

Abstract

A series of 2-sulfonyl-pyrimidinyl derivatives was developed as apoptosis inhibitors. These represent the first class of apoptosis inhibitors that function through stabilizing mitochondrial respiratory complex II. Starting from a phenotypic screen hit with micromolar activity, we optimized the cellular apoptosis inhibition activity of 2-sulfonyl-pyrimidinyl derivatives to picomolar level (compound 42, also named as TC9-305). The therapeutic potential of these new apoptosis inhibitors was further demonstrated by their neuroprotective effect on an ischemic animal model.
Keywords: Apoptosis inhibitors, phenotypic screen, structure−activity optimization, ischemia treatment, mitochondria protection
Excessive apoptosis is closely associated with immunodeficiency diseases, liver diseases, ischemia-associated injury, and neurodegenerative diseases, including Alzheimer’s syndrome, Parkinson’s syndrome, Huntington’s syndrome, and amyotrophic lateral sclerosis.1−7 Upon recognition of stimulus signals that initiate mitochondria-mediated apoptosis, the decision of whether or not to initiate apoptosis is determined via the complicated regulation of pro- and anti-apoptotic BCL-2 (B-cell lymphoma-2) protein family members.1 BH-3 only proteins (tBid, Bim, etc.) trigger the conformational activation of Bax and/or Bak, which in turn oligomerize and translocate to the mitochondrial outer membrane and lead to mitochondrial permeability changes.1,8 These changes enable the release of pro-apoptotic proteins such as cytochrome c and Smac to the cytoplasm.1,8,9 These pro-apoptotic factors then mediate the activation of caspases and ultimately lead to cell death.
The great majority of research into apoptosis inhibitors has focused on targeting caspases or proteins of the pro-apoptotic BCL-2 family.10 Blocking caspase activity can stop the final execution step of apoptosis, but by this stage, cells have already suffered considerable damage that typically induces other types of cell death, such as necrosis.11,12 Bax and Bid inhibitors have been developed to target apoptosis.13−16 However, these apoptosis inhibitors have only moderate potency, with EC50 values above or around the micromolar level. Additionally, blocking a single pro-apoptotic BCL-2 protein may have only limited effects because other pro-apoptotic BCL-2 family proteins are known to have complementary functions.1,8,9 Therefore, there is an urgent need to develop more efficient apoptosis inhibitors.
In this work, we report the medicinal chemistry effort in the development of a series of 2-sulfonyl-pyrimidinyl derivatives as highly potent apoptosis inhibitors. Starting from a phenotypic screen hit compound with micromolar activity, we optimized the cellular apoptosis inhibition activity in a series of derivative compounds to picomolar level. We also revealed the neuroprotective effect of this class of compounds in a transient focal cerebral ischemia model, further demonstrating their therapeutic potential in treating diseases related to excessive apoptosis. To our knowledge, this class of apoptosis inhibitors is by far the most potent apoptosis inhibitors reported.
Besides, by chemical genetic methods using one of these compounds (compound 33, also named compound A), we recently reported the identification of the succinate dehydrogenase subunit B (SDHB) of mitochondrial respiratory complex II as the target protein, which is also a new target for apoptosis inhibition.17 Compound 33 inhibits apoptosis by stabilizing mitochondrial respiratory complex II and protecting the integrity of the mitochondrial electron transport chain.17 Together, we revealed a class of new and potent apoptosis inhibitors that act on a novel target in the apoptosis signaling pathway, and demonstrated their therapeutic potential in excessive apoptosis-related disease models.
A chemical library containing 200,000 small molecules was screened for compounds that block apoptosis in a cell line (U2OS_Bim) where overexpression of a pro-apoptotic BCL-2 member Bim is induced by Doxycycline (DOX).18 Several active hits were identified and further confirmed as having EC50 values below 20 μM for maintaining cell survival following Bim-induced apoptosis. All screening hits were resynthesized, and their bioactivities were reconfirmed. Of these hit compounds, N-(2-hydroxy-5-methylphenyl)-4-((4-(thiophen-2-yl)-6-(trifluoromethyl) pyrimidin-2-yl)sulfonyl)butanamide (Figure 1a, EC50 = 4.0 μM) could prevent mitochondrial cytochrome c release (Figure 1b), which could not be achieved by caspase inhibitor zVAD. This indicated the 2-sulfonyl-pyrimidinyl compound functions upstream of caspase activation, and between BCL-2 family member regulation and cytochrome c release. Therefore, this compound was selected for further study.
Figure 1.
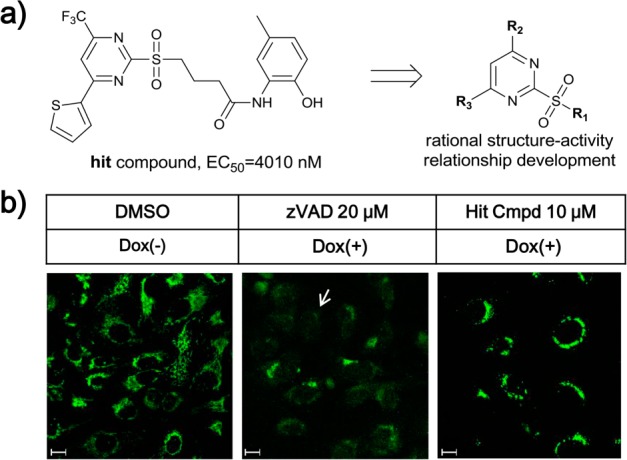
Hit compound identified from a phenotypic screen of apoptosis inhibition can block mitochondrial release of cytochrome c after Dox induced apoptosis. (a) (a) Structure and apoptosis inhibition activity of the hit compound, and the side groups (R1, R2, and R3) targeted in SAR development. (b) Immunofluorescence imaging of cytochrome c after treatment with the indicated compounds. The arrow on the middle column indicated an example of cytochrome c release. Scale bar: 20 μm.
