

Abstract
Background & objectives:
Human herpes simplex virus 1 (HSV-1) is the most common cause of sporadic encephalitis in humans that contributes to >10 per cent of the encephalitis cases occurring worldwide. Availability of limited full genome sequences from a small number of isolates resulted in poor understanding of host and viral factors responsible for variable clinical outcome. In this study genetic relationship, extent and source of recombination using full-length genome sequence derived from a newly isolated HSV-1 isolate was studied in comparison with those sampled from patients with varied clinical outcome.
Methods:
Full genome sequence of HSV-1 isolated from cerebrospinal fluid (CSF) of a patient with acute encephalitis syndrome (AES) by inoculation in baby hamster kidney-21 (BHK-21) cells was determined using next-generation sequencing (NGS) technology. Phylogenetic analysis of the newly generated sequence in comparison with 33 additional full-length genomes defined genetic relationship with worldwide distributed strains. The bootscan and similarity plot analysis defined recombination crossovers and similarities between newly isolated Indian HSV-1 with six Asian and a total of 34 worldwide isolated strains.
Results:
Mapping of 376,332 reads amplified from HSV-1 DNA by NGS generated full-length genome of 151,024 bp from newly isolated Indian HSV-1. Phylogenetic analysis classified worldwide distributed strains into three major evolutionary lineages correlating to their geographic distribution. Lineage 1 containing strains were isolated from America and Europe; lineage 2 contained all the strains from Asian countries along with the North American KOS and RE strains whereas the South African isolates were distributed into two groups under lineage 3. Recombination analysis confirmed events of recombination in Indian HSV-1 genome resulting from mixing of different strains evolved in Asian countries.
Interpretation & conclusions:
Our results showed that the full-length genome sequence generated from an Indian HSV-1 isolate shared close genetic relationship with the American KOS and Chinese CR38 strains which belonged to the Asian genetic lineage. Recombination analysis of Indian isolate demonstrated multiple recombination crossover points throughout the genome. This full-length genome sequence amplified from the Indian isolate would be helpful to study HSV evolution, genetic basis of differential pathogenesis, host-virus interactions and viral factors contributing towards differential clinical outcome in human infections.
Keywords: Encephalitis, genetic characterization, herpes simplex virus type 1, next-generation sequencing
Central nervous system (CNS) infections are of public health concern worldwide because of their high morbidity and mortality as well as considerable economic loss1. Among the CNS infectious aetiologies, viruses are most commonly associated with acute encephalitis syndrome (AES)2. Paediatric population forms the major susceptible age group among AES cases resulting in 0-11 per cent mortality3. Viruses belonging to the Herpesviridae family are mainly associated with the sporadic cases. Depending on the infecting strain, a broad range of clinical manifestations ranging from no disease to lethal encephalitis have been documented in patients4. Herpes simplex virus (HSV) is the most common cause of fatal sporadic acute encephalitis [herpes simplex encephalitis (HSE)] that contributes to about 10-20 per cent of the total cases of viral encephalitis worldwide5. Among the HSE cases, about 90 per cent are caused by HSV type 1 (also known as human herpesvirus type 1, HSV-1), whereas seven per cent of the cases are caused by HSV type 26. In India, association of HSV with sporadic AES cases has been reported from different regions of the country, but the estimate on HSE cases varies in different studies probably due to investigation of a limited number of cases. Epidemiological data on incidence, prevalence and affected age group are not available except the investigations on human clinical samples by demonstrating HSV-1 infection in 3.33 and 10.5 per cent of the sporadic cases investigated7,8.
HSV genome is made up of 152 kb double-stranded DNA encoding at least 84 polypeptides. It represents a microcosm of the features found in eukaryotic and bacterial genomes such as presence of abundant short sequence repeats (SSRs), histone modifications, splice sites and microRNAs, high G/C content (65-75%) and high frequencies of recombination in the large inverted repeats9,10. Recombination between HSV-1 genomes is a major event in the replication process that allows creation of new genetic assortments and thus may play an important role in the evolutionary process. Evolution wise, the genetic diversity of globally circulating HSV-1 strains is significantly linked to the high frequency of recombination, and it is estimated that most of the strains are recombinants11,12. The large genome size of HSV-1 has probably influenced the generation of full-length genome sequence of multiple strains by Sanger's sequencing method. High-throughput sequencing techniques have enabled substantial inroads into novel pathogen discovery and genetic characterization of different pathogens13. Considering the variability and complexity of HSV-1 genomes, next-generation sequencing (NGS) technology is being explored as a more appropriate tool for full-length genomic characterization14. This study was undertaken to determine the full-length genome sequence of an Indian clinical HSV-1 strain isolated from cerebrospinal fluid (CSF) collected from an AES case using the NGS platform Ion Torrent and appropriate data analysis pipeline. The complete genome sequence generated from the newly isolated HSV-1 strain was further explored to determine its evolutionary relationship with other HSV-1 strains and presence of any recombination events which may have occurred during its evolution.
Material & Methods
This study was conducted in the National Institute of Virology (NIV), Pune, India, after prior approval from the institutional biosafety as well as ethical committees.
