

Abstract
Background
Glioblastoma (GBM), a highly aggressive brain tumor, contains a subpopulation of glioblastoma stem-like cells (GSCs) that play roles in tumor maintenance, invasion, and therapeutic resistance. GSCs are therefore a promising target for GBM treatment. Our group identified the cellular prion protein (PrPC) and its partner, the co-chaperone Hsp70/90 organizing protein (HOP), as potential target candidates due to their role in GBM tumorigenesis and in neural stem cell maintenance.
Methods
GSCs expressing different levels of PrPC were cultured as neurospheres with growth factors, and characterized with stem cells markers and adhesion molecules markers through immunofluorescence and flow cytometry. We than evaluated GSC self-renewal and proliferation by clonal density assays and BrdU incorporation, respectively, in front of recombinant HOP treatment, combined or not with a HOP peptide which mimics the PrPC binding site. Stable silencing of HOP was also performed in parental and/or PrPC-depleted cell populations, and proliferation in vitro and tumor growth in vivo were evaluated. Migration assays were performed on laminin-1 pre-coated glass.
Results
We observed that, when GBM cells are cultured as neurospheres, they express specific stemness markers such as CD133, CD15, Oct4, and SOX2; PrPC is upregulated compared to monolayer culture and co-localizes with CD133. PrPC silencing downregulates the expression of molecules associated with cancer stem cells, upregulates markers of cell differentiation and affects GSC self-renewal, pointing to a pivotal role for PrPC in the maintenance of GSCs. Exogenous HOP treatment increases proliferation and self-renewal of GSCs in a PrPC-dependent manner while HOP knockdown disturbs the proliferation process. In vivo, PrPC and/or HOP knockdown potently inhibits the growth of subcutaneously implanted glioblastoma cells. In addition, disruption of the PrPC-HOP complex by a HOP peptide, which mimics the PrPC binding site, affects GSC self-renewal and proliferation indicating that the HOP-PrPC complex is required for GSC stemness. Furthermore, PrPC-depleted GSCs downregulate cell adhesion-related proteins and impair cell migration indicating a putative role for PrPC in the cell surface stability of cell adhesion molecules and GBM cell invasiveness, respectively.
Conclusions
In conclusion, our results show that the modulation of HOP-PrPC engagement or the decrease of PrPC and HOP expression may represent a potential therapeutic intervention in GBM, regulating glioblastoma stem-like cell self-renewal, proliferation, and migration.
Electronic supplementary material
The online version of this article (doi:10.1186/s13287-017-0518-1) contains supplementary material, which is available to authorized users.
Keywords: Cellular prion protein, Hsp70/90 organizing protein, Glioblastoma, Stem cells, Proliferation
Background
Glioblastoma (GBM) is the most common and aggressive type of central nervous system (CNS) tumor and is one of the most lethal human cancers [1]. This aggressive behavior has been attributed to a highly proliferative subset of cells called glioblastoma stem-like cells (GSCs) that contribute to tumor initiation and therapeutic resistance [2]. These cells are thought to be responsible for maintaining GBM tumors after therapy and repopulating them after total resection [3]. In addition, they are involved in tumor angiogenesis [4] and immune evasion [5], promoting tumor progression.
The cellular prion protein (PrPC) and the Hsp70/Hsp90 organizing protein (HOP) modulate several stem cell functions. PrPC regulates proliferation and self-renewal of neural stem cells (NSCs) [6], while HOP participates in the maintenance of pluripotent stem cells [7]. Previous data from our group show that PrPC modulates NSC proliferation and self-renewal through its interaction with HOP [6]. Both PrPC and HOP also modulate tumorigenesis, affecting the progression and maintenance of different types of cancers [8]. PrPC associates with a poor clinical outcome and survival in pancreatic ductal adenocarcinoma and melanoma [9, 10], and with invasion and metastasis in gastric and breast cancers [8, 11, 12]. Depleting PrPC inhibits growth, promotes programmed cell death in gliomas [13], and sensitizes tumor cells to cytotoxic drugs [14]. Likewise, HOP expression correlates with tumor progression [15], and is associated with proliferation [16, 17], invasion [17], and poor patient prognosis [18], and HOP has been described as an important regulator of tumor maintenance [19–21]. Recent findings show that HOP-PrPC binding modulates migration and invasion of colorectal cancer cells [22]. Erlich et al. [16] reported that the interaction of PrPC with HOP modulates proliferation in glioma cell lines, and data from our group showed that higher expression of both proteins is correlated with greater tumor proliferation and lower survival in patients with GBM [15]. In addition, blocking the HOP-PrPC complex decreases tumor growth and increases survival in an animal model [15], making it a potential target for GBM therapy.
While the HOP-PrPC complex participates both in GBM tumorigenesis and in the maintenance of NSCs—which are believed to originate GSCs—their role in GSC biology is still unclear. Therefore, this study aimed to elucidate the role of the HOP-PrPC complex in the regulation of GSCs by downregulating the expression of PrPC in a human GBM cell line cultured as GSCs and by testing how proliferation and self-renewal properties were affected in the presence of exogenous HOP; we also wished to evaluate the therapeutic potential of targeting this complex using synthetic peptides in attempt to alter GSC biology. Additionally, we investigated whether PrPC may be used as a novel biomarker for GBM by studying its role in GSC stemness.
Methods
Proteins and peptides
Mouse recombinant HOP was purified as previously described [23]. Human pepHOP230–245 (ELGNDAYKKKDFDTAL) and C-terminal pepHOP422–437 (GCKTVDLKPDWGKGYS) peptides were synthesized by GenScript (Piscataway, NJ, USA).
Cell culture
The human U87 and U251 glioblastoma cell lines (ATCC) were cultured in DMEM-F12, supplemented with B27 (Cat No. 17504-044; Gibco, Gaithersburg, MD, USA; 1:50) in the presence of 20 ng/ml epidermal growth factor (EGF; Cat No. E4127; Sigma Aldrich, St. Louis, MO, USA) and basic fibroblast growth factor (bFGF; Cat No. F0291; Sigma Aldrich) at 37 °C under 5% CO2 to form neurospheres. The medium was replaced every 2 days. After 1 week, cells were treated with 0.25% trypsin (Cat No. 25200-056; Gibco) in HBSS (Cat No. 14170-112; Gibco) for 5 min at 37 °C. Trypsin was washed out and the cells were mechanically dissociated and plated for distinct assays.
PrPC/HOP silencing
U87 cells were infected with lentiviral particles carrying two constructions targeted to the human PrPC sequence [15]. Efficiency was low for shRNA-PrP1 and high for shRNA-PrP2. Therefore, shRNA-PrP1 was used to silence PrPC to intermediary levels, while shRNA-PrP2 was used for low PrPC expression. Replication-deficient lentiviral particles were produced in HEK293FT cells using the ViraPower Lentiviral Expressing System (Invitrogen) according to the manufacturer’s instructions. Dr. Andrew Hill (La Trobe University, Australia) kindly provided the constructs for shRNA-PrPC sequences. The following pLenti constructs were used for the shRNA-PrPC sequence:
shRNA-PrP1: 5′-caccgcgtcaatatcacaatcaagccgaagcttgattgtgatattgacgc-3′
shRNA-PrP2: 5′- caccagaacaacttcgtgcacgactcgaaaagtcgtgcacgaagttgttc-3′
Stable silencing of HOP was performed using MISSION® shRNA (Sigma Aldrich) according to the manufacturer’s instructions using the following sequences: TRC 2.0; NM_006819 – Mission – SIGMA/TRCN0000243096 and TRCN0000243099.
CRISPR/Cas9 for PrPC knockout
The human PrPC gene sequence (NM_000311.3) was used to design the guide RNA using the Optimized CRISPR Design (http://crispr.mit.edu/). gRNA sequences selected were: Hu PrPC (Top1) CACCGgctgggggcagccgatacccg/Hu PrPC (Bottom1) AAACcgggtatcggctgcccccagcC. The gRNAs were phosphorylated, annealed, and cloned into px330-U6-GFP vector (Addgene) according to the Addgene website instructions. The construct was sequenced, transfected into the U251 cell line with lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions, and the clones were isolated by serial dilution.