To obtain more potent compounds to be used as both target identification tools and potential drug candidates, we undertook rational structure–activity relationship (SAR) optimization. We started by optimization of fragment R1 (Table 1). In each case, replacement of R1 with aliphatic groups of varying sizes increased the apoptosis inhibition activity by 3–5-fold (compounds 1–3). Aromatic substitutes were also tested (compounds 4, 5), and their activities were similar to that of the hit compound. These results indicate that fragment R1 was able to tolerate a relatively large change in its structure and maintain the activity. Because compound 1 had the strongest apoptosis inhibition activity, we replaced the original structure of R1 with a methyl group for further SAR optimization of other parts of the molecule (R2 and R3, Figure 1a).
Table 1. SAR Study of Compounds 1–20.
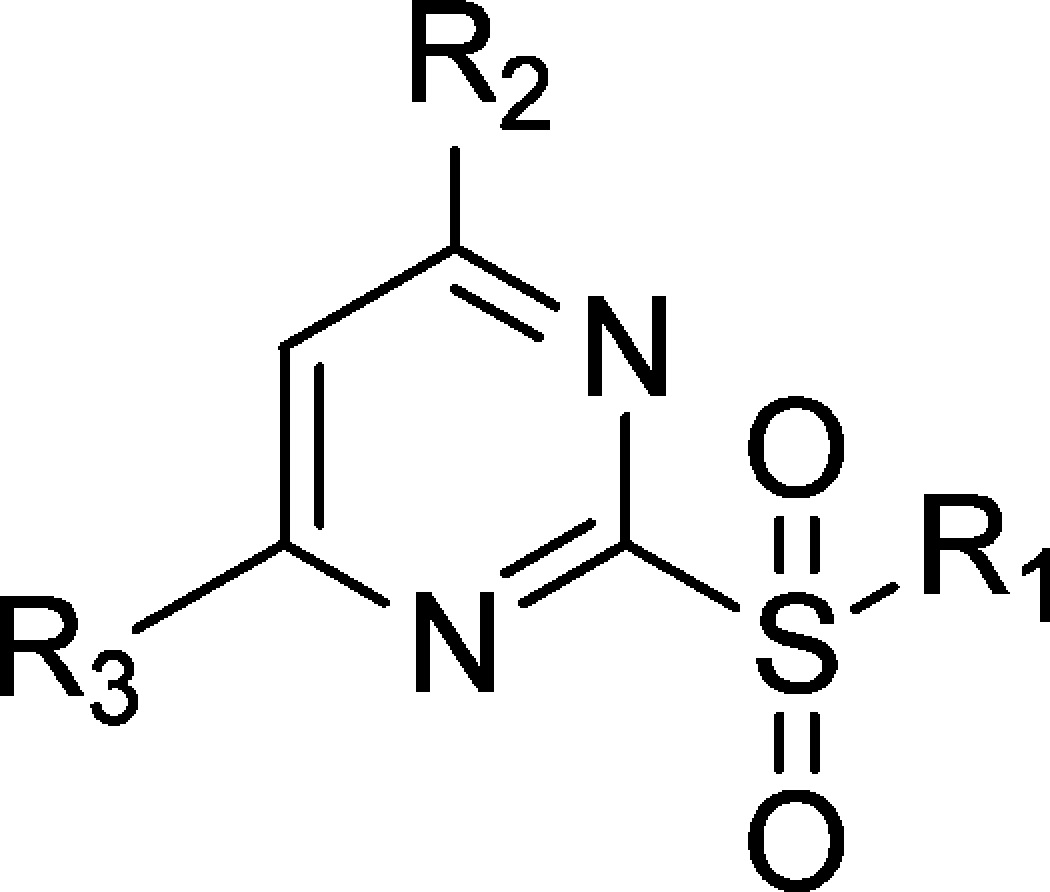
EC50: cellular apoptosis inhibition on the U2OS_Bim cell line.
We next substituted the −CF3 group at the R2 position with different groups (compounds 6–9); all such substitutions led to the total loss of activity (Table 1). This may be related to either a constrained binding environment or the necessity for the strong electron-withdrawing property of the original −CF3 group.
We next conducted SAR optimization of fragment R3 (Table 1). Although the replacement of the original thiophene group with phenyl rings (compound 10) resulted in decreased activity, it offered more options for testing the influence of substituents on different positions of the ring. Compounds with −CH3 and −COOCH3 substitutions at the 2-, 3-, and 4-positions of the phenyl ring had divergent activities (compounds 11–16). Substitutions at both the 2- and 4-positions (compounds 11, 13, 14, and 16) led to decreased activity compared with compound 10, whereas substitutions at the 3-position (compounds 12, 15) resulted in EC50 values similar to those for 10. This suggested more open space around the 3-position of the phenyl group. Compound 17, with a −CON(CH3)2 group, had a complete loss of activity, probably because of its polarity or unfavorable conformation. Though compound 18, which has a 3-oxybenzene group, had a 3-fold decrease in activity compared with compound 10, the result agreed with our assumption of more open space in this direction and encouraged us to do further SAR morphing at this position. Considering the convenience of modification and synthesis, we chose to replace the original thiophene group at the R3 position with the pyridone ring (compound 19) as an intermediate for further SAR exploration of the 3-position. Compound 19 had no activity, possibly because of the polarity of the pyridone ring. However, by adding a benzyl group to the N atom of the pyridone, we obtained compound 20, of which the activity increased by 2-fold compared with 10. This result further confirmed that a hydrophobic pocket has been reached by the addition of a benzyl group.