Clinical specimen: A previously healthy 15 year old patient belonging to Pune (Maharashtra) had a history of high-grade fever, lethargy and headache for eight days. The patient experienced neck stiffness and two episodes of generalized seizures gradually leading to altered sensorium. The patient was hospitalized on day 10 (post onset of fever) after deterioration of consciousness resulting in coma. The CSF collected on hospitalization (ID. 0116209, 2011) was referred to the NIV for virological investigations. The CSF was directly processed for routine panel of diagnostic reverse transcriptase polymerase chain reaction (RT-PCR)/PCR assays detecting Japanese encephalitis virus (JEV), chandipura virus (CHPV), herpes simplex virus (HSV) types 1 and 2, cytomegalovirus (CMV) and Epstein-Barr virus (EBV) infections along with IgM antibody capture ELISA for diagnosis of JE and CHP infections15,16. Viral nucleic acid isolated from CSF using PureLink Viral RNA/DNA Kit (Life Technologies, India) was directly processed for viral RNA amplification and detection by standard assays, whereas diagnosis of HSV types 1 and 2, CMV and EBV in CSF was performed using quadruplex PCR17.
Virus isolation: The CSF (100 μl) was filtered through a 0.22 μm Millipore filter and processed for virus isolation by inoculation in baby hamster kidney (BHK-21) cells as described earlier15. Infected cells were observed for cytopathological effects up to seven days of infection along with uninfected cells as negative controls and repeatedly passaged three times. The resultant cell culture stocks were maintained at -80°C until use.
DNA preparation, PCR and Sanger sequencing: The viral DNA from cell culture-grown virus was prepared using QIAamp DNA Mini Kit (Qiagen, USA) as per the manufacturer's instructions. The HSV PCR using nucleic acid prepared from the CSF and cell culture supernatants was carried out to amplify a fragment from glycoprotein D gene of HSV-1 using Platinum Taq DNA polymerase (Life Technologies). PCR amplifications and sequencing of 200-300 bp long fragments from different regions of HSV-1 genome were performed using primers (Primer Express software Version 3.0; Applied Biosystems, USA) flanking the target sequences. The primers used for PCR amplification and sequencing along with their genomic location and region amplified are given in the (Table I) Using the available sequence from both the ends of gaps, the primers were designed and synthesized commercially by Integrated DNA Technologies (USA). The PCR reactions were analyzed on agarose gel, and the purified PCR products were sequenced using a BigDye Terminator Cycle Sequencing Ready Reaction Kit and an automated sequencer (ABI Prism 310 Genetic Analyzer; Applied Biosystems) as described earlier18.
Table.
Primers and their genomic locations used for editing of the uncovered genomic regions of herpesvirus 1 strain
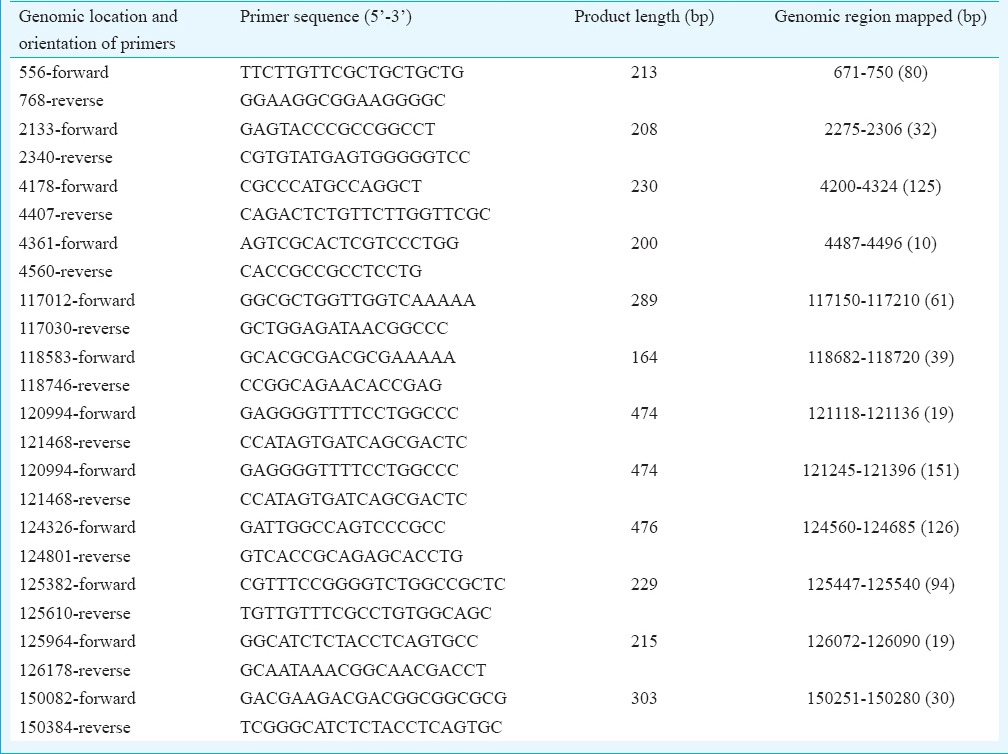
DNA library and template preparation: Quantity of HSV-1 DNA was determined by DNA Assay Kits (Life Technologies) using a Qubit Fluorometer (Life Technologies) as per the instructions and directly used for PCR and library preparation. The library of 1 μg DNA extracted from cell culture-grown virus was prepared using the Ion Fragment Library Kit (Life Technologies) as per the instructions. Both the ends (5’ and 3’) of DNA fragments (100-250 bp size) generated by mechanical shearing were ligated to the Ion Torrent specific adapters, nick translated with amplification primers and analyzed using Agilent DNA 1000 Kit (Agilent Technologies, USA) on the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Amplified DNA library containing about 4.9 × 108 molecules was clonally amplified using Ion Xpress Template 200 Kit (Life Technologies, USA) as per the instructions. The clonally amplified template-positive Ion Sphere Particles (ISPs) were enriched using the Ion Template Kit and processed for sequencing.