Flow cytometry analysis
Cells (106) were dissociated, washed twice with phosphate-buffered saline (PBS), and incubated with anti-PrPC [23], anti-CD133 (Cat No. 130-090-852; Miltenyi, Auburn, CA, USA), CD15 (Cat No. FCMAB182F; Millipore, Temecula, CA, USA), anti-E-cadherin (Cat No. 610181; BD Bioscience, San Diego, CA, USA), and anti-integrin α6 (Cat No. ab97760; Abcam, Cambridge, UK) antibodies, all at 1:50 dilution in 0.5% bovine serum albumin (BSA) in PBS for 60 min at 4 °C. After washing, samples were incubated with anti-mouse IgG Alexa-488/PE (Cat No. A21200; Invitrogen, Carlsbad, CA, USA; 1:200) or with anti-rabbit IgG Alexa-488/405 (Cat No. A21441; Invitrogen; 1:200) antibodies for 60 min at 4 °C. For PrPC internalization assays, cells were pre-incubated for 40 min in 250 μM solution of CuSO4 in PBS plus 5% BSA at 37 °C, followed by antibody incubation. Only secondary staining was used for the negative control. Cells were analyzed by flow cytometry for forward scatter, side scatter, and fluorescence (FACSCanto II; BD Biosciences).
Immunoblotting assays
For the analysis of neurosphere protein extracts, cells were cultured overnight (2 × 105 cells/well), starved for 24 h, treated with serum or recombinant HOP for 15 min, and washed with cold PBS. Protein extracts were prepared in RIPA buffer with protease/phosphatase inhibitors, centrifuged (10,000 × g), and then loaded (5 μg) onto a 10% SDS-PAGE gel, followed by immunoblotting with polyclonal anti-HOP (1:10,000) [23], anti-phospho Erk1/2 (Cat No. 4370S; Cell Signaling, Danvers, MA, USA; 1:4000), anti-Erk1/2 (Cat No. 4695S; Cell Signaling; 1:4000) or anti-PrPC [23]. Anti-GAPDH (Cat No. G9545; Sigma Aldrich) or anti-actin antibodies (Cat No. A2103; Sigma Aldrich) were used as protein loading controls.
Immunofluorescence staining
For PrPC and Oct4 staining, whole neurospheres were harvested, fixed in 4% paraformaldehyde, and paraffin-embedded. For Ki67 experiments, xenografts (Balb/c nude mice) were resected, fixed in 4% paraformaldehyde, and paraffin-embedded. Slides with 3-μm sections were prepared for immunofluorescence. Sections were incubated in xylol at 60 °C for paraffin removal and immersed in citrate buffer, pH 6.0, for 1 h at 96 °C for antigen retrieval. Sections were then blocked for 1 h at room temperature (RT) in 5% BSA in PBS and incubated overnight at RT with anti-PrPC (1:50) [23] or anti-Oct4 (Cat No. 2840S; Cell signaling; 1:50) in 1% BSA in PBS. After washing, slides were incubated for 1 h at RT with anti-mouse Alexa-488 (Cat No. A21202; Invitrogen; 1:1000) or anti-rabbit Alexa-546 (Cat No. A10040; Invitrogen; 1:1000), and stained with TO-PRO (Cat No. T3605; Molecular Probes, Eugene, OR, USA) for nuclei. For other markers, whole neurospheres were harvested, plated on coverslips previously treated with poly-l-lysine and fixed in 4% paraformaldehyde. Coverslips were blocked for 1 h at RT with 5% BSA plus 0.3% triton in PBS. Coverslips were incubated overnight at RT with anti-PrPC (1:100) and anti-HOP (1:100) [23], anti-nestin (Cat No. N5413; Sigma Aldrich; 1:100), anti-Musashi1 (Cat No. 5663P; Cell Signaling; 1:100), anti-Sox2 (Cat No. ab75485; Abcam; 1:100), anti-βIII tubulin (Cat No. 5568S; Cell Signaling; 1:100), anti-β catenin (Cat No. ab32572; Abcam; 1:100), anti-CD133 (Cat No. MAB4399; Millipore; 1:100), anti-GFAP (Cat No. Z0334; Dako, Cambridge, UK; 1:100) or anti-E-cadherin (Cat No. 610181; BD Bioscience; 1:100) in 1% BSA 0.1% triton in PBS. After washing, coverslips were incubated for 1 h at RT with anti-mouse Alexa-488 (Cat No. A21202; Invitrogen; 1:1000) or anti-rabbit Alexa-546 (Cat No. A10040; Invitrogen; 1:1000), and stained with TO-PRO (Cat No. T3605; Molecular Probes) or DAPI (Cat No. D1306; Invitrogen) for nuclei. Cells were imaged on a Leica TCS SP2 II laser scanning confocal system.
Cell proliferation assay
Whole neurospheres were harvested and plated on coverslips previously treated with poly-l-lysine in DMEM-F12 supplemented with B27 at 37 °C. Control cells were treated only with the growth factors (20 ng/ml) EGF (Cat No. E4127; Sigma Aldrich) and bFGF (Cat No. F0291; Sigma Aldrich), and for HOP treatments cells were treated with growth factors and recombinant HOP and/or HOP peptides (1 μM) for 24 h. Cells received a 60-μM BrdU pulse 3 h prior to fixation in 4% paraformaldehyde. Fixed cells were treated with HCl 2 N for 30 min, washed with borate buffer, and permeabilized with 0.3% Triton X-100 in PBS for 15 min. Cells were blocked in 0.3% Triton 5% BSA in PBS for 1 h and stained with biotin-conjugated anti-BrdU (Cat No. MAB3262B; Millipore; 1:100), Strepta-AlexaFluor-546 (Cat No. S11225; Molecular Probes; 1:500), and DAPI (Cat No. D1306; Invitrogen; 1:500) for nuclei. Images were taken from at least four microscopic fields (Zeiss AxioVertA1) for each duplicate per treatment and analyzed on ImageJ software (NIH). The percentage of BrdU-positive nuclei in the total number of nuclei (DAPI) was calculated. HOP-silenced populations were plated 24 h prior to performing the transfection and proliferation assays (as described above). For colorimetric assays, cells were treated with growth factors and recombinant HOP and/or HOP peptides (1 μM) for 24 h and the Cell Proliferation ELISA BrdU kit (Cat. No. 11647229001; Roche, Indianapolis, IN, USA) was utilized according to the manufacturer’s instructions.
Clonal density assay
Neurospheres were treated with 0.25% trypsin (Cat No. 25200-056; Gibco) in HBSS (Cat No. 14170-112; Gibco) for 20 min at 37 °C. Trypsin was washed out and cells were mechanically dissociated. In each well (96-well plate), 200 cells were plated in triplicates per condition (control or treatment with 500nM recombinant HOP and/or HOP peptides). Cells were treated every 48 h for 1 week. Images were acquired through light microscopy (Zeiss PrimoVert) and the number and size of neurospheres were evaluated and compared between conditions with the ZEN software (Zeiss). The optimal concentration of HOP for self-renewal or proliferation assays was chosen based on previous experiments of dose-response curve (data not shown) or according to previous data [6], respectively.
Migration assay
Neurospheres were plated on coverslips previously treated with poly-l-lysine and laminin-1 (5 μg/ml) in DMEM-F12 supplemented with B27, and the growth factors EGF (Cat No. E4127; Sigma Aldrich) and bFGF (Cat No. F0291; Sigma Aldrich) and 2% fetal bovine serum (FBS; Lot No. 003/14; Vitrocell, Campinas, Spain), and cultured for 24 h at 37 °C. Images were acquired through light microscopy (ZeissPrimoVert) and the halo of migration was compared to neurosphere radius to evaluate cell migration using ZEN software (Zeiss). For cell scratch assays, neurospheres were dissociated and plated 0.2 × 106 per well (six-well plate), the scratch was performed, and images were acquired at 0 h and 24 h after the scratch. Images of three experimental replicates were acquired using Zeiss PrimoVert microscope and the distance of each scratch closure after 24 h was measured by comparing with the images at time 0 h using ZEN software (Zeiss).
In vivo tumor growth
Institutional guidelines for animal welfare were followed and the study approved by the Animal Ethics Committee of the Institute of Biomedical Sciences/University of Sao Paulo (book 03, page 15, protocol number 002 of 04/03/2014). Neurospheres (1 × 106) as single cells were injected subcutaneously into the flank of female Balb/C nude mice (12 weeks old) in PBS. Tumor growth was measured every 2 days and euthanasia by CO2 saturation was performed on day 10 after tumor detection. Tumors were resected and fixed in paraformaldehyde 4%.