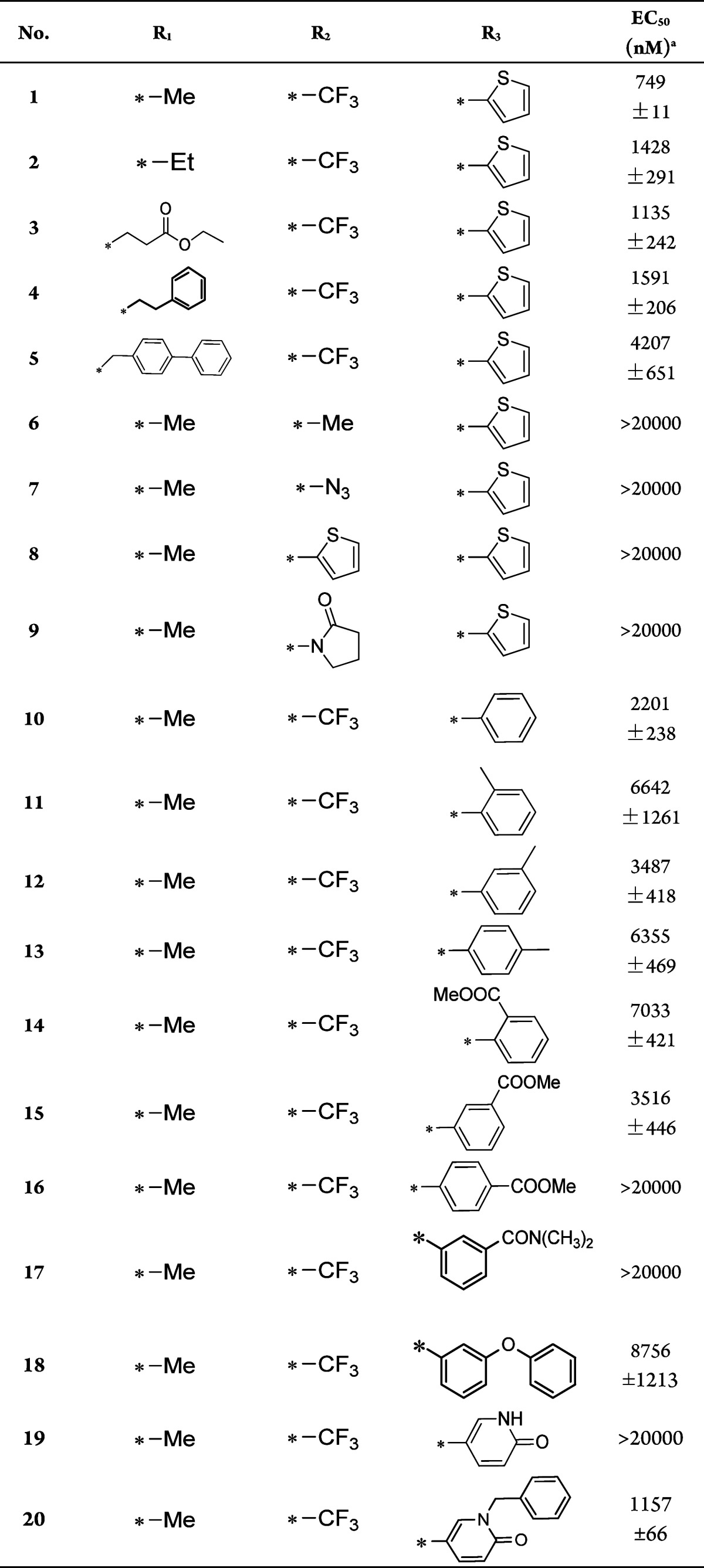
Encouraged by the result, SAR exploration on the benzyl ring was then performed in more detail (Table 2). The effects of chloro-substitution at different positions of the benzyl ring (compounds 21–23) were evaluated. The 3- and 4-chloro substitutions of the benzyl ring (compounds 22 and 23) led to increased apoptosis inhibition activity. We next focused on substitutions at these two positions. Compound 24, which has a 3-OH substitution, had the same activity as compound 20 and was less potent than the 3-chloro compound 22, which suggested a hydrophobic environment around the benzyl ring. The 3-OMe substitution of the benzyl ring (compound 25) increased activity by about 20-fold compared with compound 20. The −OEt- and −OPr-substituted compounds 26 and 27 displayed similar activities to that of compound 25. We subsequently examined the influence of 4-OMe substitution (compound 28) and found that it increased activity by 12-fold compared with the case of compound 20, similar to the increase observed for compound 25. In addition to oxyalkyl substitution, we also tested the activities of ethynyl and cyano groups at the 3- and 4-positions of the benzyl ring (compounds 29–32). Only compound 30, which has an ethynyl substitution at the 4-position of the benzyl ring, had an increase in activity compared with compound 20, and this increase was mild. Ethynyl substitution at the 3-position and cyano substitution at both positions led to diminished activity. Based on these results, we next combined the 3- and 4-position–OMe substitutions in compound 33 (compound A), which showed yet a further increase in apoptosis inhibition activity, with an EC50 as low as 25 nM. Thus, compound 33 is 150-fold more potent than the initial hit compound.