High-throughput sequencing: The template-positive ISPs and control Ion Spheres (internal quality controls) were mixed with the annealing buffer and precipitated by centrifugation. The sequencing primer annealed to the ISPs by incubation for two minutes each at 95 and 37°C and mixed with the sequencing DNA polymerase from Ion sequencing 200 Kit (Life Technologies). The reaction was incubated at room temperature for five minutes and loaded onto the 316 Ion Chip (Life Technologies) which in turn was loaded on to the pre-initialized Ion Torrent Personal Genome Machine (Applied Biosystems) for sequencing.
Bioinformatics pipeline for NGS data analysis: Sequence data obtained from DNA prepared from cell culture-grown HSV-1 were transferred to the Torrent Server for analysis. Raw signals from each well of the ion chip containing a single template-positive ISP were converted into a base call for each flow of nucleotides to produce nucleotide sequence reads in the SFF and FASTQ file formats. Pre-processing of the data including removal of adapters and low quality reads and trimming of the 3’ ends, etc., were performed using the Ion Torrent Software Suite. Primary alignment of the sequence reads generated from each ISP was achieved through the Torrent Mapping Alignment Program (TMAP-V4.0) available on Torrent Server. Mimicking Intelligent Read Assembly (MIRA-V4.0) software (http://www.chevreux.org/) was used for mapping, and de novo assembly of the reads that uses algorithms to find overlapping information between reads, leading to the generation of large sequence blocks of continuous sequence (contigs). BLAST search of individual contigs was performed to identify the query sequence from the NCBI database (ftp://ftp.ncbi.nih.gov/blast/db/). The reference sequence was selected on the basis of identity of multiple contigs through BLAST search uploaded on the Torrent Software Suit (V4.0), and the data were realigned to the reference sequence to define the coverage of sequence generated during the run. Confirmatory analysis of the sequence data was performed on SeqMan NextGen software of DNASTAR (Lasergene Genomics Suit, Madison, USA) by reference-guided assembly of the contigs. This software uses distance information from the paired-read sequences (library sizes) to link short contigs into larger scaffolds. Alignment of all contigs with the reference sequence to define the coverage was visualized in the Integrative Genome Viewer (IGV-2.3) software (http://www.broadinstitute.org/igv/). Sequence data containing a large number of sequences were curated and were mapped against the different genome data sets corresponding to eukaryotic, human (GRch37/hg19), 2352 bacterial and 3735 viral genomes available in NCBI database. The genomes that mapped to the maximum number of reads post-alignment were further reanalyzed to generate the sequence data.
Phylogenetic and recombination analysis: Multiple alignments using complete genome sequences were carried out with Clustal X1.8319. The full-length genome sequence-based phylogenetic tree was constructed using maximum likelihood (ML) statistical method and general time reversible (GTR) nucleotide substitution model with gamma correction available in MEGA620. The best-fit nucleotide substitution GTR model for phylogenetic reconstruction was selected on the basis of smallest Akaike information criteria score of 0.1 obtained for the present data set in MEGA6. The phylogenetic tree was constructed with the GTR model with 500 bootstrap replications. The rates of site-specific variations for each site were estimated using Gamma distribution model with the nearest neighbour interchange ML heuristic method20. The initial ML trees were prepared using the NJ/BioNJ distance-based tree with very strong filter. The genetic distances were estimated using maximum composite likelihood method implemented in MEGA6 with 1000 replicates. Phylogenetic tree was constructed using the present isolate of HSV-1 and 33 other representative HSV-1 full genomic sequences of globally isolated strains available in GenBank. Phylogenetic trees of the DNA sequence between crossover points obtained by SimPlot analysis were generated by neighbour joining method in MEGA6 with 1000 bootstrap replicates through maximum composite likelihood method20 for distance estimation to support the recombination events.
Phylogenetic relatedness generated in the ML algorithm was further validated using a NeighborNet distance transformation and equal angle splits transformation as implemented in SplitsTree4 version 4.13.1 (http://en.bio-soft.net/tree/SplitsTree.html). The split network is helpful to evaluate the impact of recombination and is ideally used for computing recombination networks21. The split network was generated with 1000 bootstrap replicates with the split threshold values ranging from 0.15 to 0.30. The hyperdimensional boxes in the networks represent areas with incompatible splits. The degree of denseness of boxes in a network reflects the intensity of contradictory evidence for grouping certain taxa, and the length of an edge is determined by the weight assigned to it21,22. The full genome DNA sequences of global HSV-1 isolates were also analyzed to define recombination across entire genome using SimPlot Program version 3.5.123. All the 34 trimmed sequences of worldwide distributed HSV-1 isolates were used for the SimPlot analysis using the present Indian HSV-1 as the query sequence. In addition, the similarity plot between the Asian subgroup of strains was derived using Indian HSV-1 as query sequence. The bootscan and similarity plot analysis were performed using a window size of 3 kb with the step size of 200 bp in ML model with 1000 bootstrap replicates, transition/transversion ratio of 2 and GapStrip on scan option.
