 (Thr-222)). Secondary antibodies were obtained from Dako (Glostrup, Denmark).
Cell culture
Isolation and culture of primary mouse hepatocytes were performed according to the standard operating procedures described previously (32). The use of mice for hepatocyte isolation was approved by the local animal care committees. Animals were handled and housed according to specific pathogen-free conditions.
Briefly, primary mouse hepatocytes were isolated from the livers of 8- to 12-week-old C57BL/6 wild-type or MK2-deficient (33) male mice by collagenase perfusion of the liver, subjected to centrifugal separation, and plated on collagen-coated dishes (34–37). 106 hepatocytes/6-cm dish were cultivated in William's medium E complemented with 10% FCS, 2 mm l-glutamine, 100 mg·ml−1 penicillin, 100 mg·ml−1 streptomycin, and 100 nm dexamethasone. After 3 h, the culture was rinsed with PBS to remove debris. Hepatocytes were cultivated in FCS-free medium overnight. 3 h prior to the respective experimental procedure, cells were rinsed with PBS and cultivated in dexamethasone-free medium. For concentration and time course experiments, cells were treated with 0–40 ng·ml−1 IL-1β and 0–5 μm of the p38MAPK inhibitor SB203580 as outlined in the respective figure legends.
Murine bone marrow-derived macrophages were prepared, characterized, and cultivated as described previously (38). Cells were seeded at 1.5 × 106 cells/dish on 6-cm dishes.
All cells were maintained in a 5% CO2 humidified atmosphere at 37 °C. Cells were lysed in Triton lysis buffer (136 mm NaCl, 20 mm Tris-HCl, 10% glycerol, 2 mm EDTA, 50 mm β-glycerophosphate, 20 mm sodium pyrophosphate, 0.2 mm Pefabloc, 5 μg·ml−1 aprotinin, 5 μg·ml−1 leupeptin, 4 mm benzamidine, 1 mm sodium orthovanadate, and 1% Triton X-100 (pH 7.4)). The protein concentration was assayed using Bradford reagent.
Cloning, expression, and purification of GST-p38MAPK and GST-MK2
Total RNA was isolated and transcribed in cDNA by using the Transcriptor First Strand cDNA synthesis kit (Roche). cDNA was used to amplify the transcript encoding murine p38 and murine MK2. Amplicons were cloned into the pGEX-6P-3 expression vector system (GE Healthcare). Vectors were transformed into Escherichia coli BL21 cells (GE Healthcare) for protein expression. The recombinant proteins were purified by affinity chromatography using glutathione-Sepharose 4B (GE Healthcare). The identity of the proteins was validated, and the concentration was assessed by immunoblotting applying monoclonal antibodies, Coomassie Brilliant Blue-stained SDS-PAGE, and mass spectrometric analysis.
In vitro phosphorylation of recombinant proteins
Purified recombinant GST-p38MAPK was incubated for 30 min in the presence of constitutively active human MKK6 (Sigma), whereas, for in vitro phosphorylation of recombinant GST-MK2, constitutively active human p38MAPK (R&D Systems, Minneapolis, MN) was used. Incubation was performed in a buffer containing 20 mm Hepes, 60 mm NaCl, 2 mm MgCl2, 5 mm EGTA, and 100 mm ATP according to the recommendations of the manufacturer.
Protein identification was validated by mass spectrometry and subsequent analysis by MASCOT (Matrix Science, London, UK). Assessment of protein concentration was performed by Coomassie Brilliant Blue staining following SDS-PAGE and co-separation of a serial dilution of purified bovine serum albumin of known concentration (supplemental Fig. S1, C and D). Mass spectrometric analysis was performed using a direct coupling of nanoUPLC (nanoAcquity, Waters) with an Orbitrap XL mass spectrometer (Thermo). Data evaluation for measurement of the degree of phosphorylation of p38MAPK from these data was performed as described in detail elsewhere (39).
Immunoblot analysis and establishment of an internal standard
For the detection of phosphorylated p38MAPK, total p38MAPK, phosphorylated MK2, and total MK2, 50 μg of whole cell lysate was subjected to SDS-PAGE. For the detection of large-scale series, a customized electrophoresis system for a maximum of 43 lanes/gel was used, and gels were transferred to a nitrocellulose membrane. Immunoblots were incubated with the respective antibodies. Proteins were detected by chemiluminescence (Amersham Biosciences ECL Plus, GE Healthcare) on a Kodak Image Station 4000 MM, and then signal intensity was quantified. To avoid systematic blotting errors, probes were randomly applied to SDS-PAGE, and values were rearranged using the MATLAB-based tool Gelinspector (40).