Table 2. SAR Study of Compounds 21–33.
| No. | R4 | EC50(nM)a |
|---|---|---|
| 21 | 2-Cl | 1143 ± 235 |
| 22 | 3-Cl | 257 ± 37 |
| 23 | 4-Cl | 66 ± 4.2 |
| 24 | 3-OH | 1821 ± 371 |
| 25 | 3-OMe | 57 ± 1.7 |
| 26 | 3-OEt | 110 ± 25 |
| 27 | 3-OPr | 69 ± 9.7 |
| 28 | 4-OMe | 94 ± 9.6 |
| 29 | 3-ethynyl | 4477 ± 1040 |
| 30 | 4-ethynyl | 817 ± 189 |
| 31 | 3-cyano | 4060 ± 477 |
| 32 | 4-cyano | 3489 ± 184 |
| 33b | 3,4-dimethoxy | 25 ± 0.57 |
EC50: cellular apoptosis inhibition in the U2OS_Bim cell line.
Compound A.
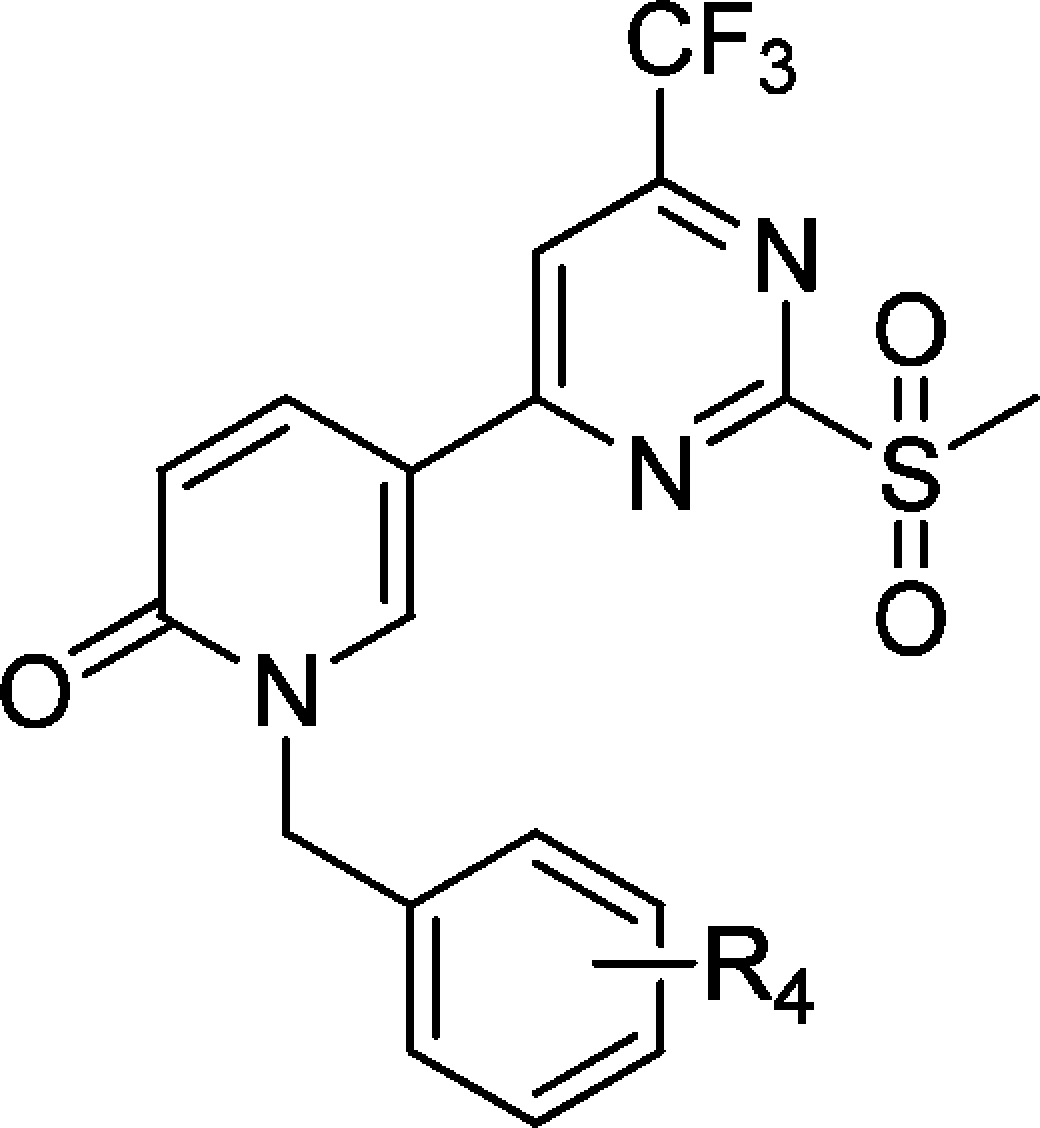
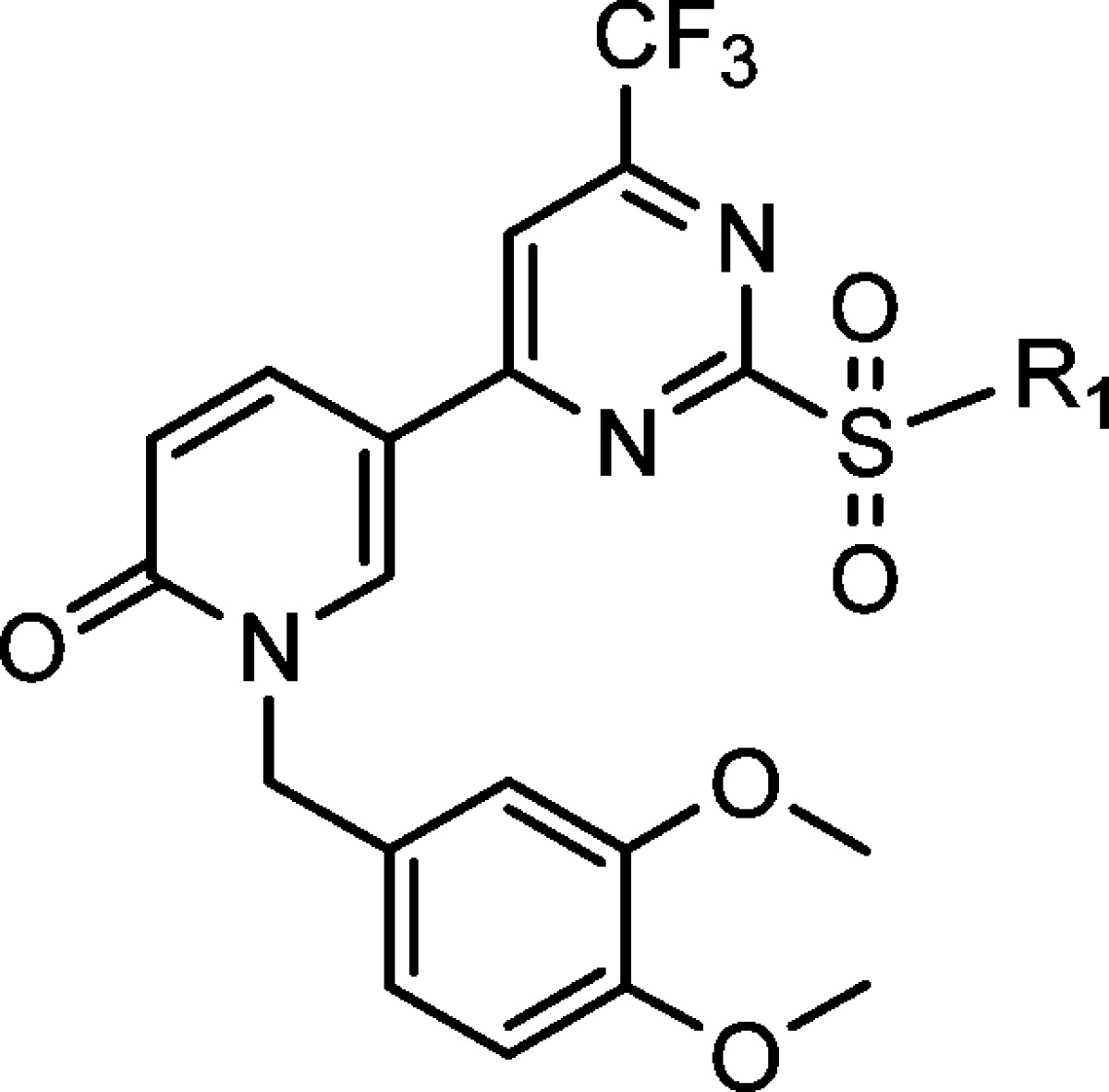
To better understand the structural requirements for activity and to further optimize potency, we undertook a second round of SAR optimization at the original R1 position (Table 3). Because the previous SAR analysis suggested a large tolerance for various structures, we focused on testing if extension of the R1 fragment could improve activity.
Table 3. SAR Study of Compounds 34–42.
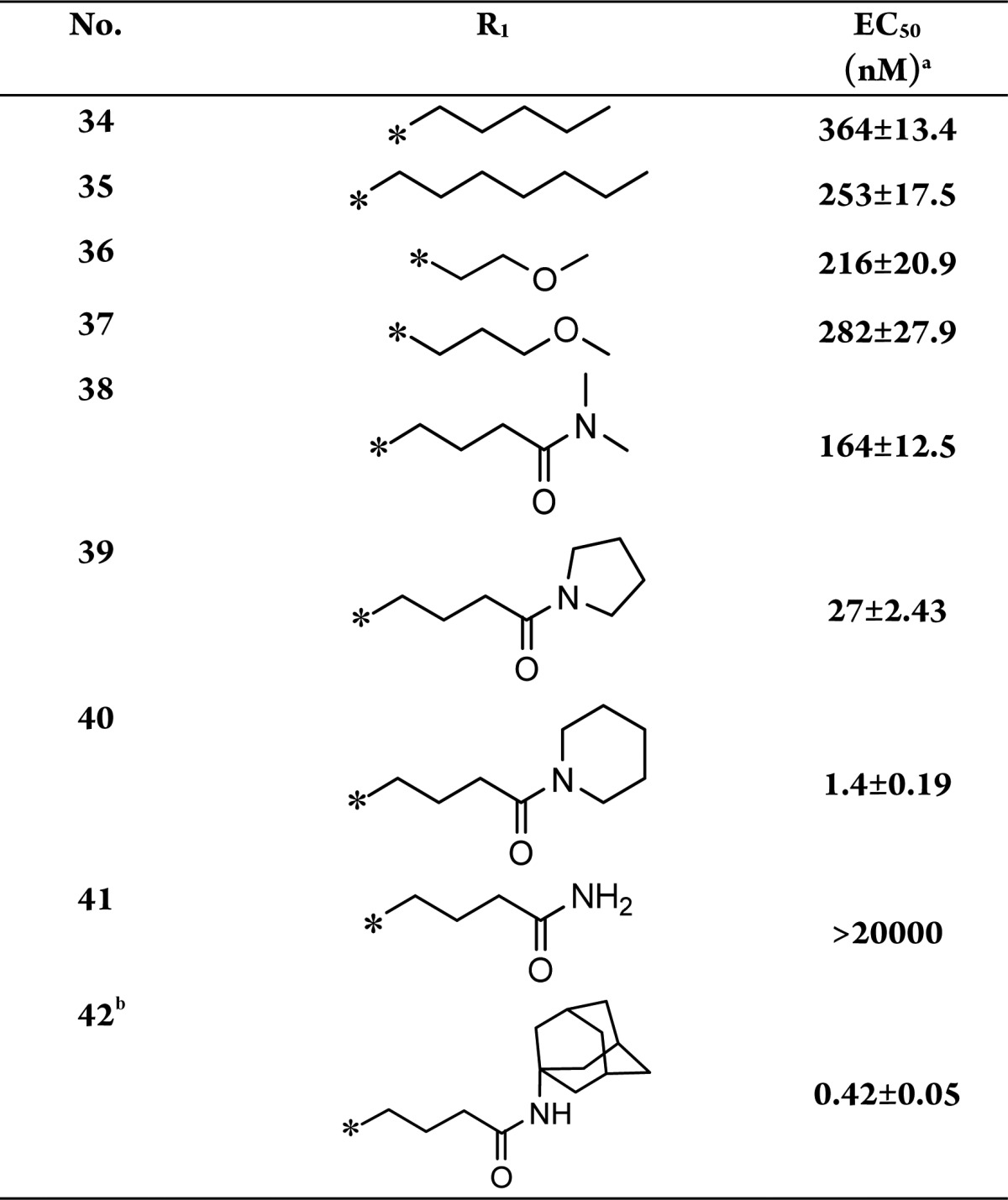
EC50: cellular apoptosis inhibiton activity.
Compound 42 is also named TC9-305.
We first replaced the methyl group in compound 33 with n-pentyl and n-heptyl groups (compounds 34 and 35), but lengthening the alkyl chain largely reduced cellular activity. Ether structures (compounds 36 and 37) were then tried; however, these did not improve the cellular activity much, either. An amide structure was then tested. Compound 38, which has N, N-dimethylbutyramide at the R1 position, had an EC50 of 164 nM; this is a mild increase compared with the activities of compounds 36 and 37. Replacement of the dimethylamine with pyrrolidine (39) further increased the activity by ∼5-fold compared with compound 38. We then tried the piperidine at the amide bond (compound 40), and activity increased dramatically to an EC50 of 1.4 nM, which is much higher than that observed for compounds 33 and 39. These observations indicate that large aliphatic moieties at the amide position