Results
Virus isolation: The CSF sample of the suspected AES patient tested negative by anti- JEV IgM antibody capture ELISA and RT-PCR for JEV and CHPV infection. The diagnostic PCR yielded HSV-1 specific 271 bp product which upon sequencing showed 100 per cent identity with the partial glycoprotein D (US6) gene of HSV-1 strain KOS (GB# JQ780693). The BHK-21 cells inoculated with CSF showed a few foci after 48 h of inoculation in the first passage, and by the end of 96 h, >50 per cent of the cells were floating in comparison to the uninfected cell cultures. Repeated passage of tissue culture fluid collected from the first passage showed rounding and floating of all the cells within 72 h post-infection. Amplification and sequence analysis of the HSV-1 specific PCR product generated from the DNA isolated from infected cell cultures confirmed virus isolation. The stock of HSV-1 isolate (HSV-1/0116209/India/2011) was used for further genomic characterization.
Genome assembly: Amplification and sequencing of the HSV-1 DNA generated a total of 376,332 reads (total 47,512,313 bases) of the lengths ranging between 25 and 202 bp. Reads with the mean length of 122.13 bp were selected for analysis while short and duplicate reads were filtered and excluded from further analysis. BLAST analysis of the reads showed identity mainly with the viral, human and other higher eukaryotic genomes. Direct mapping of the reads through reference-guided assembly with the complete genome sequence of HSV-1 strain KOS (GB# JQ780693) mapped a total of 149,966 bases out of the 151,024 bp complete genome sequence resulting in 99.3 per cent sequence coverage. A total of 26 per cent reads were (98631/376,332) generated from HSV-1 DNA which mapped every base from both the ends with the exception of uncovered 3240 bases throughout the genome length resulting due to 29-220 base long gaps at 23 different locations (Fig. 1). Of the total 376,332 reads, 60,213 (16%) were mapped with human, 3198 (0.85%) with bacterial and 139,242 reads (37%) with viral genome data sets available in NCBI, whereas 173,679 reads (46%) were not mapped with any of these data sets. The de novo assembly of the 139,242 reads mapped to viral genome database generated 142 contigs of size ranging from 350 to 5000 bases. Of these, 83 exclusively mapped to HSV-1 genome while the remaining mapped to human, Mus musculus, Rattus rattus and hamster genome data sets. Mapping of the 83 contigs with HSV-1 full-length genome sequence generated 150,093 bp complete genome sequence (99.38% coverage) with the exception of 931 bp sequence due to the presence of 12 gaps of 10-151 bases at different locations. Sequence of 12 unmapped fragments at different genomic locations was generated by PCR amplification using primers designed against the highly conserved genomic termini to obtain PCR products which were sequenced directly using Sanger sequencing (Table). The 151,024 bp long full-length genome sequence of Indian HSV-1 isolate (HSV-1/0116209/India/2011) generated in this study is available in GenBank (Accession number KJ847330).
Fig. 1.
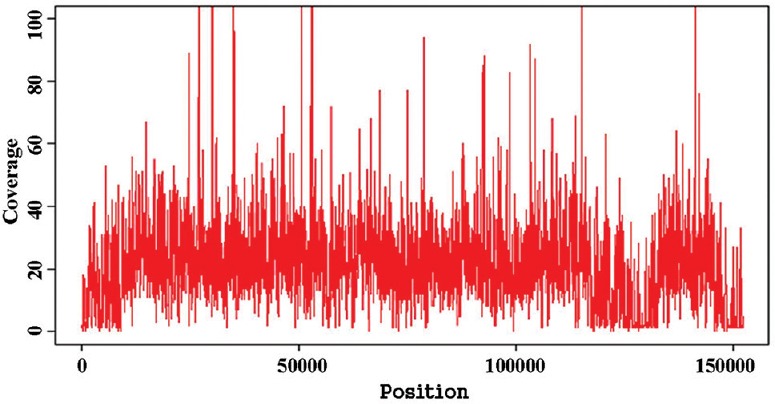
Coverage histogram by the HSV-1-specific reads generated by next-generation sequencing (NSG) mapped to complete genome sequence.
