

Abstract
The identification and discrimination of four epigenetic modifications to cytosine in the proposed active demethylation cycle is demonstrated at the single-molecule level, without the need for chemical pre-treatment or labeling. The wild-type protein nanopore α-hemolysin is used to capture individual DNA duplexes containing a single cytosine-cytosine mismatch. The mismatch is held at the latch constriction of α-hemolysin, which is used to monitor the kinetics of base flipping at the mismatch site. Base flipping and the subsequent interactions between the DNA and the protein are dramatically altered when one of the cytosine bases is replaced with methyl-, hydroxymethyl-, formyl-, or carboxylcytosine. As well as providing a route to single-molecule analysis of important epigenetic markers in DNA, our results provide important insights into how the introduction of biologically-relevant, but poorly understood, modifications to cytosine effect the local conformational dynamics of a DNA duplex in a confined environment.
Graphic Abstract
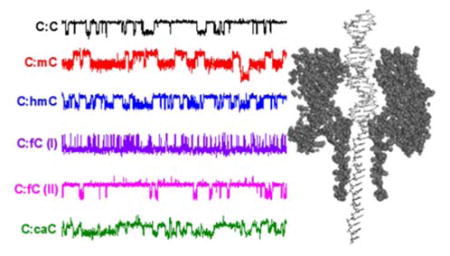
Introduction
Epigenetic modifications to the nucleobase cytosine control gene regulation in human cells and have implications in the development of cancer and other diseases. 1–2 The most common modification is the enzyme-catalyzed addition of a methyl group to the carbon-5 position of cytosine to generate methylcytosine (mC).3 Methylation of cytosine usually occurs at “CpG sites” in which a cytosine base is immediately proceeded by a guanine base when reading in the 5′ to 3′ direction. Typically, 70 – 80% of CpG sites in mammalian cells are methylated,4–6 and mC accounts for 1% of all DNA bases in the human genome.5–6
The process of removal of a methyl group from cytosine, i.e., demethylation, remains an active and current field of research. Since the direct reversal of methylation is energetically unfavorable, pathways to demethylation that involve oxidative intermediaries of mC have been proposed.7 Recent research has led to the discovery of three other epigenetic modifications to cytosine; hydroxymethyl- (hC),8 formyl- (fC),9–10 and carboxyl- (caC)10–11 that together comprise a feasible pathway for the active reversal of cytosine methylation via sequential oxidation, base excision, and subsequent repair (Figure 1).7, 12 Furthermore, hC, fC, and caC have all been found to naturally occur in mammalian embryonic stem (ES) cells,13–14 indicating that these bases are stable and may themselves have some role in gene regulation.
Figure 1.

The proposed pathway for methylation and demethylation of cytosine. Image adapated from refs. 9 & 34. The protein DNA methlytrasferase (DNMT) methylates cytosine at the C5 position to produce mC. Subsequent enzyme-catalysed oxidation by ten eleven translocation (TET) proteins produces sequentially the bases hmC fC and caC. The bases fC and caC can be excised by thymine DNA glycosylase (TDG) and replaced with cytosineby the base excision repair pathway (BER).
The ability to discriminate between C, mC and the oxidized derivatives of mC that comprise the active demethylation cycle is of clear biological importance in the quest to understand how genes regulate cell function and development. While cytosine and mC can be readily discriminated with high precision using bisulfite sequencing,15 the development of suitable assays for discriminating the products of mC oxidation remains a significant challenge. Variations of bisulfite sequencing, in which the target of identification (hmC, fC, or caC) is first selectively modified through chemical or enzymatic reaction have been presented,16–22 but in order to be completely reliable the conversion reactions require an unfeasible 100% reaction yield. This is especially important given the relatively low abundance of oxidative products of mC, where hmC, fC, and caC are found at levels of just ~0.5%, ~0.002%, and 0.0003%, respectively, of all cytosine in mouse ES cells.23
Nanopore devices have received attention as an alternative approach to identifying epigenetic markers in DNA sequences due to their potentially high sensitivity. Variants of the protein nanopore Mycobacterium smegmatis porin A (MspA) have been used to identify all five cytosine variants, with accuracies of up to 98%.24–26 There have also been recent demonstrations of the detection of mC and hmC with the protein pore α-hemolysin (αHL),27–31 but detection of all five epigenetic cytosine variants with this pore has not previously been demonstrated.