are favorable for activity, and we speculate that the extended R1 moiety possibly interacts hydrophobically with the target protein. Consistent with our assumption, the introduction of an unsubstituted amide, a butyramide structure, at R1 (compound 41), was detrimental to activity. Therefore, to increase the hydrophobicity of the R1 fragment, we synthesized compound 42 (also named as TC9-305), which has a highly hydrophobic adamantane moiety. The activity of this compound increased to an EC50 of 0.42 nM, which is 50-fold higher than that of the parent compound 33.
In addition to testing the inhibition activity of the 2-sulfonyl-pyrimidinyl derivative compounds on Bim overexpression induced apoptosis, we also evaluated if they could block apoptosis induced by the overexpression of tBid, another pro-apoptotic protein of the Bcl-2 family. Most compounds displayed similar apoptosis inhibition activities, and compound 42 had an EC50 of 0.23 nM (Table S1).
By chemical genetic methods using a biotinylated derivative of compound 33 as bioaffinitive probe, we previously reported that compound 33 covalently binds the 243 cysteine of SDHB through an SN2 substitution at the sulfone position.17 Sulfone can serve as a good leaving group in SN2 substitution reactions when linked to electron-withdrawing aromatic rings.19−21 In the SAR study, we observed that the replacement of −CF3 at the R2 position with less powerful electron-withdrawing groups totally abolished activity (Table 1, compounds 6–9). In model reactions with different nucleophilic reagents (cysteine, lysine, and glutathione) (Figure S1, S2), we observed that compounds 1, 33, and 42 reacted with cysteine and glutathione but not lysine, whereas compound 6, of which the electron-withdrawing group −CF3 was replaced by an electron-donating CH3, did not react with any of them. This indicated the reactivity of the sulfone group in the tested compounds was consistent with the cellular activity of the corresponding compounds. We