Phylogenetic and recombination analysis: To determine the genetic relationships of Indian HSV-1 isolate (HSV-1/0116209/India/2011), 34 representative complete or nearly complete genome sequences of strains originated in the USA, the UK, South Korea, Kenya, Japan and China available in GenBank were used. All those sequences formed three distinct genetic lineages, in which the strains sampled from Asian countries i.e., 0116209 (India), CR38 (China), R11 and R62 (South Korea), and S23 and S25 (Japan) clustered together with the KOS and RE strains from the USA in lineage 2 (Fig. 2). Lineage 1 was composed of the strains isolated in the UK (17) and the USA (F, H129, CJ970, OD4, CJ394, TFT401, 17, McKrae, 134, CJ311 and CJ360). All the HSV-1 strains isolated from Kenya (Africa) clustered into two distinct groups within lineage 3. The full genome sequences of worldwide isolated HSV-1 strains were also analyzed with SplitsTree, which is an alternative algorithm for the analysis and visualization of evolutionary data that is not always best represented by a standard phylogenetic tree. The SplitsTree graph showed clear separation of all the worldwide isolated HSV-1 strains into two distinct splits, in which the European, American and Asian strains clustered within the same network, whereas the African strains separated into two splits (Fig. 3). Both the ML (Fig. 2) and SplitsTree (Fig. 3) derived phylogenetic trees exhibited similar clustering pattern for the worldwide distributed HSV-1 strains. The genetic divergence between all the three lineages ranged from 0.65 to 1.93 per cent. Among all the sequences, maximum of 99.75 ± 0.01 per cent nucleotide identity (PNI) was documented between R62 (South Korea) and S23 (Japan), whereas the minimum of 98.27 ± 0.01 PNI shared between S23 (Japan) and E03 (Kenya). The RE strain clustered with the Asian strain showed highest sequence similarities of 99.43 ± 0.02 PNI with CR38 and KOS strains, followed by 99.40 ± 0.01 PNI with the Indian isolate. The RE strain shared 99.30-99.40 PNI with all the strains isolated from Europe and America, whereas it shared 98.80-99.28 PNI with the African strains. The Indian HSV-1 isolate 0116209 shared the maximum of 99.67 ± 0.01 PNI with the KOS strain, followed by 99.59 ± 0.01 PNI with CR38 and 99.27-99.30 PNI with Japanese, South Korean and American strains clustered in lineage 2. HSV-1 strains clustered within one genetic lineage showed 0.09-0.73 per cent divergences from each other whereas the strains clustered in different lineages showed 0.75-1.93 per cent divergence. The KOS strain clustered in Asian lineage shared 99.58 ± 0.21 PNI with CR38, 99.39 ± 0.30 PNI with R11, 99.38 ± 0.30 PNI with R62, 99.32 ± 0.34 PNI with S25 and 99.30 ± 0.38 PNI with S23, respectively. The comparison of nucleotide changes between the 0116209 (India) and KOS (USA) strains which were genetically closely related showed 481 nucleotide changes throughout the genome resulting in 144 amino acid changes. The maximum of 23 amino acid changes occurred in UL36 coding gene, followed by 4-7 amino acid changes occurred in RL2, US4, UL5, UL8, UL24, UL27, UL32, UL37, UL39, UL47 and UL49 coding genes.
Fig. 2.
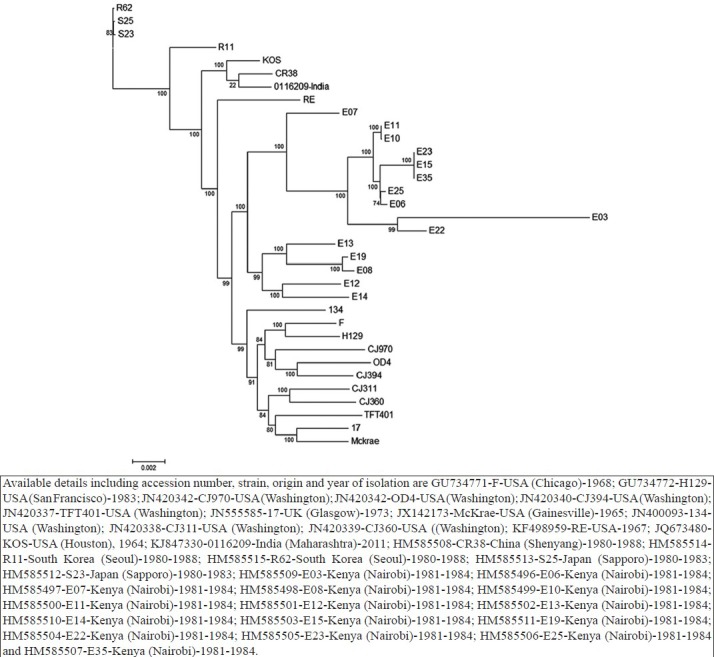
HSV-1 complete genome sequence-based phylogenetic analysis using maximum likelihood method. Details of GenBank accession numbers, strain names and geographic origin (if available) of sequences used in this analysis are mentioned in the tree. The full genome sequence of additional 34 HSV-1 strains available in GenBank used in the analysis.
Fig. 3.
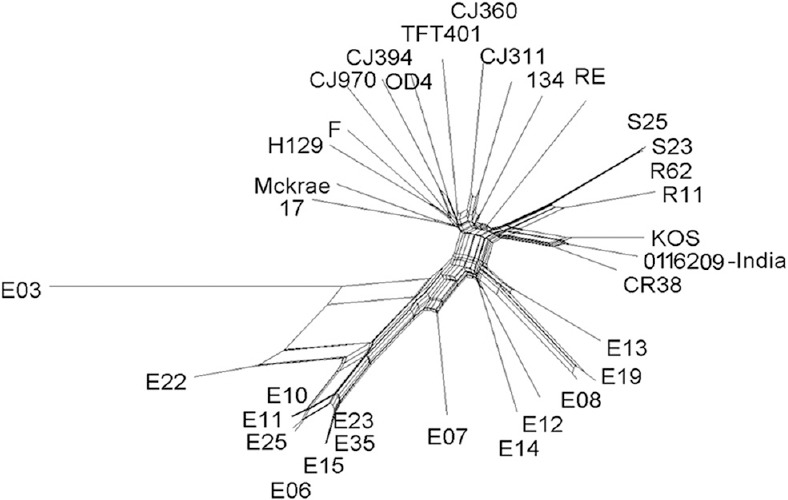
HSV-1 complete genome sequence-based splits tree analysis confirming the authenticity of maximum likelihood-based phylogenetic tree.