In our recent work, we have demonstrated that the 2.6 nm latch constriction of αHL is able to measure the kinetics of localized conformational changes at a mismatched base-pair in DNA, which we have attributed to a single base flipping in and out of the helix at the mismatch site.32–33 Here, we show that the kinetics of base flipping of a cytosine-cytosine pair situated at the latch constriction of αHL is significantly altered when one of the cytosine bases in the mismatch is modified at the carbon-5 position. Measuring the base flipping kinetics with a molecule-by-molecule approach, we are able to discriminate between duplexes containing a single mC, hmC, fC, or caC base. Our method does not require labelling, and unambiguously identifies the modification without recourse to complex statistical analysis. Our data also provides new fundamental insights into how epigenetic modifications to cytosine alter the local conformational dynamics of DNA and the effect of sequence context on such dynamics, for example, pointing towards the existence of the hydrated form of formyl cytosine in aqueous conditions.
Results & Discussion
Measuring the dynamics of a DNA mismatch site one molecule at a time
We used a model sequence, 23 bases in length from a section of the KRAS gene to demonstrate base flipping analysis at the single molecule level. In addition to being well-characterized with our nanopore system, modifications to the KRAS gene have been implicated in uncontrolled cell growth and formation of human carcinomas.35 A homogeneous single-stranded tail, 24 thymine bases in length, was added to the sequence to ease threading of the duplex into the αHL protein pore.36 Hybridization of the probe sequence, which is fully complementary except at the 9th base as counted from the 3′ terminus, generates a single cytosine-cytosine mispair that is specifically placed to align with the latch constriction of αHL when the DNA is captured by the pore, as shown in Figure 2A.
Figure 2.

Trapping of DNA and analysis of base flipping at a C:C mismatch site within a DNA duplex. (A) The DNA duplex is driven into the αHL nanopore under an applied potential where it is held for up to 20 s and then ejected by reversing the applied bias. While resident within the pore, the C:C mismatch site is aligned with the latch constriction of αHL. (B) Modulating current signatures are observed while DNA resides within the nanopore, where I1 corresponds to a confirmation where all bases are intrahelical and I2 corresponds to a conformation where one of the cytosine bases at the mismatch site is extrahelical. Intra- and extrahelical lifetimes are given by t1 and t2, respectively. Uninterrupted current-time traces demonstrating sequential capture and release of multiple DNA duplexes are shown in Figures S1 and S2.
Upon capture of the DNA duplex, attenuation of the measured current is observed due to an immediate decrease in the ion flux through the pore. Proximity of the C:C mismatch to the latch constriction when the DNA resides inside the pore leads to distinct modulation of current between two states (Figure 2B). The two states that comprise the modulating signature are separated by approximately 1.6 pA in amplitude and have a modulation periodicity on the order of 10 ms. We have previously obtained evidence that the observed modulation between two distinct states is a result of one of the cytosine bases in the unstable mismatch flipping in and out of the DNA helix.32–33 The less-blocking state (approx. -10 pA) is assigned to the intrahelical conformation because the same current amplitude (and an absence of current modulation) is observed when the mismatch at the latch constriction is replaced by a stable complementary (C:G) basepair.33
DNA with a double-stranded component is unable to pass the 1.4 nm central constriction within the pore36–37 (Figure 2A), and under an applied bias will remain within the protein vestibule before unzipping into its constituent components.38–39 How long the DNA remains within the pore prior to unzipping, i.e., the characteristic ‘residence lifetime’, is dependent primarily on the applied bias and the DNA composition.39 Residence lifetimes range from a few milliseconds for shorter duplexes to tens of seconds for longer duplexes. Lower voltages increase the residence time and higher voltages decrease the residence time.
At an applied voltage of 100 mV, the majority (>80%) of the 23 base-paired duplexes utilized in the experiments reported here can be held within the pore for 20 seconds or longer, with the base flipping in and out of the helix around 200 times in this period. Under such conditions, it is possible to capture the duplexes containing a C:C mismatch one at a time, hold them within the pore for 20 seconds, and then release by reversing the bias and driving the DNA back out into bulk solution (Figure 2B). Each duplex captured is thus analyzed individually to determine the base flipping kinetics at the C:C mismatch site at the single molecule level.
