

Abstract
Context:
A subpopulation of obese individuals remains insulin sensitive (ISO). They represent a unique human model to investigate factors underlying insulin resistance (IR) without the confounding effect of major differences in weight/adiposity. Altered fatty-acid (FA) metabolism in sc adipose tissue (SAT) contributes to obesity-associated IR.
Objective:
To test the hypothesis that ISO and body mass index-matched insulin-resistant obese (IRO) patients demonstrate differential SAT expression profiles of genes involved in glycerolipid-FA metabolism and that weight loss-induced improvement of IR ameliorates these changes.
Design and Setting:
A cross-sectional and longitudinal study.
Patients and Intervention:
Thirty-eight nondiabetic obese women were stratified into ISO (n = 25) or IRO (n = 13) groups based on hyperinsulinemic-euglycemic clamp results. Subjects were studied before and after a 6-month hypocaloric diet intervention.
Main Outcome Measures:
mRNA (quantitative RT-PCR) and protein (mass spectrometry and immunoblots) levels were measured in SAT biopsies.
Results:
Despite having age, body mass index, and fat mass similar to ISO individuals, IRO patients had lower insulin sensitivity and glucose tolerance (P < .05). Baseline SAT mRNA and protein levels of genes involved in both the synthesis and lipolysis of glycerolipid-FAs were higher in IRO individuals (P < .05), even when groups were matched for visceral adipose tissue content. The dietary intervention resulted in approximately 6% weight loss in both the IRO and ISO groups (P < .05) but only ameliorated insulin sensitivity in IRO individuals (P < .05). Likewise, the intervention reduced the expression of most glycerolipid-FA metabolism genes (P < .05), with expression levels in IRO individuals being restored to ISO levels.
Conclusions:
Increased SAT expression of genes involved in both the synthesis and hydrolysis of glycerolipid-FAs is closely associated with IR in obese women. The results suggest that enhanced glycerolipid-FA cycling in SAT contributes to obesity-associated IR.
Despite the strong correlation observed between obesity and insulin resistance, the mechanism(s) leading to obesity-associated insulin resistance and type 2 diabetes are still unclear, and not all obese patients develop such metabolic syndrome-associated diseases.
Accordingly, a subpopulation of obese individuals referred to as “metabolically healthy but obese” or “insulin-sensitive but obese” (ISO) (1–5) appears to be relatively protected from developing metabolic complications (6). They could represent as much as 30–40% of obese adults in the United States (7). Despite having similar body mass index (BMI) and fat mass, these ISO individuals typically show normal insulin sensitivity, glucose tolerance, serum lipid levels, and favorable hormonal and inflammatory profiles compared to insulin-resistant obese (IRO) patients (2). Although the underlying factors that lead to this protective phenotype remain largely unknown, these individuals provide an excellent human model to delineate key factors that either contribute to or prevent the development of insulin resistance without the confounding effect of major differences in body fat mass.
Adipose tissue (AT) dysfunction plays a key role in the development of obesity-associated metabolic diseases (8). One of the main functions of AT is to store lipids and release them in the form of nonesterified fatty acids (NEFAs) as a fuel for other tissues in times of need. In obese subjects, several lines of evidence suggest that defective fatty acid (FA) disposal in sc AT (SAT) leads to the spillover of fat into ectopic tissues (such as liver, skeletal muscle, and visceral AT [VAT]), resulting in lipotoxicity and insulin resistance (9). It is well established that less ectopic fat accumulation occurs in ISO patients compared to IRO patients, suggesting that spillover could be involved even when patients are matched for body fat mass (1). However, the molecular factors underlying impaired FA disposal in SAT of IRO patients remain unknown.
Intracellular lipid pools are dynamic and in constant turnover in a process termed glycerolipid-FA cycling, with a high percentage of NEFAs released to the cytoplasm upon lipolysis being immediately re-esterified, even at steady-state levels of triacylglycerol and NEFAs (10). Storage of fat in AT thus results from several interrelated steps of FA metabolism (reviewed in Refs. 11 and 12), including: 1) FA uptake, facilitated by molecules such as CD36 and FA transport protein 1 (FATP1); 2) their acylation by enzymes such as acyl-coenzyme A synthetase long-chain family member 1 (ACSL1) and FATP1; 3) their esterification into diacylglycerol and triacylglycerol by enzymes such as glycerol-3-phosphate acyltransferases (GPATs), 1-acylglycerol-3-phosphate O-acyltransferases (AGPATs), and diacylglycerol O-acyltransferases (DGATs); and 4) the opposing effects of glycerolipid hydrolysis, which is regulated at several steps by proteins such as adipose triglyceride lipase (ATGL), abhydrolase domain containing 5 (ABHD5), hormone-sensitive lipase (HSL), monoglyceride lipase, perilipin (PLIN), adipocyte fatty acid-binding protein (FABP4), and aquaporins (AQPs). Fat storage in AT is also regulated by the adipogenic transcription factor peroxisome proliferator-activated receptor γ (PPARγ), the lipid droplet-associated protein cell death activator CIDE-3 (CIDEC) (13), and the de novo lipogenesis enzymes fatty acid synthase (FASN) and acetyl-coenzyme A carboxylase (ACC)-1 and transcription factor sterol regulatory element-binding protein 1c (SREBP-1c) (11). Thus, efficient FA disposal into AT requires the coordinated action of key enzymes, transport proteins, and regulatory molecules that control the activity of the lipogenic and lipolytic arms of glycerolipid-FA cycling.