The immunoblots were quantified using an internal standard with known quantities of total p38MAPK and MK2 and a known total degree of phosphorylation of the activation motif of p38MAPK (Thr-180/Tyr-182) and MK2 (Thr-222), which was co-separated on each gel as a dilution series. Aliquots of this internal standard were established from a total protein lysate stock generated from primary mouse hepatocytes treated for 20 min with 20 ng·ml−1 of IL-1β. The concentration of total p38MAPK, total MK2, and respective phosphorylated variants in the internal standard was assessed by calibration to phosphorylated recombinant GST-p38MAPK and GST-MK2, which was generated as described above.
Assessment of cell volume by fluorescence microscopy
Murine bone marrow-derived macrophages or starved primary mouse hepatocytes were rinsed in PBS and incubated for 5 min with 5 μm CellMask Orange (Molecular Probes). Spinning disk microscopy was performed using a Vivatome-coupled Zeiss Observer A1. Images were analyzed using Axiovision and ImageJ software. Optical slices were binarized, and positive (black) pixels were calculated and multiplied by a device-dependent scaling factor to obtain cell volume. This technique was validated by applying fluorescent microspheres (Molecular Probes) of known size.
Mathematical modeling
To quantitatively analyze the kinetics of p38MAPK/MK2 signaling, a mathematical model based on ODE was formulated. For mathematical modeling, the MATLAB-based software tool Data2Dynamics was used (20). The initial conditions of the ODE model were obtained assuming that the system is in steady state with a given total concentration of p38MAPK and MK2. The output of the ODE system was linked to the experimental data with an observation function depending on scaling and calibration parameters. To assess the measurement noise, we used an error model with log-normal distributed noise (41).
The unknown model, scaling, calibration, and error parameters were simultaneously estimated in a multiexperiment fit by maximizing the likelihood. Because the logarithm of log-normal distributed data is normally distributed, maximizing the likelihood is equivalent to a least-squares problem. The optimization was performed with the trust region algorithm LSQNONLIN, which is provided by the MATLAB Optimization Toolbox (MathWorks).
To ensure that we identified a global maximum, we performed multiple optimization runs initialized with starting parameters chosen by Latin hypercube sampling. Parameter and prediction confidence intervals were calculated with the profile likelihood for each parameter (21).
Author contributions
A. K., J. G. B., J. T., A. R., and R. E. conceived and coordinated the study and wrote the paper, which was critically revised by U. K., M. G., W. D. L., and D. H. R. E., A. R., J. T., and A. K. formulated the ODE model. A. K., J. G. B., R. E., A. R., and U. K. designed and analyzed the experiments, which were performed by A. K. and in part also by C. E. and U. A. B. H. and W. D. L. designed, analyzed, and performed the mass spectrometry and subsequent analysis by MASCOT. U. A. and A. K. cloned, expressed and purified GST-MK2 and GST-p38. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Carina Franek and Marijana Suzanj for excellent technical assistance and Bernd Münstermann and Bernhard Kump for the customized manufacturing of a SDS-PAGE chamber for the separation of 43 probes.
This work was mainly supported by the Virtual Liver Network funded by the German Federal Ministry of Education and Research (BmBF) and in part by the collaborative research center SFB 974 funded by the Deutsche Forschungsgemeinschaft. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Fig. S1 and Information S2.
- MK
- MAPK-activated protein kinase
- pl
- picoliter
- ODE
- ordinary differential equation(s)
- MKP
- MAPK phosphatase(s).