We found that the lifetimes of the two modulating states from a single duplex are well described by first-order rate kinetics, and the distribution of state lifetimes can be used to extract characteristic lifetime constants τ1 and τ2, (Figure 3A), which represent the intrahelical (less blocking, I1) and extrahelical (more blocking, I2) conformations at the mismatch site. Representative intra and extrahelical lifetime constants were found to vary from duplex to duplex of the same composition. The analysis of approximately 40 individual duplexes demonstrates a Gaussian-like distribution (Figure 3B), from which average lifetime constants for a population of duplexes of the same composition, measured with the same protein, can be calculated (τ1 (mean) and τ2 (mean)). This Gaussian-like distribution indicates the stochastic variation in base flipping kinetics for different DNA duplexes captured with a single protein channel. Repeating the same experiment with DNA of the same composition and under the same conditions, but with a different protein channel, returns (within error) the same values for τ1 (mean) and τ2 (mean), as shown in Figure 3C and Figure S3. The mean values from three unique protein channels (i.e., three unique experiments) were found to be 13.8, 13.1, and 14.1 ms for τ1 (mean), and 41.6, 43.0, and 42.2 ms for τ2 (mean).
Figure 3.

Reproducible analysis of base flipping at the αHL latch in individual DNA duplexes. (A) Representative lifetime histograms for states I1 (intrahelical) and I2 (extrahelical), for a single molecule of DNA, from which lifetime constants can be extracted. (B) Distribution of lifetime constants for states I1 and I2 across a sample of 35 individual duplexes, measured with a single protein channel. (C) Scatter plot of intra- and extrahelical lifetime constants τ1 and τ2 for individual DNA duplexes measured across three independent αHL channels (black squares, red circles, blue triangles). Distribution of lifetime constants for proteins 2 and 3 are presented in Figure S3.
Modifications to cytosine alter the base flipping kinetics
We synthesized DNA identical to that shown in Figure 2A, with the exception of a mC, hmC, fC, or caC base replacing one of the cytosines in the duplex at the 9th position in the sequence as counted from the 3′ terminal of the shorter (23 base) strand. Initially, we replaced the cytosine in the shorter probe strand, to generate a C:X mismatch (where X is either mC, hmC, fC, or caC) in proximity to the latch constriction of αHL upon capture by DNA.
Replacing the cytosine base on the probe strand in the mismatch pair results in significant changes to the observed current modulation when DNA resides inside αHL (Figure 4). Most striking is the clear change to the intrahelical and extrahelical lifetimes (states I1 and I2, respectively). There are also clear changes to the relative current noise associated with each of the states, and in the case of mC, modulation to a previously unseen, less blocking third state (I3).
Figure 4.

Substituting the cytosine base at the mismatch site in the shorter (23-mer) strand for methylcytosine (mC) or one of is oxidative derivatives changes the base flipping kinetics. (A) Representative current time traces from a 6 second window of a single DNA capture event demonstrating measurement of base flipping at a C:X mismatch site where X is mC, hmC, fC or caC. Note that two event types (I and II) are observed for fC, with type I comprising 80% of events. This topic is addressed later in the text. Uninterrupted current-time traces demonstrating sequential capture and release of multiple DNA duplexes for each modification are shown in Figures S4 – S7.
For the C:mC duplex, state I1 becomes significantly longer relative to the C:C duplex, and it is characterized by a higher noise level, particularly in the intrahelical state. This is consistent with our proposed model of base flipping, because two recent reports have suggested that the incorporation of mC into a base-pair stabilizes the intrahelical state relative to the extrahelical state.40–41 The C:hmC, C:fC, and C:caC base pairs all present modulating current signatures between two states, but the lifetimes of each state are dramatically altered relative to the C:C duplex. For C:hmC, the extrahelical lifetime significantly decreases relative to C:C, while for C:caC, the intrahelical lifetime increases, but not to the same extent of C:mC. Of particular curiosity is the C:fC duplex, which exhibits two distinct event types. In type I events, the extrahelical lifetimes are extremely short relative to duplexes with the C:C base pair, and in type II events, the extrahelical lifetimes are extremely long relative to duplexes with the C:C base pair. The ratio of type I to type II events is approximately 5:1, and leads to the intriguing implication that duplexes containing the fC base, or the fC base itself, may exist in two uniquely identifiable forms. We discuss this topic in detail later.