Statistical analysis
One-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test was used for multiple comparisons. A p value <0.05 was considered statistically significant. The non-parametric Student’s t test was also used in migration assays. Mean values represent at least three independent data sets; error bars represent standard errors of the mean (SEM).
Results
Characterization of neurosphere culture from GBM cell line
We compared the expression of several stem cell markers in monolayer and neurosphere cultures of the U87 glioblastoma cell line. Neurospheres showed higher expression of the stem cells markers CD15, CD133, Oct4, Musashi-1, and Sox2, suggesting enrichment in the number of stem-like cells and thus supporting the use of the neurosphere-formation assay as a model to study GSCs (Fig. 1a–d). However, U87 monolayer cultures and neurospheres presented similar expression of the neural precursor marker nestin (Fig. 1c).
Fig. 1.
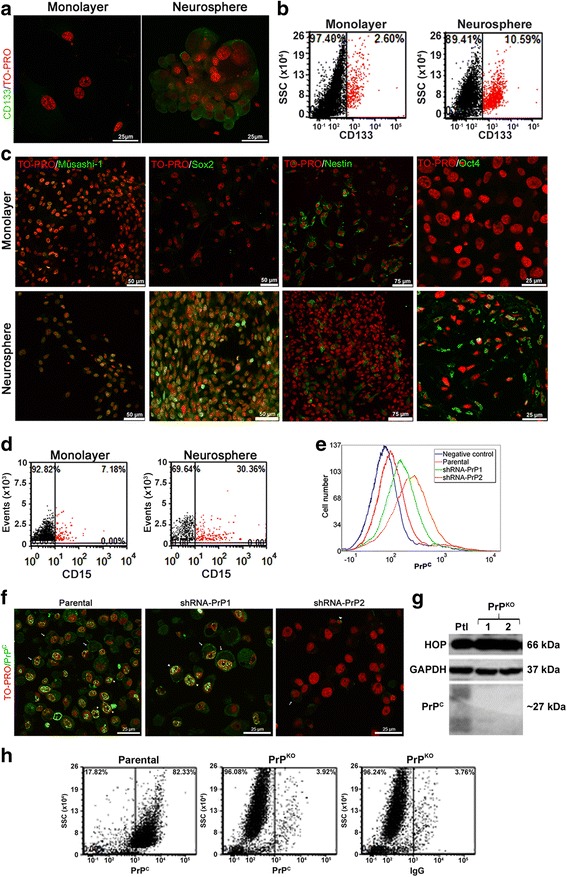
Characterization of glioblastoma U87 and U251 neurospheres. a Immunofluorescence for CD133 (green) in U87 cultured as monolayer plus serum (left) or neurospheres (right). Nuclei staining (TO-PRO) shown in red. b Dot plot for CD133 expression in monolayer cultured with serum (left) and neurospheres (right). CD133+ cells shown in red and CD133– cells in black . c Immunostaining for the stem cells markers Oct4, Musashi-1, Sox2, and Nestin (green) in monolayer (upper) and neurospheres (lower), with nuclei staining (TO-PRO) shown in red. d Dot plot graph for CD15 expression in monolayer cultured with serum (left) and as neurospheres (right). CD15+ shown in red and CD15– in black. e Cellular prion protein (PrP C) expression assessed by flow cytometry in parental (orange), shRNA-PrP1 (green), or shRNA-PrP2 (red) populations. Negative control shown in blue (only secondary antibody staining). f Immunofluorescence for PrPC (green), in parental (left), shRNA-PrP1 (middle), or shRNA-PrP2 (right) populations. Arrow indicates staining on the cell surface and arrowhead in the perinuclear region. Nuclei staining (TO-PRO) shown in red. g Immunoblot for Hsp70/90 organizing protein (HOP) (top) and PrPC (bottom) expression in U251 knockout (PrP KO) clones (1 and 2) compared to the parental (Ptl) population. GAPDH was used as the loading control. Note that the smear for PrPC immunostaining is due to the different glycosylated isoforms. h Flow cytometry for PrPC expression in the U251 populations parental (left) and U251 PrP-knockout clone 2 (PrPKO) (middle). IgG isotype (right) was used as the negative control
Effect of PrPC modulation on the expression of stem-cell markers
Due to their roles in GBM and NSCs, PrPC and HOP were proposed to modulate essential functions in GSC biology. To better understand these functions, an shRNA lentivirus system was used in the U87 cell line to silence PrPC to intermediary (shRNA-PrP1) and low (shRNA-PrP2) levels (Fig. 1e and f). The CRISPR/Cas9 technique was used to generate PrPC knockout U251 cells (Fig. 1g). U251 clone 2 presented clear depletion of PrPC expression (PrPKO) and was selected to perform functional experiments (Fig. 1g and h). Both monolayer populations (shRNA-PrP1and shRNA-PrP2), previously used in [15], and also PrPKO were cultured as neurospheres.
Compared to monolayer cells, PrPC is upregulated in neurospheres (Fig. 2a); therefore, the detection of its expression may indicate tumor enrichment in stem-like populations. Additionally, the stem cells marker CD133 was co-expressed with PrPC and co-localized partially on the plasma membrane of neurospheres (Fig. 2b), being internalized with PrPC in the presence of copper presenting a similar endocytosis profile (Fig. 2c and d), suggesting that these molecules can form a functional complex on the membrane domain.
Fig. 2.
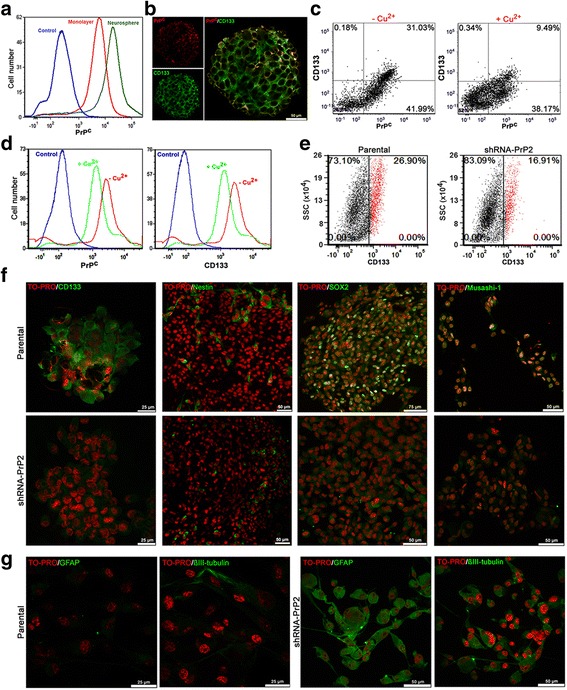
Stem cells marker expression in cellular prion protein (PrP C)-depleted neurospheres. a PrPC expression assessed by flow cytometry in parental monolayer (red) and neurosphere (green) cultures. Negative control shown in blue (only secondary antibody staining). b Immunofluorescence for PrPC (red) and CD133 (green) in parental neurospheres shows co-localization on the cell surface. c Dot plot of CD133 and PrPC expression in parental neurospheres in the absence (–Cu2+) or presence (+Cu2+) of CuSO4 250 μM. d Histogram for PrPC and CD133 in the absence (–Cu2+) and presence (+Cu2+) of CuSO4 250 μM. Negative control shown in blue (only secondary antibody staining). e Dot plot of CD133 expression in parental (left) and shRNA-PrP2 (right) neurospheres. CD133+ shown in red and CD133– shown in black. f Immunofluorescence for the stem cells markers musashi-1, nestin, Sox2, and CD133 (green) in parental (upper) and shRNA-PrP2 (lower) neurospheres. Nuclei staining (TO-PRO) shown in red. g Immunofluorescence for the cell differentiation markers GFAP and βIII-tubulin (green) in parental (left) and shRNA-PrP2 (right) neurospheres after 5 days of serum treatment. Nuclei staining (TO-PRO) shown in red
The stem cell marker CD133 expression is decreased in the U87 PrP2 population compared to the parental population (Fig. 2e), and Sox2 had higher expression in the parental population than in the PrP2 population (Fig. 2f). Musashi-1, on the other hand, presented different cellular locations: nuclei for parental population and cytoplasm for PrP2 (Fig. 2f). No significant difference in expression of the neural precursor marker nestin was observed between populations (Fig. 2f). Cellular differentiation markers, such as GFAP and βIII-tubulin, were highly expressed in PrP2 cells compared to parental neurospheres after serum stimulation (Fig. 2g). These findings reveal a potential role for PrPC in the regulation of the GSC multipotent status.