To analyze the degree of recombination and crossovers resulting in shifting phylogenetic relationships between worldwide distributed strains, the bootscan and SimPlot methods were used with Indian HSV-1 isolate as the query sequence. Similarity plot analysis demonstrated the presence of extensive recombination events occurred throughout the tree even in the strains circulating in similar geographic clusters. The SimPlot analysis suggested multiple recombination points in the Indian HSV-1 genome mostly resembling the KOS and CR38 isolates. The analysis demonstrated higher sequence similarities between the Indian and KOS isolates in the genomic regions ranging from 1 to 38,000 and 114,900 to 151,000 nucleotides. The genome between 38,000 and 114,900 nucleotides showed multiple crossover points from matching to different worldwide distributed strains (Fig. 4 A, B). The longest collinear area of similarity between Indian HSV-1 and KOS strain was about 74 kb, of which about 38 kb was distributed from 1 to 38,000 bp and 36 kb was distributed from 115,000 to 151,000 bp. Further confirmation of these findings was obtained by analysis of strains clustered in the Asian group. The Asian strain-based analysis reconfirmed genetic similarities between the Indian and KOS strains throughout the 74 kb region while the genomic region between 38,000 and 114,900 nucleotides mostly resembled to the CR38 and KOS strains. The phylogenetic trees generated from selected regions from Asian group with 1000 bootstrap supports confirmed these findings (Fig. 5 A-C).
Fig. 4.

HSV-1 recombination analysis. Recombination analysis using Bootscan and SimPlot. Panels A and B illustrate the bootscan and SimPlot analysis using Indian HSV-1 strain 0116209 as the query sequence. Bootscan plots demonstrate highly fragmented genomes as a result of recombination. Similarity plots demonstrate the sequence similarity between the Indian HSV-1 0116209 as query sequence and the other Asian strains sequences.
Fig. 5.

Phylogenetic analysis of 0116209 sequence suggesting recombination due to mixing of multiple strains from Asian lineage. Phylogenetic trees generated from selected regions from Asian group with 1000 bootstrap support suggests genetic similarities between 0116209 and KOS strain; (A) 1 to 38000 and (B) 38801 to 151000 nucleotides and between 0116209 and CR38 strain; (C) 115001 to 151024 nucleotides.
Discussion
HSV genome represents one of the complex viral genomes. Limitations of first-generation sequencing tools to resolve the complex genomes appear from the fact that while determining full-length genome sequences of H129, 17 and F strains, the lengths of 14 major SSRs were not resolved and were instead set to match the reference genomes9. Most of the currently resolved full-length genome sequences of HSV-1 strains are generated using different NGS platforms. As the sequence analysis of diagnostic PCR product showed maximum identity with HSV-1 KOS strain, the 376,332 reads amplified from the viral DNA library were primarily mapped against it through reference-guided and de novo assemblies. The unmapped gaps in the assembled sequence were further covered by PCR amplification, and sequencing of the unmapped regions to generate the full-length genome sequence of 151,024 bp. A previous phylogenetic study demonstrated that herpesviruses have co-evolved with their hosts and sorted according to their geographic origin24. Phylogenetic analysis of worldwide distributed HSV-1 strains classified them into three distinct groups strictly correlating with their geographic lines of sampling except the North American KOS strain that was clustered in the East Asian lineage25. Due to non-availability of HSV-1 full genome sequences of strains sampled from different geographic regions of Asia, conclusions drawn on Asian lineage are mainly based on representative strains from China, Japan and South Korea. In India, molecular evolution studies on HSV-1 are mainly based on glycoprotein G and I coding sequences amplified from clinical isolates of HSV-1 revealed the existence of novel genotypes and subgenotypes26. The existing genetic classification based on shorter sequences may not represent an accurate picture due to a high degree of recombination throughout the genome of HSV11,12,24.
In our study, phylogenetic analysis based on ML algorithm using 34 complete genome sequences of worldwide distributed strains classified them into three distinct lineages. The strain distribution appropriately followed their geographic origin as Europe/North American, Asian and the African lineages, which were further divided into two different groups. The KOS and RE strains originally isolated in the USA were clustered in the Asian lineage along with the Indian and other Asian strains. Since all the HSV-1 strains used in this analysis grouped according to their geographic origins, clustering of KOS and RE strains in Asian lineage with higher nucleotide identity with Asian strains hints their origin in Asian countries. The SplitsTree produced a clear separation of the worldwide distributed HSV-1 strains into distinct networks correlating with their geographic origin except the KOS (USA) strain clustered with the Asian strains and the RE (USA) placed in between the Asian and Europe/American strains. The observed split network pattern correlates with the topologies obtained in ML-based phylogenetic tree. As the phylogenetic grouping of HSV-1 strains approximately correlates with a geographic region of their origin, higher sequence similarities between RE and the Asian strains raises a concern about the actual geographic origin of the RE strain. Further studies on the evolutionary history of RE strain and comparative analysis with additional strains sampled from Asian and American countries would be helpful to define any impact of the higher rate of mixing of strains from different geographic regions due to the increased human movements.