In most cases, visual inspection of the current-time trace is sufficient to observe which epigenetic modification to cytosine is present at the mismatch site within the duplex. While duplexes containing different epigenetic modifications are difficult to differentiate from just one parameter, for example, C:C, C:caC, and C:mC containing duplexes all have similar extrahelical (τ2) lifetimes, the use of both the intra- and extrahelical lifetime parameters together permits ready identification of all epigenetic modifications to cytosine. The base flipping kinetics of each modification are sufficiently different to allow unambiguous identification of C:C, C:mC, C:hmC, or c:fC at the single molecule level (Figure 5). Plotted as τ2 versus τ 1, the data are resolved into clusters that in most cases do not overlap and are readily distinguished. While some overlap is seen for C:mC and C:caC, the former can be readily differentiated from the latter based on its unique three-state modulation signature and distinctly higher noise in state I1 relative to I2 (Figure 4).
Figure 5.
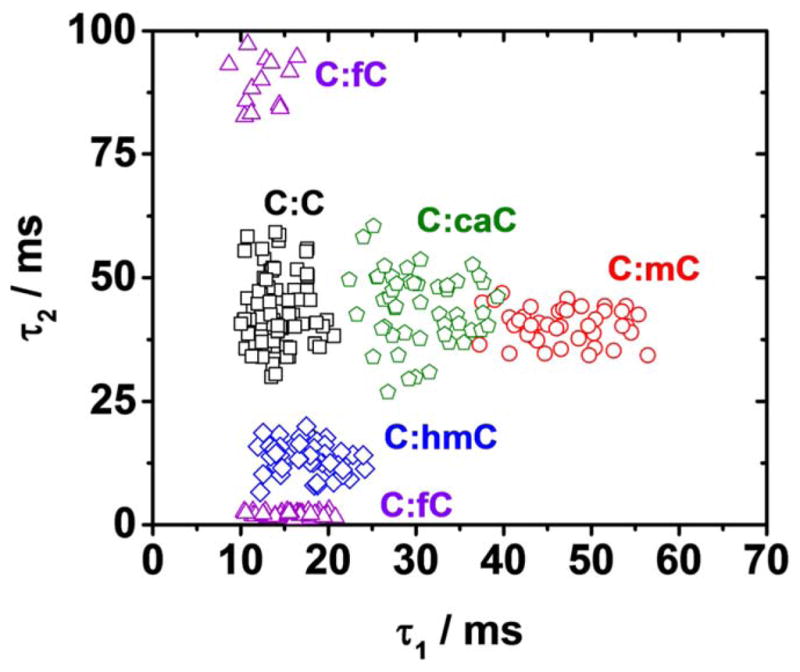
Identification of all epigenetic modifications to cytosine in the proposed methylation/active demethylation cycle. Scatter plot of intrahelical (τ1) vs. extrahelical (τ2) lifetime constants for duplexes C:C (black squares), C:mC (red circles), c:hmC (blue diamonds), c:fC (purple triangles), and C:caC (green pentagons). Each data point represents a base flipping measurement for a single DNA molecule. Distribution of intra and extrahelical lifetime constants for duplexes containing each modification are presented in Figures S8 and S9.
The distinct kinetics for the different modifications can be readily used to determine the identity of an individually captured duplex from a mixed sample, and thus used to determine the ratio of duplex concentrations (Figure S10). The method we present here can thus be used to determine the percentage of a particular cytosine variant (mC, hmC, fC or caC) at a specific site within a known DNA sequence. In one envisaged application, fragmented genomic DNA from cells would be captured by a probe DNA strand that would generate a CC mismatch at a known methylation site. The ratio of event types could then be used to determine the percentage of the cytosine that has been modified with the mC, hmC, fC or CaC variants.