The PrPC-HOP complex modulates GSC proliferation
Recently, our group described that the expression of PrPC and HOP associates with GBM malignancy [15] and, considering that GBM contains stem cells implicated in tumor development, we evaluated the role of the PrPC-HOP interaction in GSC biology. We observed a clear co-localization of PrPC and HOP on the cell surface of neurospheres (Fig. 3a) and both proteins are highly expressed in neurospheres (Fig. 3b). In addition, GSC neurospheres from both parental and shRNA-PrPC populations present equivalent endogenous (Fig. 3c and d) and secreted (Fig. 3e and f) HOP levels, indicating that the expression pattern of HOP is not affected by PrPC ablation in GSCs.
Fig. 3.
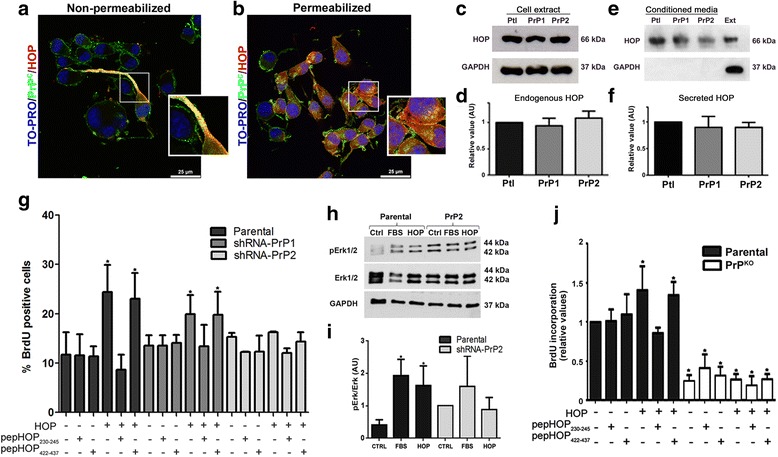
Hsp70/90 organizing protein (HOP) promotes proliferation of neurospheres dependent on cellular prion protein (PrP C) by activating the Erk1/2 signaling pathway. Immunofluorescence for PrPC (green) and HOP (red) in parental neurospheres shows co-localization on the cell surface in non-permeabilized cells (a) and expression of both proteins in the cytoplasm in permeabilized cells (b). Nuclei staining (TO-PRO) in shown in blue. A higher magnification field is shown in the inset. c Immunoblot for HOP in parental (Ptl), shRNA-PrP1 (PrP1), and shRNA-PrP2 (PrP2) neurospheres. GAPDH was used as the loading control. d Densitometry analysis of immunoblot; values of three independent experiments are expressed relative to control (parental). e Immunoblot of conditioned medium of parental (Ptl), shRNA-PrP1 (PrP1), and shRNA-PrP2 (PrP2) PrPC-depleted populations. Total protein extract (Ext). GAPDH immunodetection was used as cell lysis control. f Densitometry analysis of immunoblot; values of three independent experiments are expressed relative to control (parental). g U87 neurospheres treated with HOP and/or peptides pepHOP230–245 or pepHOP422–437 (1 μM), combined or alone, compared to untreated control. Percentage of BrdU-positive cells in all conditions in relation to total number of cells (n = 6, *p < 0.005). h Immunoblot of phosphorylated Erk1/2 and total Erk1/2 in parental and shRNA-PrP2 neurospheres for basal levels (Ctrl), fetal bovine serum (FBS), or recombinant HOP treatments (HOP) for 20 min. GAPDH was used as the loading control. i Densitometry of the relative values of ERK1/2 activation are represented by the ratio of p-ERK and total ERK1/2 in parental and shRNA-PrP2 neurospheres after treatment with recombinant HOP or FBS. j U251 neurospheres of parental and U251 knockout (PrP KO) populations treated with HOP and/or peptides pepHOP230–245 or pepHOP422–437 (1 μM), combined or alone, compared to untreated control. BrdU-positive cells detected by spectrophotometry (n = 4, p < 0.05, ANOVA followed by Tukey post-hoc test)
We evaluated the effect of exogenous (recombinant) HOP and synthetic peptides (pepHOP230–245 which mimics the PrPC binding site, and peptide pepHOP422–437 used as the control) on the proliferation of parental, PrP1, and PrP2 neurospheres. After 24 h of treatment, there was a significant increase in proliferation of parental and of PrP1 neurospheres treated with HOP, compared to PrPC silenced cells (PrP2) (Fig. 3g). On the other hand, populations pre-treated with pepHOP230–245 were unable to proliferate in the presence of HOP (Fig. 3g), suggesting that pepHOP230–245 is able to block the PrPC-HOP interaction and impair GSC proliferation. Moreover, we observed that HOP activates the Erk1/2 pathway only in neurosphere cultures expressing high levels of PrPC (Fig. 3h and i). PrPC-depleted cells have increased basal levels of phospho-Erk1/2 (Fig. 3h and i), corroborating our previous data in primary cultures from PrPC knockout mice and literature data in different cell types [24–26]. Similar results were obtained using an additional glioblastoma cell line (U251). As shown in Fig. 3j the positive HOP effect upon proliferation was also abrogated by HOP230–245 peptide in U251 PrPC-expressing cells, as well as being observed with U87 (Fig. 3g). Neurospheres from the PrPC knockout U251 cell line present a very low proliferation rate when compared to parental cells (Fig. 3j). Together these findings indicate that PrPC-HOP interaction is able to sustain the proliferation of GSCs of distinct glioblastoma cell lines.
HOP silencing impairs GSC proliferation
To determine the contribution of HOP towards GSC proliferation, we silenced HOP expression in parental and PrP2 populations. Decreased expression of HOP was confirmed by immunoblotting (Fig. 4a and b). The expression of PrPC was also addressed in HOP knockdown cells (HOPKD) and showed a slight decrease compared to the parental population (Fig. 4c). The proliferative basal levels were affected in both HOPKD and PrP2/HOPKD populations (Fig. 4d); however, the treatment with recombinant HOP is able to rescue the proliferation phenotype in PrPC-positive cells (Fig. 4d). Together, these results indicate both PrPC and HOP as key players in the regulation of GSC proliferation.
Fig. 4.
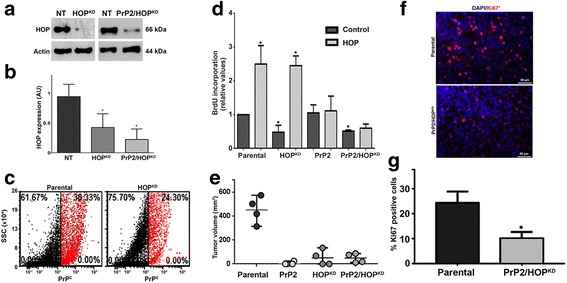
Cellular prion protein (PrP C) and/or Hsp70/90 organizing protein (HOP) knockdown suppresses cell proliferation and tumor growth in vivo. a Immunoblot for HOP expression in U87 non-target (NT), shRNA-HOP (HOP KD), or shRNA-PrP2/HOP (PrP2/HOP KD) populations. Actin was used as the protein loading control. b Immunoblot densitometry analysis; values of three independent experiments are expressed relative to control (parental). c Dot plot for PrPC expression of U87 parental and HOPKD populations. PrPC+ cells shown in red and PrPC– cells shown in black. d. Colorimetric BrdU incorporation assay in U87 parental, HOPKD, PrP2, or PrP2/HOPKD populations. Values of four independent experiments are expressed relative to control (parental). *p < 0.05, ANOVA followed by Tukey post-hoc test. e U87 neurospheres cells from parental, PrP2, HOPKD, or PrP2/HOPKD populations (1 × 106 cells) were implanted into the flank of nude mice and the tumor growth was monitored daily. Data represent tumor volume on day 10 after tumor detection (n = 4, *p < 0.05, ANOVA followed by Tukey post-hoc test). f Tumors were resected, fixed, paraffin embedded, and immunostained for Ki67. Representative images of Ki67 labeling (red) and DAPI (nuclei, blue). g Values represent the percentage of Ki67-positive cells relative to total number of cells (nuclei, DAPI staining)
PrPC and HOP depletion decreases GSC tumorigenicity in vivo
Neurosphere cells expressing different levels of PrPC and/or HOP were injected subcutaneously into Balb/c nude mice flanks and tumor growth was evaluated. As depicted in Fig. 4e, cells expressing PrPC grew rapidly, while ablated cells for PrPC (PrP2) and/or HOP (HOPKD) showed slower or no tumor formation. Histological sections of resected tumors showed that PrPC or HOP silencing caused cell proliferation reduction, as depicted by Ki-67 nuclear immunostaining (Fig. 4f and g). These data support our in vitro assays and highlight the importance of PrPC and HOP in tumor maintenance through the modulation of GSC proliferation.