also confirmed that active 2-sulfonyl-pyrimidinyl derivative compounds shared the covalent binding property using a cellular wash/no-wash assay (Table S2).22 Before apoptosis induction and cell viability determination, two substets of cells were incubated with tested compounds, and then one subset of cells was washed several times to eliminate the unbound test compounds while the other subset of cells was not washed. Tested compounds all had similar EC50 values under these two conditions, indicating irreversible binding modes for these compounds.
In addition to blocking mitochondrial-mediated apoptosis, this class of 2-sulfonyl-pyrimidinyl derivative apoptosis inhibitors could also block the dysfunction of mitochondria that occurs following the induction of apoptosis. To evaluate mitochondrial function, we measured changes in mitochondrial membrane potential under different conditions using tetramethylrhodamine methyl ester (TMRM), a membrane potential sensitive red fluorescent dye. Dox induction of Bim overexpression is known to result in the loss of mitochondrial membrane potential and thus diminish TMRM enrichment during apoptosis.23 All of the tested compounds maintained TMRM enrichment and mitochondria membrane potential, an outcome that cannot be achieved with the caspase inhibitor zVAD (Figure 2a). The mitochondrial protective effect of the 2-sulfonyl-pyrimidinyl derivative apoptosis inhibitors was further supported by measurements of reactive oxygen species (ROS) levels taken following the induction of apoptosis. ROS levels are known to increase the following interruption of the electron transport chain and the resulting changes in membrane permeability that occur during apoptosis. Such ROS increases are understood to be stimulative factors for apoptosis induction and to cause further damage to cells.24 Treatment with the novel apoptosis inhibitor compounds maintained normal ROS levels, even upon apoptosis induction, in a dose-dependent manner (Figure 2b).