Base flipping kinetics are dependent on the flanking bases for mC and hmC containing duplexes
Base flipping kinetics, and indeed the stability of a mismatch site, have been shown previously in some cases to be dependent on the identity of the flanking base pairs.42–43 As a simple extension of our work to check for sequence context effects, we synthesized a new series of duplexes in which the modified cytosine base at the mismatch site is now placed on the longer target strand rather than the probe strand. When incorporated into the probe strand, the modified base at the C:X mismatch is flanked by a 5′G and a 3′T, and in the target strand, the modified base at the X:C mismatch is flanked by a 5′ A and a 3′C. The position of the mismatch site relative to the latch constriction of αHL remains unchanged, while the pore itself is seven-fold symmetric.37
A series of experiments with duplexes containing the modified cytosine flanked by 5′A and 3′C revealed changes to the base flipping kinetics of a population relative to the duplexes containing the modified cytosine flanked by 5′G and 3′T for the cases of mC, hmC, and fC (Figure 6). While a determination of the bases that flank the modified cystoine cannot be made at the single molecule level, our preliminary experiments do reveal a statistically significant sequence context effect. For example, the C:mC mismatch has average state lifetime constants τ1 (mean), and τ2 (mean) of 46.5 and 41.3 ms, respectively, while the mC:C mismatch has τ1 (mean), and τ2 (mean) values of 59.1 and 43.8 ms. Changing the context of the mC base from A(mC)C to G(mC)T results in a 27% increase in τ1 (mean). Changes to the time constant of the third state, τ3 (mean), are also observed, with a significant decrease when mC is placed in the A(mC)c context (Figure S18). The increase in τ1 (mean), indicates that an A and C either side of the methylcytosine base work to stabilize the intrahelical state relative to flanking T and G pairs.
Figure 6.
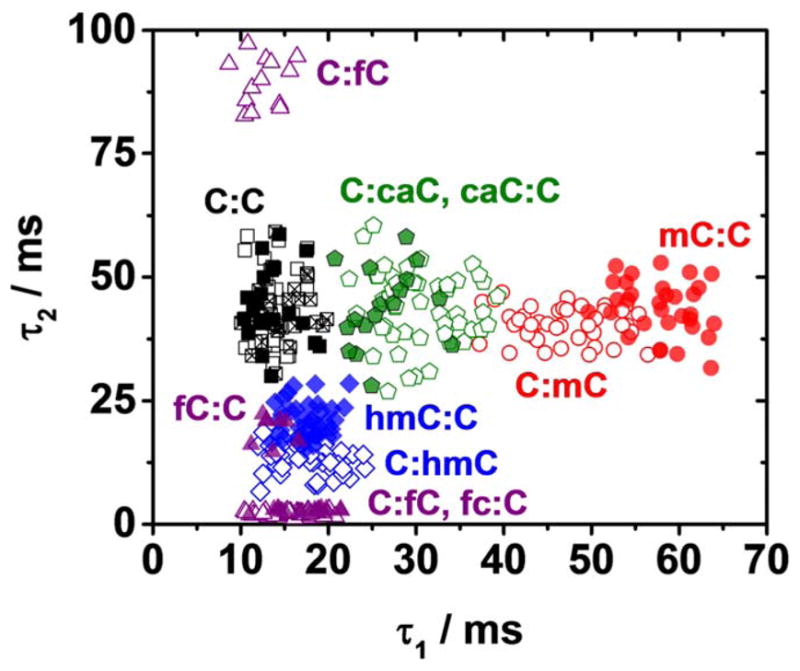
Base flipping kinetics at a mismatch site within the αHL latch are sequence dependent. When the flanking basespairs of the cytosine modifications mC (red cicles), hmC (blue diamonds) and fC (purple triangles) are changed from 5′A and 3′C (hollow symbols, data from Fig. 5) to 5′G and 3′T (solid symbols), the population centres of the lifetime constants τ1 and τ2 are shifted. Changes to the lifetime constant when chanigmn the sequence context of fC are observed only for the minor event type. No changes are observed for caC. Three independent measurements, i.e., with 3 different protein channels (hollow, solid, and hatched squares) for the C:C duplex highlight the negligible variation in population centres expected from experiment to experiment with DNA of the same composition. Representative current time traces and distributions of intra- and extrahelical lifetime constants for duplexes containing each modification are presented in Figures S12 – S17.