Self-renewal of GSCs depends on PrPC expression and its interaction with HOP
Since the proposal that NSCs originate GSCs, important data describing the induction of NSC self-renewal by HOP-PrPC interaction has emerge, supporting a function for PrPC and HOP in GSC biology [6]. Thus, self-renewal was evaluated by formation of secondary neurospheres in clonal density assays. Cells expressing lower PrPC levels (PrP2 population) formed fewer colonies when compared to parental and PrP1 populations (Fig. 5a). HOP increased the number of neurospheres in parental and PrP1 populations when compared to untreated cells. However, any HOP effect was observed in the PrP2 population that lacked PrPC (Fig. 5a). These results indicate that PrPC expression is required for self-renewal and is also necessary to mediate HOP activity. Neurosphere formation mediated by HOP-PrPC interaction was abrogated in the presence of pepHOP230–245, which mimics and competes for the PrPC binding site. The control peptide pepHOP422–437 (Fig. 5a) did not have any effect.
Fig. 5.
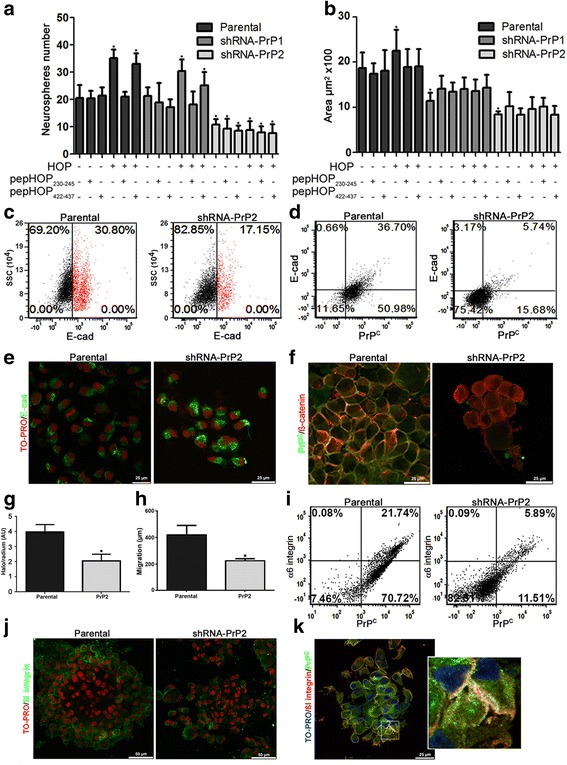
Cellular prion protein (PrP C) promotes GSC self-renewal binding HOP and modulates cell surface adhesion molecule stability. Neurosphere number (a) or size (b) after 1 week treatment every 48 h with Hsp70/90 organizing protein (HOP) and peptides pepHOP230–245 and pepHOP422–437 (1 μM), combined or alone (500nM), compared to control (n = 6, *p < 0.05, ANOVA followed by Tukey post-hoc test). c Dot plot of E-cadherin expression in parental and shRNA-PrP2 neurospheres. E-cad+ cells shown in red and E-cad– cells shown in black. d Dot plot of E-cadherin and PrPC expression in parental and shRNA-PrP2 neurospheres. e Immunofluorescence for E-cadherin (green) in parental and shRNA-PrP2 neurospheres, showing expression on the cell surface (parental) and in the perinuclear region (shRNA-PrP2). Nuclei (TO-Pro) stain shown in red. f PrPC (green) and β-catenin (red) expression and co-localization (yellow) of parental and shRNA-PrP2 neurospheres. g Migration assay, ratio between cell migration distance (halo), and neurosphere size for parental and shRNA-PrP2 neurospheres 24 h after plating on laminin-1 (n = 3, *p < 0.05). h Cell scratch assay; images of three experimental replicates were acquired and the distance of each scratch closure after 24 h was measured by comparing with the images at time 0 h for parental and shRNA-PrP2 neurospheres plated on laminin-1 (n = 4, *p < 0.05). i Dot plot of α6 integrin and PrPC expression in parental and shRNA-PrP2 neurospheres. j Immunofluorescence for β1 integrin (green) of parental and shRNA-PrP2 neurospheres. Nuclei (TO-PRO) stain shown in red. k. PrPC (green) and β1 integrin (red) expression and co-localization (yellow) of parental and shRNA-PrP2 neurospheres. Nuclei (TO-PRO) stain shown in blue; a higher magnification is shown in the inset
As previously described, PrPC ablation decreases neurosphere number (Fig. 5a) probably by downregulating essential molecules involved in stem cell maintenance (Fig. 2), supporting its previously suggested role in stemness. The smaller size of neurospheres formed by PrPC-depleted cells (compared to their counterparts) suggests a putative role for PrPC in GSC cell adhesion maintenance (Fig. 5b; Additional file 1: Figure S1A). To address this issue, the expression of E-cadherin and β-catenin, key components of adherens junctions, was evaluated in PrP2 neurospheres and parental control populations. The expression of E-cadherin is associated with PrPC on the cell surface; this was more obvious for the parental population, where there was higher expression of E-cadherin (Fig. 5c) and a clear double staining for both proteins in neurosphere cells (Fig. 5d). Although the cell membrane of parental cells had high E-cadherin expression, PrP2 had abundant cytoplasmic E-cadherin (Fig. 5e). Parental and PrP2 neurospheres expressed β-catenin abundantly at similar levels. However, there was an intense co-localization of β-catenin and PrPC in the same membrane domain of parental neurospheres, while in PrP2 neurospheres the β-catenin expression was diffuse (Fig. 5f). This may suggests that PrPC plays a role in E-cadherin recruitment to the cell membrane and, consequently, on β-catenin engagement.
Due to its putative function as a scaffold protein and multiprotein assembly platform on the cell surface, we also tested if additional anchorage-dependent cell processes, such as migration, was altered in PrPC-depleted cells. Additionally, migration on laminin is impaired in PrPC-silenced cells compared to parental neurospheres (Fig. 5g and h; Additional file 1: Figure S1B). Moreover, the surface expression of integrin α6β1, a laminin receptor, was verified. Integrin α6 expression is clearly more evident in parental neurospheres compared to PrP2 (Fig. 5i) and is co-expressed with PrPC, while integrin β1 expression is apparently similar in both populations (Fig. 5j). However, a high co-localization of integrin β1 and PrPC was observed (Fig. 5k), suggesting that PrPC plays a key function in enhancing cell surface stability of cell adhesion molecules, thereby modulating the invasive process.
Discussion
In this study, the role of the PrPC-HOP complex in the regulation of GSC biology was evaluated. First, we demonstrated that our model of neurospheres expressed several markers of stemness. The expression of CD15, CD133, Oct4, Musashi-1, and Sox2 was increased in neurospheres compared to a non-stem condition (monolayer), validating our experimental model (Fig. 1). PrPC expression was higher in neurospheres than in monolayer cultures (Fig. 2a) and, since the enrichment of GBM cultures with stem cells leads to more malignant tumors in vivo [27], these data support previous results from our group which show that PrPC expression is correlated with tumor aggressiveness [6, 15].