Origin of KOS strain in the Asian countries and its dissemination to North America through migration has been estimated by Kolb et al24. Evidence on origin of KOS strain in Asian continent also came from the report of Grose27 suggesting that the KOS strain was originated in Korea during the early 1950s instead of the USA. Our study on phylogenetic analysis using full genome sequence of worldwide distributed strains confirmed the origin of KOS strain in the Asian continent. Indian HSV-1 strain 0116209 shared close genetic relationship with the KOS strain as compared to the Chinese, Japanese and South Korean strains clustered in the Asian lineage. These observations are supported by the fact that the KOS strains shared highest nucleotide identity with Indian, followed by Chinese, Japanese and Korean strains. These findings support single evolutionary ancestor of KOS, 0116209 and CR38 strains in the central Asian countries.
HSV-1 genomes are mosaic genomes and appear to undergo frequent recombination, at a much higher rate12. We examined the presence of recombination in the entire genome of Indian HSV-1 strain 0116209 through SimPlot analysis. The 0116209 strain genome was compared with worldwide generated 34 HSV-1 sequences to identify putative recombination sites. The crossover points were identified and mapped with sequence data sets exclusively generated from Asian strains. Multiple recombination and crossover points in Indian HSV-1 genome, especially resembling the Asian strains suggested that it was a recombinant strain and indicated possibilities of recombination among strains circulating in the same geographic region. Recombination analysis among the strains clustered in Asian lineage further confirmed possibilities of genetic transfer in the Indian isolate through other Asian strains. The recombination pattern observed in SimPlot analysis was confirmed for recombination signals using SplitsTree while the phylogenetic analysis of selected recombinant fragments in HSV-1 0116209 genome resembling to the sequence of KOS and CR38 strains was further confirmed by neighbour joining phylogenetic trees generated for each recombinant fragments. These findings indicated multiple recombination events between different HSV-1 strains circulating in similar geographic regions which might favour the evolution of newer recombinant strains. The comparatively higher levels of genetic conservation between the KOS, 0116209 and CR38 strains clustered in the Asian lineage suggested that these strains might share a single evolutionary source. Quantitative analysis of genomic polymorphisms among worldwide distributed HSV-1 strains also suggested that the evolutionary pattern was conserved among strains from a particular geographic region, and the Asian strains were comparatively less variable than the non-Asian strains27.
The HSV-1 KOS strain isolated from labial lesion is comparatively less virulent than other HSV-1 strains of American origin (Mckrae and 17)28,29,30,31. Studies to determine the virulence nature of Indian strain 0116209 sharing maximum genetic identity with the less virulent KOS strain isolated from an encephalitis patient will be helpful to investigate the impact of genetic changes documented in both these strains. Generation of sequence database from newer strains isolated worldwide will be helpful to define the potential and frequency of recombination occurring between genetically divergent strains. These studies will be crucial in the development of a protective vaccine candidate and effective therapeutics against herpesviruses.
In conclusion, this study generated full-length genome sequence of an HSV-1 isolate obtained from a patient suspected to have AES using the NGS approach and demonstrated its close genetic relationship with the KOS strain belonging to the Asian lineage. Considering the worldwide association of HSV with AES cases, availability of additional full-length genome sequence data of HSV-1 strains sampled from different regions of the country will be helpful in defining mode of evolution, structure-function analysis of viral proteins and identification of recombination in different strains, genetic basis of virulence and genetic markers for pathogenesis.
Acknowledgment
Authors acknowledge Shrimati D. Pavitrakar and R. Gunjikar, Shriyut V. M. Ayachit and G. Gunjal for technical support during virus isolation and data analysis. The authors thank the Director, NIV, for the constant intellectual support throughout the work. Financial support by the Indian Council of Medical Research to the first author (VPB) is acknowledged.
Footnotes
Conflicts of Interest: None.