Figure 2.

Mitochondrial protective effects of a series of apoptosis inhibitors. (a) TMRM staining of mitochondria in US2OS-Bim cells with (1) no apoptosis induction and treatment with DMSO, and with apoptosis inducted by the addition of DOX in the presence of the following treatments: (2) DMSO; (3) zVAD (10 μM); (4) compound 20 (10 μM); (5) compound 22 (1 μM); (6) compound 23 (1 μM); (7) compound 33 (100 nM); (8) compound 42 (3 nM). Scale: 100 μm. (b) Mean cellular ROS levels in U2OS-Bim cells with (red bars) and without (blue bars) Dox induced apoptosis under the indicated compound treatments. Top: ROS levels under treatment with various apoptosis inhibitor compounds, DMSO, and zVAD. Bottom: ROS levels under treatment with different concentrations of compound 42, DMSO, and zVAD (20 μM). The error bars represent the standard error of results from two separate experiments. P values are based on comparisons of each sample to the DMSO-treated apoptosis induction group. *: P < 0.05; **: P < 0.01; ***: P < 0.001.
It is now firmly established that excessive apoptotic cell death is prominent in neurological disorders, such as Alzheimer’s disease and Parkinson’s disease, and it is also a main cause for cerebral ischemic stroke.25 Following cerebral ischemia, BH3-only proteins are upregulated and activate the intrinsic apoptosis pathway.26 Compound 33 has already been shown to promote cell survival in a Parkinson’s disease model.17 Here, we tested the potential efficiency of this series of apoptosis inhibitor compounds in a rat transient focal ischemia model.27 A nylon suture was introduced to occlude the origin of the middle cerebral artery (MCA). A 5 μL solution of different concentrations of compound 33 or 42 was injected into the left striatum after suture insertion. The suture was removed 1 h after insertion to allow reperfusion. After 24 h, animals were sacrificed. Evaluation of the brain infarct volume in different groups showed that both compounds 33 and 42 had obvious protective effects on brain tissues and reduced the brain infarct volume due to induction of focal cerebral ischemia (Figure 3). Consistent with the SAR study, at injection concentrations of 50 μM and 200 μM, compound 42 had a better protective effect than compound 33.
Figure 3.

Compound 33 (a) and compound 42 (b) have protective effects against ischemia-reperfusion injury. (left) Representative 2,3,5-triphenyltetrazolium chloride (TTC) stained sections from middle cerebral artery occlusion (MCAO) brains injected with 5 μL of the indicated concentrations of compounds 33 and 42. The white color represents the infarct, and the red color represents the normal tissue. (right) Quantification of relative infarct volume. The numbers 1–7 on the x-axis correspond to the treatments in the top panel. Bars represent means ± standard deviation, n = 3–6. P values are based on comparisons of each sample to the DMSO-treated ischemia-reperfusion (+) group. *: P, 0.05; **: P < 0.01; ***: P < 0.001.
In conclusion, using medicinal chemistry, we systematically developed a novel and highly potent series of apoptosis inhibitors. Based on an initial hit compound from phenotypic screening, we improved the cellular apoptosis inhibition activity of 2-sulfonyl-pyrimidinyl derivatives from low micromolar to picomolar levels through SAR optimization, and verified their covalent binding mode. This series of compounds showed mitochondrial protective effect as well as apoptosis inhibition. Following our previous work on the identification of the SDHB subunit of mitochondrial respiratory complex II as the interacting target of compound 33 and the demonstration of the protective effect on the Parkinson model, we further showed that compounds 33 and 42 are also highly efficacious on the transient focal cerebral ischemia model, indicating the great therapeutic potential in the development of novel therapies to treat apoptosis-related diseases.
Acknowledgments
We thank Zhenhua Zhang (China Agricultural University) for providing several azide reagents (synthesized in the National Key Technologies R&D Program of China, 2015BAK45B01, CAU) and Qinsi Ji for his synthesis work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00489.
Detailed experimental procedure, supporting figures and tables, and synthesis and characterization of all compounds and intermediates (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
This work is supported by National High Technology Project 973 (2011CB504300).
The authors declare no competing financial interest.