PrPC expression seems to be associated with stem-like properties, since its silencing led to a differentiated expression of stem cell markers. Compared to parental neurospheres, CD133 expression decreased and Sox2 expression became undetectable in PrPC-depleted populations (Fig. 2e and f). Previous studies have shown that CD133 and Sox2 are exclusively expressed at perinecrotic and perivascular regions associated with stem-like cell pools, and that nestin and Musashi-1 are homogeneously expressed across the tumor, identifying precursors [28]. PrP2 neurospheres expressed Musashi-1 in the cytoplasm in contrast to parental neurospheres, where it segregated preferentially to cell nuclei (Fig. 2f). Nuclear expression of Musashi-1 has been associated with activation of the Notch pathway in gliomas [29, 30] which, in turn, may lead to increased tumor malignancy via induction of proliferation and therapy resistance [31]. The expression of nestin was similar in parental and PrP2 neurospheres (Fig. 2f). Expression of differentiation markers is more evident in PrP2 neurospheres compared to the parental population after serum stimuli (Fig. 2g). Together, our results suggest that parental neurospheres have a stem-like phenotype with some precursor cells, and that the PrP2 population has precursors and cells committed to a specific phenotype. Indeed, the function of PrPC in stem cell biology has been broadly studied. PrPC has been described as an important molecule for neural commitment and for the proliferation of precursors [32]. In tumor stem-like cells, PrPC promotes proliferation and migration [11]. PrPC interacts with the cell surface protein CD44, a marker for several types of cancer stem-like cells [33] that associates with tumor-initiating and metastatic capacities and promotes epithelial-mesenchymal transition (EMT) and tumor growth after resection [34].
A possible interaction between PrPC and CD133 was also observed given their co-expression and localization on the cell surface of parental neurospheres (Fig. 2b and c). PrPC and CD133 were previously shown to localize to the same membrane domains (lipid rafts), modulating differentiation and stemness, respectively [35]. Additionally, a reduction in cell surface expression of CD133 associated with PrPC was observed after copper stimulus, suggesting PrPC as a carrier for CD133 internalization (Fig. 2c and d). PrPC is constitutively endocytosed via clatrin-coated pits [36] and copper ions reversibly stimulate this endocytic pathway [37]. CD133 has been shown to affect the clathrin-endocytosis process [38] and trafficking down the endosomal and lysosomal pathway for degradation [39]. Remarkably, the cytoplasmatic domain of CD133 binds β-catenin and the downregulation of CD133 increases β-catenin degradation and impairs tumor growth in vitro and in vivo [39]. Indeed, the β-catenin localization was perturbed in PrPC-depleted cells (Fig. 5f), suggesting the central role of PrPC in the stabilization of the signaling module on the cell surface.
PrPC-depleted populations formed less secondary neurospheres (Fig. 5a), suggesting that withdrawing PrPC impairs self-renewal ability. This supports the hypothesis that PrPC can act as an important player in stemness maintenance and that its dowregulation induces cell line commitment, corroborating recent evidence that showed a less oncogenic phenotype in PrPC-depleted GSCs [40]. Thus, it is possible that PrPC acts as an essential molecule for GSC biology and is capable of maintaining an undifferentiated state in this GBM subpopulation and, since its expression may indicate tumor enrichment with stem-like cells, it may be used as a tumor progression marker. Interesting, the effect of temozolomide, a common chemotherapeutic agent for brain tumors, is enhanced in PrPC-depleted glioma cells, supporting PrPC as an effective target for GBM [41].
As a number of studies show that PrPC acts as a scaffold protein, assembling signaling platforms on the plasma membrane to elicit several biological processes including in stem cells (reviewed in [8, 42]), we looked for alterations in the expression of cell adhesion proteins on the cell surface of GSCs. We observed a decrease in E-cadherin and integrin α6 expression on the cell surface of PrPC-silenced populations (Fig. 5c, d and i), and detected E-cadherin in the cytoplasm (Fig. 5e). The expression of these proteins was also associated with PrPC, as E-cadherin- or integrin α6-positive cells were almost exclusively positive for PrPC (Fig. 5d and i). Integrin β1 expression was detected in both parental and PrP2 neurospheres, and co-localized with PrPC on the cell surface (Fig. 5j and k). In addition, GSC migration on laminin was impaired in PrPC-depleted cells (Fig. 5g and h). Indeed, it has been demonstrated that PrPC participates in E-cadherin recruitment to the cell surface [43] with no significant differences verified in transcripts levels [43, 44]. Interestingly, it was demonstrated that PrPC-null mice present increased paracellular permeability, with lower levels of E-cadherin, desmoplakin, occluding, and other proteins related to cell-cell junctions in intestinal tissues [45]. Furthermore, it was reported that PrPC is able to regulate β1 integrin adhesiveness modulating ligand-induced changes in integrin activation [46]; however, its depletion had no effect on total β1 integrin expression levels [47]. These data suggest that PrPC may be capable of recruiting cell adhesion molecules to the cell surface of GSCs, raising the hypothesis of PrPC modulating invasion-related processes.
We also reported that GSCs have high expression of HOP and PrPC (Fig. 3b) and described the HOP-PrPC engagement on their plasma membrane (Fig. 3a). HOP expression and secretion was similar in parental, PrP1, and PrP2 populations (Fig. 3c–f), as demonstrated previously by Santos and collaborators in murine wild-type and PrPC-null neurospheres [6]. We demonstrated that HOP is able to modulate GSC proliferation (Fig. 4d) and self-renewal, depending on its interaction with PrPC (Figs. 3g, 4d, and 5a); inhibiting the PrPC-HOP interaction with a HOP peptide, which mimics the PrPC binding site, abrogates the effects of recombinant HOP (Figs. 3g and 5a). Similar effects were also observed in another glioblastoma cell line (U251) confirming the importance of these complexes in GSC biology (Fig. 3j). Another HOP peptide (anti-TPR), which inhibits the HOP-Hsp90 interaction, has been described to induce cell death in several cancer cell lines [48] and produce a cytotoxic effect in glioblastomas [49], highlighting HOP as a potential target for GBM therapy. Previous reports [15] as well as data from this study (Fig. 3e and h) also described that PrPC and secreted HOP interact on the cell surface of human glioblastoma cell lines and modulate GBM progression by promoting proliferation through activation of the Erk1/2 pathway [16]. Remarkably, a higher basal phosphorylation of Erk was found in PrPC-depleted cells when compared to their counterparts (Fig. 3h and i), confirming our prior data in retinae [24] and hippocampal neurons [25], as well as those from other authors in adult brain and cerebellum extracts [26]. This feature suggests that high activity of Erk could be a compensatory effect for prion protein ablation [42]. Indeed, the targeting of stress response proteins has remarkable potential for the development of molecular treatments. Simultaneous inhibition of expression of both HOP ligands Hsp70 and Hsp90 reduced proliferation and promoted apoptosis in GBM cell lines in vitro [50]. Furthermore, HOP performs an important function in compensatory mechanisms of stress responses in tumor cells [51], further supporting its relevance in GBM maintenance.
Finally, we observed that PrPC and/or HOP knockdown affects the proliferative and tumorigenic capacity of GSCs (Fig. 4e and f) in vivo, and supports PrPC and HOP molecules as potential new targets for developing more efficient therapeutic strategies.
Conclusions
Here, we reported that HOP promotes GSC proliferation and self-renewal by interacting with PrPC and that silencing PrPC significantly impairs GSC self-renewal. PrPC-silenced cells have lower expression of stem cell markers and increased differentiation, suggesting that PrPC plays a role in maintenance of an undifferentiated state in GSCs. HOP and PrPC ablation suppresses the malignancy of GBM cells in vivo and has potential application as a therapeutic target in glioblastoma. In addition, PrPC could play a role in the expression and cell surface localization of cell adhesion proteins, participating in cell migration mechanisms and, consequently, in invasion. Our results suggest that one mechanism by which PrPC governs GSC biology is through its role as a cell surface scaffold protein assembling a dynamic signaling platform and also interacting with soluble partners, such as HOP, to maintain stemness status (Fig. 6).
Fig. 6.