References
- 1. Cargnello M., and Roux P. P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raingeaud J., Gupta S., Rogers J. S., Dickens M., Han J., Ulevitch R. J., and Davis R. J. (1995) Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 270, 7420–7426 [DOI] [PubMed] [Google Scholar]
- 3. Gaestel M. (2006) MAPKAP kinases: MKs: two's company, three's a crowd. Nat. Rev. Mol. Cell Biol. 7, 120–130 [DOI] [PubMed] [Google Scholar]
- 4. Cuadrado A., and Nebreda A. R. (2010) Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417 [DOI] [PubMed] [Google Scholar]
- 5. Mavropoulos A., Orfanidou T., Liaskos C., Smyk D. S., Billinis C., Blank M., Rigopoulou E. I., and Bogdanos D. P. (2013) p38 mitogen-activated protein kinase (p38 MAPK)-mediated autoimmunity: lessons to learn from ANCA vasculitis and pemphigus vulgaris. Autoimmun. Rev. 12, 580–590 [DOI] [PubMed] [Google Scholar]
- 6. Kim E. K., and Choi E. J. (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 [DOI] [PubMed] [Google Scholar]
- 7. Feng Y. J., and Li Y. Y. (2011) The role of p38 mitogen-activated protein kinase in the pathogenesis of inflammatory bowel disease. J. Dig. Dis. 12, 327–332 [DOI] [PubMed] [Google Scholar]
- 8. Denise Martin E., De Nicola G. F., and Marber M. S. (2012) New therapeutic targets in cardiology: p38 α mitogen-activated protein kinase for ischemic heart disease. Circulation 126, 357–368 [DOI] [PubMed] [Google Scholar]
- 9. Han J., and Ulevitch R. J. (1999) Emerging targets for anti-inflammatory therapy. Nat Cell Biol. 1, E39–E40 [DOI] [PubMed] [Google Scholar]
- 10. Frantz S. (2002) Pocket remedy. Nat. Rev. Drug Discov. 1, 253 [Google Scholar]
- 11. Gaestel M., Kotlyarov A., and Kracht M. (2009) Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discov. 8, 480–499 [DOI] [PubMed] [Google Scholar]
- 12. Bode J. G., Albrecht U., Häussinger D., Heinrich P. C., and Schaper F. (2012) Hepatic acute phase proteins: regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 91, 496–505 [DOI] [PubMed] [Google Scholar]
- 13. Campbell J. S., Argast G. M., Yuen S. Y., Hayes B., and Fausto N. (2011) Inactivation of p38 MAPK during liver regeneration. Int. J. Biochem. Cell Biol. 43, 180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hui L., Bakiri L., Stepniak E., and Wagner E. F. (2007) p38α: a suppressor of cell proliferation and tumorigenesis. Cell Cycle 6, 2429–2433 [DOI] [PubMed] [Google Scholar]
- 15. Stepniak E., Ricci R., Eferl R., Sumara G., Sumara I., Rath M., Hui L., and Wagner E. F. (2006) c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 20, 2306–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bode J. G., Ludwig S., Freitas C. A., Schaper F., Ruhl M., Melmed S., Heinrich P. C., and Häussinger D. (2001) The MKK6/p38 mitogen-activated protein kinase pathway is capable of inducing SOCS3 gene expression and inhibits IL-6-induced transcription. Biol. Chem. 382, 1447–1453 [DOI] [PubMed] [Google Scholar]
- 17. Albrecht U., Yang X., Asselta R., Keitel V., Tenchini M. L., Ludwig S., Heinrich P. C., Häussinger D., Schaper F., and Bode J. G. (2007) Activation of NF-κB by IL-1β blocks IL-6-induced sustained STAT3 activation and STAT3-dependent gene expression of the human γ-fibrinogen gene. Cell Signal. 19, 1866–1878 [DOI] [PubMed] [Google Scholar]
- 18. Radtke S., Wüller S., Yang X. P., Lippok B. E., Mütze B., Mais C., de Leur H. S., Bode J. G., Gaestel M., Heinrich P. C., Behrmann I., Schaper F., and Hermanns H. M. (2010) Cross-regulation of cytokine signalling: pro-inflammatory cytokines restrict IL-6 signalling through receptor internalisation and degradation. J. Cell Sci. 123, 947–959 [DOI] [PubMed] [Google Scholar]
- 19. Ehlting C., Böhmer O., Hahnel M. J., Thomas M., Zanger U. M., Gaestel M., Knoefel W. T., Schulte Am Esch J., Häussinger D., and Bode J. G. (2015) Oncostatin M regulates SOCS3 mRNA stability via the MEK-ERK1/2-pathway independent of p38(MAPK)/MK2. Cell Signal. 27, 555–567 [DOI] [PubMed] [Google Scholar]
- 20. Raue A., Schilling M., Bachmann J., Matteson A., Schelker M., Schelke M., Kaschek D., Hug S., Kreutz C., Harms B. D., Theis F. J., Klingmüller U., and Timmer J. (2013) Lessons learned from quantitative dynamical modeling in systems biology. PLoS ONE 8, e74335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raue A., Kreutz C., Maiwald T., Bachmann J., Schilling M., Klingmüller U., and Timmer J. (2009) Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics 25, 1923–1929 [DOI] [PubMed] [Google Scholar]
- 22. Goldstein D. M., and Gabriel T. (2005) Pathway to the clinic: inhibition of P38 MAP kinase: a review of ten chemotypes selected for development. Curr. Top. Med. Chem. 5, 1017–1029 [DOI] [PubMed] [Google Scholar]