Supplementary Material
References
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007, 35 (4), 495–516. 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie A. H. Where, O death, is thy sting?” A brief review of apoptosis biology. Mol. Neurobiol. 2010, 42 (1), 4–9. 10.1007/s12035-010-8125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1 (2), 120–130. 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Roshal M.; Zhu Y.; Planelles V. Apoptosis in AIDS. Apoptosis 2001, 6 (1–2), 103–116. 10.1023/A:1009636530839. [DOI] [PubMed] [Google Scholar]
- Patel T.; Roberts L. R.; Jones B. A.; Gores G. J. In Dysregulation of apoptosis as a mechanism of liver disease: an overview; Seminars in liver disease; Thieme Medical Publishers, Inc.: 1998; pp 105–114. [DOI] [PubMed] [Google Scholar]
- Favaloro B.; Allocati N.; Graziano V.; Di Ilio C.; De Laurenzi V. Role of apoptosis in disease. Aging 2012, 4 (5), 330–349. 10.18632/aging.100459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W. Ischemia-induced neuronal apoptosis. Curr. Opin. Neurobiol. 1996, 6 (5), 667–672. 10.1016/S0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- Mayer B.; Oberbauer R. Mitochondrial regulation of apoptosis. Physiology 2003, 18 (3), 89–94. 10.1152/nips.01433.2002. [DOI] [PubMed] [Google Scholar]
- Gross A. BCL-2 Proteins: Regulators of the Mitochondrial Apoptotic Program. IUBMB Life 2001, 52 (3–5), 231–236. 10.1080/15216540152846046. [DOI] [PubMed] [Google Scholar]
- Reed J. C.; Huang Z. Apoptosis pathways and drug targets. Nature Rev. Drug Discov 2004, 3 (111), 895–981. [Google Scholar]
- Samali A.; Nordgren H.; Zhivotovsky B.; Peterson E.; Orrenius S. A comparative study of apoptosis and necrosis in HepG2 cells: oxidant-induced caspase inactivation leads to necrosis. Biochem. Biophys. Res. Commun. 1999, 255 (1), 6–11. 10.1006/bbrc.1998.0139. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P.; Vanden Berghe T.; Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci. STKE 2006, 2006 (358), pe44. 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- Bombrun A.; Gerber P.; Casi G.; Terradillos O.; Antonsson B.; Halazy S. 3, 6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J. Med. Chem. 2003, 46 (21), 4365–4368. 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- Hetz C.; Vitte P.-A.; Bombrun A.; Rostovtseva T. K.; Montessuit S.; Hiver A.; Schwarz M. K.; Church D. J.; Korsmeyer S. J.; Martinou J.-C. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J. Biol. Chem. 2005, 280 (52), 42960–42970. 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- Becattini B.; Sareth S.; Zhai D.; Crowell K. J.; Leone M.; Reed J. C.; Pellecchia M. Targeting apoptosis via chemical design: inhibition of bid-induced cell death by small organic molecules. Chem. Biol. 2004, 11 (8), 1107–1117. 10.1016/j.chembiol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Becattini B.; Culmsee C.; Leone M.; Zhai D.; Zhang X.; Crowell K. J.; Rega M. F.; Landshamer S.; Reed J. C.; Plesnila N. Structure–activity relationships by interligand NOE-based design and synthesis of antiapoptotic compounds targeting Bid. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (33), 12602–12606. 10.1073/pnas.0603460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Li L.; Ying Z.; Pan C.; Huang S.; Li L.; Dai M.; Yan B.; Li M.; Jiang H. A Small Molecule That Protects the Integrity of the Electron Transfer Chain Blocks the Mitochondrial Apoptotic Pathway. Mol. Cell 2016, 63 (2), 229–239. 10.1016/j.molcel.2016.06.016. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Jiang H.; Shen Z.; Wang X. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (41), 14782–14787. 10.1073/pnas.1417253111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferkorn J. A.; Lou J.; Minich M. L.; Filipski K. J.; He M.; Zhou R.; Ahmed S.; Benbow J.; Perez A.-G.; Tu M. Pyridones as glucokinase activators: identification of a unique metabolic liability of the 4-sulfonyl-2-pyridone heterocycle. Bioorg. Med. Chem. Lett. 2009, 19 (12), 3247–3252. 10.1016/j.bmcl.2009.04.107. [DOI] [PubMed] [Google Scholar]
- Litchfield J.; Sharma R.; Atkinson K.; Filipski K. J.; Wright S. W.; Pfefferkorn J. A.; Tan B.; Kosa R. E.; Stevens B.; Tu M. Intrinsic electrophilicity of the 4-methylsulfonyl-2-pyridone scaffold in glucokinase activators: role of glutathione-S-transferases and in vivo quantitation of a glutathione conjugate in rats. Bioorg. Med. Chem. Lett. 2010, 20 (21), 6262–6267. 10.1016/j.bmcl.2010.08.095. [DOI] [PubMed] [Google Scholar]
- Kathman S. G.; Statsyuk A. V. Covalent tethering of fragments for covalent probe discovery. MedChemComm 2016, 7 (4), 576–585. 10.1039/C5MD00518C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Hur W.; McDermott U.; Dutt A.; Xian W.; Ficarro S. B.; Zhang J.; Sharma S. V.; Brugge J.; Meyerson M.; Settleman J.; Gray N. A structure-guided approach to creating covalent FGFR inhibitors. Chem. Biol. 2010, 17 (3), 285–295. 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly J. D.; Grubb D.; Lawen A. The mitochondrial membrane potential (Δψm) in apoptosis; an update. Apoptosis 2003, 8 (2), 115–128. 10.1023/A:1022945107762. [DOI] [PubMed] [Google Scholar]
- Circu M. L.; Aw T. Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biol. Med. 2010, 48 (6), 749–762. 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U.; Iadecola C.; Moskowitz M. A. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999, 22 (9), 391–397. 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Niizuma K.; Yoshioka H.; Chen H.; Kim G. S.; Jung J. E.; Katsu M.; Okami N.; Chan P. H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta, Mol. Basis Dis. 2010, 1802 (1), 92–99. 10.1016/j.bbadis.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uluç K.; Miranpuri A.; Kujoth G. C.; Aktüre E.; Başkaya M. K. Focal cerebral ischemia model by endovascular suture occlusion of the middle cerebral artery in the rat. J. Visualized Exp. 2011, 48, e1978–e1978. 10.3791/1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.