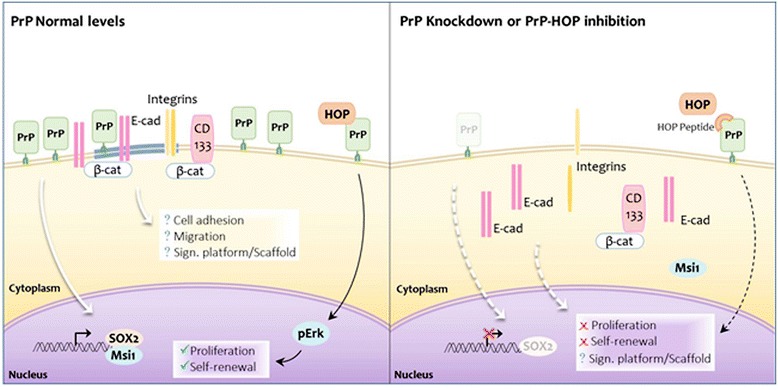
Model of prion protein (PrP) as a scaffold protein modulating GSC biology. Scheme illustrating how PrPC may act as a scaffold protein, regulating stemness (dowregulation of CD133 and Sox2 and Msi1 sub-localization altered), recruiting cell adhesion molecules (E-cadherin and integrin α6β1), binding CD133-β-catenin to the plasma membrane, and modulating GSC proliferation and self-renewal through its interaction with Hsp70/90 organizing protein (HOP). Blockage of PrPC-HOP interaction with HOP peptide impairs the binding of HOP and PrPC and, consequently, Erk1/2 activation, affecting proliferation
Major commitments for developing novel therapeutic strategies for GBM are under way, as this is an extremely aggressive type of cancer. In particular, several attempts to therapeutically eradicate GSCs have been made, as these cells have well-established characteristics in tumors. Our findings support the concept that PrPC, HOP, and their complex are important for GSC biology, regulating essential mechanisms for tumor maintenance. Therefore, they represent a novel target for developing new treatments for GBM or for improving the efficacy of current therapies by targeting GSCs.
Acknowledgements
The authors thank the Fundação de Amparo à Pesquisa do Estado de São Paulo-FAPESP (11/13906-2, 2009/14027-2, 13/19860-0, and 15/04122-9), Conselho Nacional de Desenvolvimento Científico e Tecnológico (467566/2014-3), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior for financial support. We are also very grateful to Carlos Nascimento for technical assistance, Dr. Andrew Hill (La Trobe University, Australia) for providing shRNA-PrP constructs used in this study, and Dr. Martín Roffé (A.C. Camargo Cancer Center) for excellent technical assistance with the CRISPR/Cas9 system.
Funding
Fundação de Amparo à pesquisa do Estado de São Paulo (2013/19860-0 to RPI; 2009/14027-2 to VRM; 2011/13906-2 to MHL; and 2015/04122-9 to MBP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (467566/2014-3 to VRM and TGS), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Institutional Scholarship to RPI).
Availability of data and materials
RNA design for CRISPR/Cas9 provided by Optimized CRISPR Design (http://crispr.mit.edu/) using PrPC gene sequence (NM_000311.3).
Authors’ contributions
RPI: conception and design, collection and/or assembly of data, data analysis and interpretation, and manuscript writing; MBP: collection and/or assembly of data; LC; manuscript writing; VRM: manuscript writing, provision of study material, and financial support; TGS: conception and design, data analysis and interpretation, and manuscript writing MHL: conception and design, data analysis and interpretation, manuscript writing, financial support, provision of study material, and final approval of manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethical approval
Institutional guidelines for animal welfare were followed and the study approved by the Animal Ethics Committee of the Institute of Biomedical Sciences/University of Sao Paulo (book 03, page 15, and protocol number 002 of 04/03/2014).
Abbreviations
- bFGF
Basic fibroblast growth factor
- BSA
Bovine serum albumin
- EGF
Epidermal growth factor
- GBM
Glioblastoma
- GSC
Glioblastoma stem-like cell
- HOP
Hsp70/90 organizing protein
- NSC
Neural stem cell
- PBS
Phosphate-buffered saline
- PrPC
Cellular prion protein
- RT
Room temperature
Additional file
Representative images of self-renewal and migration assays. (A) Representative image of self-renewal assay. Left panel: neurosphere growth in parental, shRNA-PrP1 (PrP1), and shRNA-PrP2 (PrP2) populations. Right panel: neurosphere size for the parental population untreated control (Ctrl) and cells treated with recombinant HOP (HOP). (B) Representative images of the migration assay of Parental (left) and shRNA-PrP2 (right) neurospheres 24 h after plating on laminin-1 (n = 4, *p < 0.05). (PPTX 205 kb)
Contributor Information
Rebeca Piatniczka Iglesia, Email: rebecapiglesia@gmail.com.
Mariana Brandão Prado, Email: maribrandaop@gmail.com.
Lilian Cruz, Email: cruz.lilianlc@gmail.com.
Vilma Regina Martins, Email: vmartins@cipe.accamargo.org.br.
Tiago Góss Santos, Email: tsantos@cipe.accamargo.org.br.
Marilene Hohmuth Lopes, Email: marilenehl@usp.br, Email: marilenehlopes@gmail.com.
References
- 1.Ellis HP, Greenslade M, Powell B, Spiteri I, Sottoriva A, Kurian KM. Current challenges in glioblastoma: intratumour heterogeneity, residual disease, and models to predict disease recurrence. Front Oncol. 2015;5:251. doi: 10.3389/fonc.2015.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Persano L, Rampazzo E, Basso G, Viola G. Glioblastoma cancer stem cells: role of the microenvironment and therapeutic targeting. Biochem Pharmacol. 2013;85:612–22. doi: 10.1016/j.bcp.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–8. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 5.Florio T, Barbieri F. The status of the art of human malignant glioma management: the promising role of targeting tumor-initiating cells. Drug Discov Today. 2012;17:1103–10. doi: 10.1016/j.drudis.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Santos TG, Silva IR, Costa-Silva B, Lepique AP, Martins VR, Lopes MH. Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells. 2011;29:1126–36. doi: 10.1002/stem.664. [DOI] [PubMed] [Google Scholar]
- 7.Longshaw VM, Baxter M, Prewitz M, Blatch GL. Knockdown of the co-chaperone Hop promotes extranuclear accumulation of Stat3 in mouse embryonic stem cells. Eur J Cell Biol. 2009;88:153–66. doi: 10.1016/j.ejcb.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Santos TG, Lopes MH, Martins VR. Targeting prion protein interactions in cancer. Prion. 2015;9:165–73. doi: 10.1080/19336896.2015.1027855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Yu S, Nakamura F, Pentikainen OT, Singh N, Yin S, et al. Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J Biol Chem. 2010;285:30328–39. doi: 10.1074/jbc.M110.147413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li QQ, Cao XX, Xu JD, Chen Q, Wang WJ, Tang F, et al. The role of P-glycoprotein/cellular prion protein interaction in multidrug-resistant breast cancer cells treated with paclitaxel. Cell Mol Life Sci. 2009;66:504–15. doi: 10.1007/s00018-008-8548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Y, Tao L, Xu J, Li Q, Yu J, Jin Y, et al. CD44/cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol Carcinog. 2014;53:686–97. doi: 10.1002/mc.22021. [DOI] [PubMed] [Google Scholar]
- 12.Pan Y, Zhao L, Liang J, Liu J, Shi Y, Liu N, et al. Cellular prion protein promotes invasion and metastasis of gastric cancer. FASEB J. 2006;20:1886–8. doi: 10.1096/fj.06-6138fje. [DOI] [PubMed] [Google Scholar]
- 13.Barbieri G, Palumbo S, Gabrusiewicz K, Azzalin A, Marchesi N, Spedito A, et al. Silencing of cellular prion protein (PrPC) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy. 2011;7:840–53. doi: 10.4161/auto.7.8.15615. [DOI] [PubMed] [Google Scholar]
- 14.Meslin F, Hamai A, Gao P, Jalil A, Cahuzac N, Chouaib S, et al. Silencing of prion protein sensitizes breast adriamycin-resistant carcinoma cells to TRAIL-mediated cell death. Cancer Res. 2007;67:10910–9. doi: 10.1158/0008-5472.CAN-07-0512. [DOI] [PubMed] [Google Scholar]
- 15.Lopes MH, Santos TG, Rodrigues BR, Queiroz-Hazarbassanov N, Cunha IW, Wasilewska-Sampaio AP, et al. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene. 2015;34:3305–14. doi: 10.1038/onc.2014.261. [DOI] [PubMed] [Google Scholar]
- 16.Erlich RB, Kahn SA, Lima FR, Muras AG, Martins RA, Linden R, et al. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia. 2007;55:1690–8. doi: 10.1002/glia.20579. [DOI] [PubMed] [Google Scholar]