References
- 1.Granerod J, Crowcroft NS. The epidemiology of acute encephalitis. Neuropsychol Rehabil. 2007;17:406–28. doi: 10.1080/09602010600989620. [DOI] [PubMed] [Google Scholar]
- 2.Gritsun TS, Lashkevich VA, Gould EA. Tick-borne encephalitis. Antiviral Res. 2003;57:129–46. doi: 10.1016/s0166-3542(02)00206-1. [DOI] [PubMed] [Google Scholar]
- 3.Glaser CA, Honarmand S, Anderson LJ, Schnurr DP, Forghani B, Cossen CK, et al. Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clin Infect Dis. 2006;43:1565–77. doi: 10.1086/509330. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy PG. Viral encephalitis: causes, differential diagnosis, and management. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 1):i10–5. doi: 10.1136/jnnp.2003.034280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitley RJ, Lakeman F. Herpes simplex virus infections of the central nervous system: therapeutic and diagnostic considerations. Clin Infect Dis. 1995;20:414–20. doi: 10.1093/clinids/20.2.414. [DOI] [PubMed] [Google Scholar]
- 6.Aurelius E, Johansson B, Sköldenberg B, Forsgren M. Encephalitis in immunocompetent patients due to herpes simplex virus type 1 or 2 as determined by type-specific polymerase chain reaction and antibody assays of cerebrospinal fluid. J Med Virol. 1993;39:179–86. doi: 10.1002/jmv.1890390302. [DOI] [PubMed] [Google Scholar]
- 7.Beig FK, Malik A, Rizvi M, Acharya D, Khare S. Etiology and clinico-epidemiological profile of acute viral encephalitis in children of western Uttar Pradesh, India. Int J Infect Dis. 2010;14:e141–6. doi: 10.1016/j.ijid.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 8.Modi A, Atam V, Jain N, Gutch M, Verma R. The etiological diagnosis and outcome in patients of acute febrile encephalopathy: a prospective observational study at tertiary care center. Neurol India. 2012;60:168–73. doi: 10.4103/0028-3886.96394. [DOI] [PubMed] [Google Scholar]
- 9.Szpara ML, Parsons L, Enquist LW. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol. 2010;84:5303–13. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGeoch DJ, Dolan A, Donald S, Brauer DH. Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res. 1986;14:1727–45. doi: 10.1093/nar/14.4.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norberg P, Bergström T, Rekabdar E, Lindh M, Liljeqvist JA. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J Virol. 2004;78:10755–64. doi: 10.1128/JVI.78.19.10755-10764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norberg P, Tyler S, Severini A, Whitley R, Liljeqvist JÅ, Bergström T. A genome-wide comparative evolutionary analysis of herpes simplex virus type 1 and varicella zoster virus. PLoS One. 2011;6:e22527. doi: 10.1371/journal.pone.0022527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One. 2011;6:e17288. doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham C, Gatherer D, Hilfrich B, Baluchova K, Dargan DJ, Thomson M, et al. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J Gen Virol. 2010;91(Pt 3):605–15. doi: 10.1099/vir.0.015891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sapkal GN, Wairagkar NS, Ayachit VM, Bondre VP, Gore MM. Detection and isolation of Japanese encephalitis virus from blood clots collected during the acute phase of infection. Am J Trop Med Hyg. 2007;77:1139–45. [PubMed] [Google Scholar]
- 16.Rao BL, Basu A, Wairagkar NS, Gore MM, Arankalle VA, Thakare JP, et al. A large outbreak of acute encephalitis with high fatality rate in children in Andhra Pradesh, India, in 2003, associated with chandipura virus. Lancet. 2004;364:869–74. doi: 10.1016/S0140-6736(04)16982-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin CH, Park GS, Hong KM, Paik MK. Detection and typing of HSV-1, HSV-2, CMV and EBV by quadruplex PCR. Yonsei Med J. 2003;44:1001–7. doi: 10.3349/ymj.2003.44.6.1001. [DOI] [PubMed] [Google Scholar]
- 18.Bondre VP, Sapkal GN, Yergolkar PN, Fulmali PV, Sankararaman V, Ayachit VM, et al. Genetic characterization of Bagaza virus (BAGV) isolated in India and evidence of anti-BAGV antibodies in sera collected from encephalitis patients. J Gen Virol. 2009;90(Pt 11):2644–9. doi: 10.1099/vir.0.012336-0. [DOI] [PubMed] [Google Scholar]
- 19.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huson DH, Kloepper TH. Computing recombination networks from binary sequences. Bioinformatics. 2005;21(Suppl 2):ii159–65. doi: 10.1093/bioinformatics/bti1126. [DOI] [PubMed] [Google Scholar]
- 22.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–67. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 23.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, et al. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1999;73:152–60. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolb AW, Ané C, Brandt CR. Using HSV-1 genome phylogenetics to track past human migrations. PLoS One. 2013;8:e76267. doi: 10.1371/journal.pone.0076267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, et al. Evolution and diversity in human herpes simplex virus genomes. J Virol. 2014;88:1209–27. doi: 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harishankar A, Jambulingam M, Gowrishankar R, Venkatachalam A, Vetrivel U, Ravichandran S, et al. Phylogenetic comparison of exonic US4, US7 and UL44 regions of clinical herpes simplex virus type 1 isolates showed lack of association between their anatomic sites of infection and genotypic/sub genotypic classification. Virol J. 2012;9:65. doi: 10.1186/1743-422X-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grose C. Korean war and the origin of herpes simplex virus 1 strain KOS. J Virol. 2014;88:3911. doi: 10.1128/JVI.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakaoka H, Kurita K, Iida Y, Takada S, Umene K, Kim YT, et al. Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J Gen Virol. 1994;75(Pt 3):513–27. doi: 10.1099/0022-1317-75-3-513. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Davido DJ, Morrison LA. HSV-1 strain McKrae is more neuroinvasive than HSV-1 KOS after corneal or vaginal inoculation in mice. Virus Res. 2013;173:436–40. doi: 10.1016/j.virusres.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perng GC, Mott KR, Osorio N, Yukht A, Salina S, Nguyen QH, et al. Herpes simplex virus type 1 mutants containing the KOS strain ICP34.5 gene in place of the McKrae ICP34.5 gene have McKrae-like spontaneous reactivation but non-McKrae-like virulence. J Gen Virol. 2002;83(Pt 12):2933–42. doi: 10.1099/0022-1317-83-12-2933. [DOI] [PubMed] [Google Scholar]
- 31.Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, et al. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 2011;7:e1002282. doi: 10.1371/journal.ppat.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]