In the present study, we examined the mRNA and protein expression levels of several genes involved in glycerolipid-FA metabolism in SAT biopsies from thoroughly phenotyped nondiabetic postmenopausal obese women to obtain better insight into the molecular factors that distinguish ISO and IRO patients from each other.
Subjects and Methods
Subjects
Thirty-eight postmenopausal obese women enrolled between 2005 and 2007 in the Complications Associated with Obesity (CAO) cohort were studied; the inclusion/exclusion criteria were reported elsewhere (14). Briefly, women were enrolled if they were: 1) obese, with BMI of 30–40 kg/m2; 2) 55–70 years old; 3) with biological confirmation of the menopause status without taking hormone replacement therapy; 4) not diabetic (fasting glucose < 7.1 mmol * L−1 and 2-h plasma glucose < 11.1 mmol * L−1 after a 75-g oral glucose tolerance test [OGTT]); 5) nonsmokers; 6) not taking medications known to interfere with metabolism except stable hypothyroidism replacement therapy; 7) free of chronic or inflammatory diseases; and 8) without any history of alcohol or drug abuse. This study conformed to the principles outlined in the Declaration of Helsinki as revised in 2000 and was approved by the Université de Montréal Ethics Committee. All subjects gave written informed consent.
Participants were classified as ISO or IRO according to insulin sensitivity thresholds predefined in a similar cohort, as assessed by their steady-state glucose disposal rate (GDR) during a 3-hour hyperinsulinemic-euglycemic clamp (HEC) as reported (2, 3). Accordingly, patients who had a GDR ≤ 9.5 mg min−1 kg−1 of fat free mass (FFM) were classified as IRO (n = 13), and those with a GDR ≥ 12.0 mg min−1 kg−1 FFM were classified as ISO (n = 25). A subgroup of patients (ISO, n = 18; IRO, n = 10) completed a medically supervised hypocaloric diet aimed at reducing body weight by 10% over a 6-month period (15) (Supplemental Data).
All pre- and postdiet anthropometric and blood measurements and SAT biopsies were obtained after an overnight fast (2, 14) (Supplemental Data).
SAT biopsies
Consent for biopsy was an optional part of the study without any other criteria required for a patient to be included in this subsample. Periumbilical SAT needle biopsies were obtained at baseline (ISO, n = 13; IRO, n = 9) and postdietary intervention (paired; ISO, n = 6; IRO, n = 6) under local anesthesia (20 mg xylocaine/mL). Samples were washed from excess blood in saline, quickly frozen in liquid nitrogen, and stored at −80°C until analysis. In a secondary analysis, the patients for whom SAT biopsies were available were stratified according to their VAT content into ISO/low VAT (n = 7), ISO/high VAT (n = 6), and IRO/high VAT (n = 8) groups. Low or high VAT content was determined according to the median VAT value of the cohort. Accordingly, patients with ≥200 cm2 VAT content were classified as high VAT, whereas patients with < 200 cm2 VAT content were classified as low VAT. Only one IRO patient was classified as low VAT and was excluded from this analysis.
Quantitative RT-PCR (qRT-PCR) and immunoblots
SAT was homogenized in lysis buffer for immunoblotting as described previously (5). RNA was extracted from SAT, followed by qRT-PCR assays with Quantitect SYBR Green Universal PCR Mix (QIAGEN) on a Rotor-Gene (Corbett Life Science, QIAGEN). Primer sequences (Supplemental Table 1) were designed with Primer-Blast (www.ncbi.nlm.nih.gov/tools/primer-blast/) to span an intron or an exon-exon junction to avoid genomic DNA amplification. The relative standard curve method was used for gene expression quantification normalized against 18s (see Supplemental Data for details).