- 17.Walsh N, Larkin A, Swan N, Conlon K, Dowling P, McDermott R, et al. RNAi knockdown of Hop (Hsp70/Hsp90 organising protein) decreases invasion via MMP-2 down regulation. Cancer Lett. 2011;306:180–9. doi: 10.1016/j.canlet.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Chao A, Lai CH, Tsai CL, Hsueh S, Hsueh C, Lin CY, et al. Tumor stress-induced phosphoprotein1 (STIP1) as a prognostic biomarker in ovarian cancer. PLoS One. 2013;8:e57084. doi: 10.1371/journal.pone.0057084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baindur-Hudson S, Edkins AL, Blatch GL. Hsp70/Hsp90 organising protein (hop): beyond interactions with chaperones and prion proteins. Subcell Biochem. 2015;78:69–90. doi: 10.1007/978-3-319-11731-7_3. [DOI] [PubMed] [Google Scholar]
- 20.Kubota H, Yamamoto S, Itoh E, Abe Y, Nakamura A, Izumi Y, et al. Increased expression of co-chaperone HOP with HSP90 and HSC70 and complex formation in human colonic carcinoma. Cell Stress Chaperones. 2010;15:1003–11. doi: 10.1007/s12192-010-0211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruckova E, Muller P, Nenutil R, Vojtesek B. Alterations of the Hsp70/Hsp90 chaperone and the HOP/CHIP co-chaperone system in cancer. Cell Mol Biol Lett. 2012;17:446–58. doi: 10.2478/s11658-012-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Lacerda TC, Costa-Silva B, Giudice FS, Dias MV, de Oliveira GP, Teixeira BL, et al. Prion protein binding to HOP modulates the migration and invasion of colorectal cancer cells. Clin Exp Metastasis. 2016;33:441–51. doi: 10.1007/s10585-016-9788-8. [DOI] [PubMed] [Google Scholar]
- 23.Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–16. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals 9. EMBO J. 2002;21:3317–26. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, et al. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci. 2005;25:11330–9. doi: 10.1523/JNEUROSCI.2313-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown DR, Nicholas RSJ, Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res. 2002;67:211–24. doi: 10.1002/jnr.10118. [DOI] [PubMed] [Google Scholar]
- 27.Lathia JD, Gallagher J, Myers JT, Li M, Vasanji A, McLendon RE, et al. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLoS One. 2011;6:e24807. doi: 10.1371/journal.pone.0024807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stieber D, Golebiewska A, Evers L, Lenkiewicz E, Brons NH, Nicot N, et al. Glioblastomas are composed of genetically divergent clones with distinct tumourigenic potential and variable stem cell-associated phenotypes. Acta Neuropathol. 2014;127:203–19. doi: 10.1007/s00401-013-1196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahlrot RH, Hansen S, Herrstedt J, Schroder HD, Hjelmborg J, Kristensen BW. Prognostic value of Musashi-1 in gliomas. J Neurooncol. 2013;115:453–61. doi: 10.1007/s11060-013-1246-8. [DOI] [PubMed] [Google Scholar]
- 30.Toda M, Iizuka Y, Yu W, Imai T, Ikeda E, Yoshida K, et al. Expression of the neural RNA-binding protein Musashi1 in human gliomas. Glia. 2001;34:1–7. doi: 10.1002/glia.1034. [DOI] [PubMed] [Google Scholar]
- 31.Gotte M, Greve B, Kelsch R, Muller-Uthoff H, Weiss K, Kharabi Masouleh B, et al. The adult stem cell marker Musashi-1 modulates endometrial carcinoma cell cycle progression and apoptosis via Notch-1 and p21WAF1/CIP1. Int J Cancer. 2011;129:2042–9. doi: 10.1002/ijc.25856. [DOI] [PubMed] [Google Scholar]
- 32.Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A. 2006;103:3416–21. doi: 10.1073/pnas.0511290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du L, Rao G, Wang H, Li B, Tian W, Cui J, et al. CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res. 2013;73:2682–94. doi: 10.1158/0008-5472.CAN-12-3759. [DOI] [PubMed] [Google Scholar]
- 34.Mehrabian M, Ehsani S, Schmitt-Ulms G. An emerging role of the cellular prion protein as a modulator of a morphogenetic program underlying epithelial-to-mesenchymal transition. Front Cell Dev Biol. 2014;2:53. doi: 10.3389/fcell.2014.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garmy N, Guo XJ, Taieb N, Tourres C, Tamalet C, Fantini J, et al. Cellular isoform of the prion protein PrPc in human intestinal cell lines: genetic polymorphism at codon 129, mRNA quantification and protein detection in lipid rafts. Cell Biol Int. 2006;30:559–67. doi: 10.1016/j.cellbi.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Shyng SL, Moulder KL, Lesko A, Harris DA. The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem. 1995;270:14793–800. doi: 10.1074/jbc.270.24.14793. [DOI] [PubMed] [Google Scholar]
- 37.Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–10. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- 38.Bourseau-Guilmain E, Griveau A, Benoit JP, Garcion E. The importance of the stem cell marker prominin-1/CD133 in the uptake of transferrin and in iron metabolism in human colon cancer Caco-2 cells. PLoS One. 2011;6:e25515. doi: 10.1371/journal.pone.0025515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012;2:951–63. doi: 10.1016/j.celrep.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corsaro A, Bajetto A, Thellung S, Begani G, Villa V, Nizzari M, et al. Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget. 2016;7(25):38638–57. doi: 10.18632/oncotarget.9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhuang D, Liu Y, Mao Y, Gao L, Zhang H, Luan S, et al. TMZ-induced PrPc/par-4 interaction promotes the survival of human glioma cells. Int J Cancer. 2012;130:309–18. doi: 10.1002/ijc.25985. [DOI] [PubMed] [Google Scholar]
- 42.Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 43.Malaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, et al. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7:e55. doi: 10.1371/journal.pbio.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solis GP, Radon Y, Sempou E, Jechow K, Stuermer CA, Malaga-Trillo E. Conserved roles of the prion protein domains on subcellular localization and cell-cell adhesion. PLoS One. 2013;8:e70327. doi: 10.1371/journal.pone.0070327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petit CS, Barreau F, Besnier L, Gandille P, Riveau B, Chatau D, Roy M, Berrebi D, Svrcek M, Cardot P, Rousset M, Clair C, Thenet S. Requirement of cellular prion protein for intestinal barrier function and mislocalization in patients with inflammatory bowel disease. Gastroenterology. 2012;143:122–32. doi: 10.1053/j.gastro.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 46.Richardson DD, Tol S, Valle-Encinas E, Pleguezuelos C, Bierings R, Geerts D, et al. The prion protein inhibits monocytic cell migration by stimulating beta1 integrin adhesion and uropod formation. J Cell Sci. 2015;128:3018–29. doi: 10.1242/jcs.165365. [DOI] [PubMed] [Google Scholar]
- 47.Loubet D, Dakowski C, Pietri M, Pradines E, Bernard S, Callebert J, et al. Neuritogenesis: the prion protein controls beta1 integrin signaling activity. FASEB J. 2012;26:678–90. doi: 10.1096/fj.11-185579. [DOI] [PubMed] [Google Scholar]
- 48.Horibe T, Kohno M, Haramoto M, Ohara K, Kawakami K. Designed hybrid TPR peptide targeting Hsp90 as a novel anticancer agent. J Transl Med. 2011;9:8. doi: 10.1186/1479-5876-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horibe T, Torisawa A, Kohno M, Kawakami K. Molecular mechanism of cytotoxicity induced by Hsp90-targeted Antp-TPR hybrid peptide in glioblastoma cells. Mol Cancer. 2012;11:59. doi: 10.1186/1476-4598-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matokanovic M, Barisic K, Filipovic-Grcic J, Maysinger D. Hsp70 silencing with siRNA in nanocarriers enhances cancer cell death induced by the inhibitor of Hsp90. Eur J Pharm Sci. 2013;50:149–58. doi: 10.1016/j.ejps.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–80. doi: 10.1016/S0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA design for CRISPR/Cas9 provided by Optimized CRISPR Design (http://crispr.mit.edu/) using PrPC gene sequence (NM_000311.3).