When the bases that flank hmC are changed from 5′A and 3′C to 5′G and 3′T τ1 (mean) remains the same, but τ2 (mean) increases by 49% from 13.8 to 20.6 ms, indicating a stabilization of the extrahelical state. The hydroxyl group of hmC will readily form hydrogen bonds, and is known to interact with neighbouring base-pairs.44 It is plausible that these interactions will play some role in determining the stability of the extrahelical conformation at the mismatch site, and by changing the flanking bases it will be possible to change the strength and or nature of these interactions.
It is noteworthy that in the cases of both mC and hmC containing duplexes, chaning the sequence context alters just one of the time constants, i.e., only τ1 (mean) for mC and only τ2 (mean) for hmC. In addition, the time constant that is altered is the same as the dominant change observed when changing from a C:C to a C:mC containing duplex or from a C:C to a C:hmC containing duplex.
For the fC-containing duplexes, changes to the base flipping kinetics when the sequence context changes are dependent on the event type. No changes are observed to the kinetics of the type I event, which retains dominance at approximately 80% of capture events. However, the average extrahelical lifetime (τ2 (mean)) of type II events decreases from 89.4 to 18.5 ms, an 80% decrease. In this sequence context, the fc:C and hmC:C duplexes have similar base-flipping kinetics and cannot be discriminated from these parameters alone at the single molecule level. It is plausible that similar overlap in the base flipping kinetics of different cytosine variant containing duplexes may be observed for other sequence contexts. In the case of caC containing duplexes, no change to the average state lifetimes (τ1 (mean) and τ2 (mean)) are observed.
The formylcytosine base can exist as a hydrate in aqueous solution
Two unique event types are observed for the formyl-cytosine containing duplex (Figures 4 and 5). A count of the number of events of each type indicates a ratio of approximately 5:1, where type I events are more prevalent, regardless of whether the fC is in the shorter (C:fC) or longer (fC:C) strand. The two distinct event types observed for the fC-containing duplex leads to the intriguing possibility that formylcytosine, within the context of our DNA duplex, exists in two unique structural forms (Figure 7), with each form having different base flipping kinetics when confined at the latch constriction of αHL.
Figure 7.
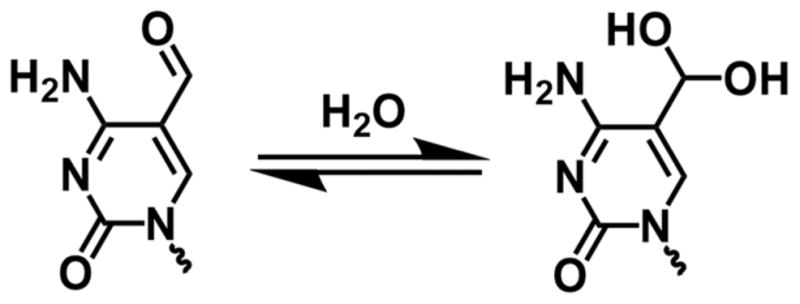
Formlycytosine undergoes nucleophilic addition of water in aqueous solution to form a stable aldehyde hydrate.
We speculate that the two event types observed for fC-containing duplexes are a result of hydration of the formyl group in aqueous solution. Aldehydes undergo nucleophilic addition in water to form hydrates, with both the hydrate and formyl structures existing in an equilibrium defined by the relative stabilities of the two structures.45 The existence of formylcytosine base in hydrate form was previously measured at very low quantities (0.5%) by Carell and co-workers via mass spectrometry.9 Our results, with the advantage that measurements are made directly in DNA’s native aqueous environment, suggest that the hydrate is potentially more abundant. Our hypothesis is supported by data for hydrate equilibrium constants for similar pyridinium aldehydes that are also electron deficient, and have previously been shown to exist in the hydrate form in significant quantities. For these types of aldehydes, the hydrate is present at levels of 1 – 20 %. (KHYD= [hydrate]/[aldehyde] = 0.2 – 0.01).45–46 Based on these prior reports, the existence of formylcytosine in the hydrate form for the DNA strands studied here is highly plausible, and we speculate that the hydrate form represents the minor (type II) events observed in our experiments.