- 23. Kukkonen-Macchi A., Sicora O., Kaczynska K., Oetken-Lindholm C., Pouwels J., Laine L., and Kallio M. J. (2011) Loss of p38γ MAPK induces pleiotropic mitotic defects and massive cell death. J. Cell Sci. 124, 216–227 [DOI] [PubMed] [Google Scholar]
- 24. Koshland D. E. Jr, Goldbeter A., and Stock J. B. (1982) Amplification and adaptation in regulatory and sensory systems. Science 217, 220–225 [DOI] [PubMed] [Google Scholar]
- 25. Grubelnik V., Dugonik B., Osebik D., and Marhl M. (2009) Signal amplification in biological and electrical engineering systems: universal role of cascades. Biophys. Chem. 143, 132–138 [DOI] [PubMed] [Google Scholar]
- 26. Hornberg J. J., Bruggeman F. J., Binder B., Geest C. R., de Vaate A. J., Lankelma J., Heinrich R., and Westerhoff H. V. (2005) Principles behind the multifarious control of signal transduction. ERK phosphorylation and kinase/phosphatase control. FEBS J. 272, 244–258 [DOI] [PubMed] [Google Scholar]
- 27. Heinrich R., Neel B. G., and Rapoport T. A. (2002) Mathematical models of protein kinase signal transduction. Mol. Cell 9, 957–970 [DOI] [PubMed] [Google Scholar]
- 28. Hendriks B. S., Hua F., and Chabot J. R. (2008) Analysis of mechanistic pathway models in drug discovery: p38 pathway. Biotechnol. Prog. 24, 96–109 [DOI] [PubMed] [Google Scholar]
- 29. Hendriks B. S., Seidl K. M., and Chabot J. R. (2010) Two additive mechanisms impair the differentiation of “substrate-selective” p38 inhibitors from classical p38 inhibitors in vitro. BMC Syst. Biol. 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Espelin C. W., Goldsipe A., Sorger P. K., Lauffenburger D. A., de Graaf D., and Hendriks B. S. (2010) Elevated GM-CSF and IL-1β levels compromise the ability of p38 MAPK inhibitors to modulate TNFα levels in the human monocytic/macrophage U937 cell line. Mol. Biosyst. 6, 1956–1972 [DOI] [PubMed] [Google Scholar]
- 31. Ligeti E., Csepanyi-Komi R., and Hunyady L. (2012) Physiological mechanisms of signal termination in biological systems. Acta Physiol. 204, 469–478 [DOI] [PubMed] [Google Scholar]
- 32. Klingmüller U., Bauer A., Bohl S., Nickel P. J., Breitkopf K., Dooley S., Zellmer S., Kern C., Merfort I., Sparna T., Donauer J., Walz G., Geyer M., Kreutz C., Hermes M., (2006) Primary mouse hepatocytes for systems biology approaches: a standardized in vitro system for modelling of signal transduction pathways. Syst. Biol. 153, 433–447 [DOI] [PubMed] [Google Scholar]
- 33. Kotlyarov A., Neininger A., Schubert C., Eckert R., Birchmeier C., Volk H. D., and Gaestel M. (1999) MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat. Cell Biol. 1, 94–97 [DOI] [PubMed] [Google Scholar]
- 34. Seglen P. O. (1972) Preparation of rat liver cells: I: effect of Ca2+ on enzymatic dispersion of isolated, perfused liver. Exp. Cell Res. 74, 450–454 [DOI] [PubMed] [Google Scholar]
- 35. Seglen P. O. (1973) Preparation of rat liver cells: II: effects of ions and chelators on tissue dispersion. Exp. Cell Res. 76, 25–30 [DOI] [PubMed] [Google Scholar]
- 36. Seglen P. O. (1973) Preparation of rat liver cells: III: enzymatic requirements for tissue dispersion. Exp. Cell Res. 82, 391–398 [DOI] [PubMed] [Google Scholar]
- 37. Berry M. N., and Friend D. S. (1969) High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J. Cell Biol. 43, 506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ehlting C., Trilling M., Tiedje C., Le-Trilling V. T., Albrecht U., Kluge S., Zimmermann A., Graf D., Gaestel M., Hengel H., Häussinger D., and Bode J. G. (2016) MAPKAP kinase 2 regulates IL-10 expression and prevents formation of intrahepatic myeloid cell aggregates during cytomegalovirus infections. J. Hepatol. 64, 380–389 [DOI] [PubMed] [Google Scholar]
- 39. Hahn B., D'Alessandro L. A., Depner S., Waldow K., Boehm M. E., Bachmann J., Schilling M., Klingmüller U., and Lehmann W. D. (2013) Cellular ERK phospho-form profiles with conserved preference for a switch-like pattern. J. Proteome Res. 12, 637–646 [DOI] [PubMed] [Google Scholar]
- 40. Schilling M., Maiwald T., Bohl S., Kollmann M., Kreutz C., Timmer J., and Klingmüller U. (2005) Computational processing and error reduction strategies for standardized quantitative data in biological networks. FEBS J. 272, 6400–6411 [DOI] [PubMed] [Google Scholar]
- 41. Kreutz C., Bartolome Rodriguez M. M., Maiwald T., Seidl M., Blum H. E., Mohr L., and Timmer J. (2007) An error model for protein quantification. Bioinformatics 23, 2747–2753 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.