Once an fC-containing duplex is captured by the αHL nanopore, no hydration or dehydration reactions are observed within the 20 s time period that the DNA is held inside the pore. Hydration and dehydration is expected to be rapid in bulk solution, catalysed by nucleophilic OH− ions in basic solutions. During a DNA capture event, the negatively-charged DNA backbone results in electrostatic exclusion of anions (including OH−) from entering the pore.38 In such circumstances, conversion between the two forms is expected to be extremely slow or impossible. The αHL nanopore is therefore capable of taking a ‘snapshot’ of the aldehyde/aldehyde hydrate equilibrium in the bulk through determination of the ratio of event types, and our results suggest an equilibrium constant of hydration for fC in our DNA of KHYD = 0.2.
Conclusions
We have demonstrated that cytosine, methylcytosine, hydroxymethlycytosine, formylcytosine, and carboxylcytosine can all be discriminated at the single molecule level based on their unique base flipping kinetics when paired opposite a cytosine base in a mismatch at the latch constriction of αHL. Discrimination is achieved without modification to the duplex and/or labelling of the DNA bases. The present findings also provide experimental evidence that formylcytosine can exist as either an aldehyde or hydrate in solution, with an equilibrium constant of hydration of 0.2. We anticipate that our methodology will be of use to researchers investigating the emerging role of cytosine derivatives in gene regulation and active demethylation.
Methods
DNA synthesis and purification, nanopore fabrication and data analysis were performed as previously reported.33 Ion channel recordings were performed using a 10 mM phosphate, 0.25 M KCl (pH 7.5) buffer at 25 °C. A 100 mV (trans vs. cis) voltage was applied across the αHL channel in all experiments. Complete experimental details are given in the Supporting Information.
Supplementary Material
Acknowledgments
RPJ acknowledges funding from a Marie Curie International Outgoing Fellowship under the EU FP7 program (Project No. 625984). This work was funded in part by a grant from the NIH (R01 GM093099). The authors thank Electronic BioSciences Inc. (San Diego, CA) for donating the ion-channel recording instrument and software.
Footnotes
Full experimental details, extended current-time traces, lifetime constant histograms, mass spectrometry of the fC containing oligomer and demonstration of capture and analysis from a mixed sample.
References
- 1.Robertson KD. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 2.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goll MG, Bestor TH. An Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 4.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. Nuc Acid Res. 1982;10:2709–2721. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bird A. Gen Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 7.Wu SC, Zhang Y. Nat Rev Mol Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kriaucionis S, Heintz N. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfaffeneder T, Hackner B, Truß M, Münzel M, Müller M, Deiml CA, Hagemeier C, Carell T. Angew Chem Int Ed. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 10.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiesser S, Pfaffeneder T, Sadeghian K, Hackner B, Steigenberger B, Schröder AS, Steinbacher J, Kashiwazaki G, Höfner G, Wanner KT, Ochsenfeld C, Carell T. J Am Chem Soc. 2013;135:14593–14599. doi: 10.1021/ja403229y. [DOI] [PubMed] [Google Scholar]
- 13.Bachman M, Uribe-Lewis S, Yang X, Burgess HE, Iurlaro M, Reik W, Murrell A, Balasubramanian S. Nat Chem Biol. 2015;11:555–557. doi: 10.1038/nchembio.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner M, Steinbacher J, Kraus TFJ, Michalakis S, Hackner B, Pfaffeneder T, Perera A, Müller M, Giese A, Kretzschmar HA, Carell T. Angew Chem Int Ed. 2015;54:12511–12514. doi: 10.1002/anie.201502722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grunau C, Clark SJ, Rosenthal A. Nuc Acid Res. 2001;29:e65. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Booth MJ, Marsico G, Bachman M, Beraldi D, Balasubramanian S. Nat Chem. 2014;6:435–440. doi: 10.1038/nchem.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Z, Dai N, Borgaro Janine G, Quimby A, Sun D, Corrêa Ivan R, Jr, Zheng Y, Zhu Z, Guan S. Mol Cell. 57:750–761. doi: 10.1016/j.molcel.2014.12.035. [DOI] [PubMed] [Google Scholar]
- 18.Lu X, Song C-X, Szulwach K, Wang Z, Weidenbacher P, Jin P, He C. J Am Chem Soc. 2013;135:9315–9317. doi: 10.1021/ja4044856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu X, Han D, Zhao BS, Song CX, Zhang LS, Dore LC, He C. Cell Res. 2015;25:386–389. doi: 10.1038/cr.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu M, Hon Gary C, Szulwach Keith E, Song C-X, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, Min J-H, Jin P, Ren B, He C. Cell. 149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 22.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C. Nat Biotech. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song CX, Yi C, He C. Nat Biotech. 2012;30:1107–1116. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wescoe ZL, Schreiber J, Akeson M. J Am Chem Soc. 2014;136:16582–16587. doi: 10.1021/ja508527b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiber J, Wescoe ZL, Abu-Shumays R, Vivian JT, Baatar B, Karplus K, Akeson M. Proc Acad Nat Sci. 2013;110:18910–18915. doi: 10.1073/pnas.1310615110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, Bartlett JJ, Pavlenok M, Gundlach JH. Proc Acad Nat Sci. 2013;110:18904–18909. doi: 10.1073/pnas.1310240110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng T, Liu L, Li T, Li Y, Gao J, Zhao Y, Wu HC. Chem Sci. 2015;6:5628–5634. doi: 10.1039/c5sc01436k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wallace EVB, Stoddart D, Heron AJ, Mikhailova E, Maglia G, Donohoe TJ, Bayley H. Chem Comm. 2010;46:8195–8197. doi: 10.1039/c0cc02864a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li WW, Gong L, Bayley H. Angew Chem Int Ed. 2013;52:4350–4355. doi: 10.1002/anie.201300413. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Luan BQ, Yang Z, Zhang X, Ritzo B, Gates K, Gu LQ. Sci Rep. 2014;4:5883. doi: 10.1038/srep05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding Y, Fleming AM, White HS, Burrows CJ. ACS Nano. 2015;9:11325–11332. doi: 10.1021/acsnano.5b05055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson RP, Perera RT, Fleming AM, Burrows CJ, White HS. Faraday Discuss. 2016;193:471–485. doi: 10.1039/c6fd00058d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson RP, Fleming AM, Beuth LR, Burrows CJ, White HS. J Am Chem Soc. 2016;138:594–603. doi: 10.1021/jacs.5b10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. [accessed 5/10/2016];DNA Methylation. http://www.whatisepigenetics.com/dna-methylation/
- 35.Pfeifer G, Besaratinia A. Hum Gen. 2009;125:493–506. doi: 10.1007/s00439-009-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin Q, Fleming AM, Johnson RP, Ding Y, Burrows CJ, White HS. J Am Chem Soc. 2013;135:19347–19353. doi: 10.1021/ja410615d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 38.Johnson RP, Fleming AM, Burrows CJ, White HS. J Phys Chem Lett. 2014;5:3781–3786. doi: 10.1021/jz502030e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson RP, Fleming AM, Jin Q, Burrows CJ, White HS. Biophys J. 2014;107:924–931. doi: 10.1016/j.bpj.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bianchi C, Zangi R. J Phys Chem B. 2013;117:2348–2358. doi: 10.1021/jp312145b. [DOI] [PubMed] [Google Scholar]
- 41.Bianchi C, Zangi R. Biophys Chem. 2014;187–188:14–22. doi: 10.1016/j.bpc.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Coman D, Russu IM. Biophys J. 2005;89:3285–3292. doi: 10.1529/biophysj.105.065763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Folta-Stogniew E, Russu IM. Biochem. 1994;33:11016–11024. doi: 10.1021/bi00202a022. [DOI] [PubMed] [Google Scholar]
- 44.Wang R, Ranganathan SV, Valsangkar VA, Magliocco SM, Shen F, Chen A, Sheng J. Chem Comm. 2015;51:16389–16392. doi: 10.1039/c5cc06563a. [DOI] [PubMed] [Google Scholar]
- 45.Hilal SH, Bornander LL, Carreira LA. QSAR Combi Sci. 2005;24:631–638. [Google Scholar]
- 46.Huang S, Miller AK, Wu W. Tet Lett. 2009;50:6584–6585. doi: 10.1016/j.tetlet.2009.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.