Quantitative proteomics
Mass spectrometry-based selected reaction monitoring (SRM) assays were developed for eight proteins implicated in glycerolipid-FA metabolism and the housekeeping protein β-actin (Supplemental Data). Monitored transitions (ie, pairs of parent and fragment ions specific to a peptide) are listed in Supplemental Table 2. Proteotypic peptides representing target proteins were selected from SAT liquid chromatography- tandem mass spectrometry data, and synthetic heavy isotope-labeled peptides were used to design, optimize, and normalize assays (Supplemental Figure 1). Tryptic digests of SAT proteins were analyzed on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Scientific). Raw data were analyzed using Pinpoint 1.3.0 (Thermo Scientific). Abundance values reported are normalized against β-actin.
Statistical analysis
Graphs were prepared with GraphPad Prism 5.04 for Windows (GraphPad Software, Inc). Results were analyzed with Statview version 5.0.1 or GraphPad Prism 5.04 for Windows and are presented as means ± SD or SEM. Two-tailed nonparametric Mann-Whitney U tests, Wilcoxon signed rank tests, Kruskal-Wallis test, or two-way ANOVA with repeated measures, as appropriate, were used to determine statistical significance of continuous variables. Dunn's multiple comparison post hoc test was used in the event of a significant probability value. For all analyses, P ≤ .05 was considered statistically significant.
Results
Baseline clinical characteristics of ISO and IRO subjects
The ISO and IRO groups had similar age, BMI, percentage body fat and FFM, abdominal SAT area (measured at L4–L5), waist and hip circumferences, blood pressure (BP), blood lipid profile, total energy expenditure, and total intakes of energy and macronutrients (Table 1).
Table 1.
Baseline Clinical Parameters of ISO and IRO Subjects
| ISO | IRO | P Value | |
|---|---|---|---|
| n | 25 | 13 | |
| Anthropometric parameters | |||
| Age, y | 57.3 ± 4.0 | 54.9 ± 3.8 | .068 |
| BMI, kg/m2 | 34.1 ± 3.1 | 35.4 ± 3.7 | .39 |
| Weight, kg | 90.1 ± 12.7 | 95.4 ± 16.0 | .47 |
| Height, m | 1.6 ± 0.1 | 1.6 ± 0.1 | .72 |
| Fat mass, % | 48.3 ± 3.7 | 47.0 ± 3.6 | .30 |
| FFM, % | 51.7 ± 3.7 | 53.0 ± 3.6 | .30 |
| VAT (L4–L5), cm2 | 185.7 ± 46.6 | 236.8 ± 53.7 | .005 |
| SAT (L4–L5), cm2 | 532.4 ± 118.0 | 538.5 ± 122.4 | 1.00 |
| Muscle fat content, cm2 | 46.8 ± 3.2 | 48.7 ± 2.8 | .058 |
| Waist circumference, cm | 104.1 ± 10.1 | 107.7 ± 9.5 | .21 |
| Hip circumference, cm | 119.0 ± 10.5 | 119.0 ± 10.7 | 1.00 |
| Hemodynamic parameters | |||
| Systolic BP, mm Hg | 126.0 ± 12.5 | 128.4 ± 15.8 | .91 |
| Diastolic BP, mm Hg | 78.6 ± 8.8 | 78.9 ± 8.1 | .82 |
| Metabolic parameters | |||
| GDR, mg min−1 kg−1 FFM | 15.0 ± 2.6 | 7.7 ± 1.8 | <.001 |
| HOMA-IR | 3.1 ± 1.1 | 6.2 ± 3.1 | <.001 |
| Fasting insulin, μIU/mL | 13.0 ± 4.0 | 23.9 ± 10.4 | <.001 |
| Fasting glucose, mm | 5.4 ± 0.5 | 5.7 ± 0.58 | .096 |
| 2-h OGTT 2-h insulin, μIU/mL | 58.48 ± 31.86 | 163.14 ± 89.1 | <.001 |
| 2-h OGTT 2 h-glucose, mm | 6.4 ± 1.5 | 8.2 ± 1.4 | .002 |
| OGTT glucose AUC | 885 ± 167 | 1123 ± 199 | .001 |
| OGTT insulin AUC | 7920 ± 2943 | 15300 ± 7483 | .009 |
| Total cholesterol, mm | 5.3 ± 1.0 | 5.7 ± 0.9 | .39 |
| LDL-C, mm | 3.2 ± 0.9 | 3.5 ± 0.8 | .33 |
| HDL-C, mm | 1.6 ± 0.3 | 1.4 ± 0.3 | .140 |
| Triacylglycerol, mm | 1.2 ± 0.5 | 1.8 ± 1.2 | .069 |
| ApoB, g/L | 0.9 ± 0.2 | 1.0 ± 0.2 | .139 |
| NEFA, mm | 0.6 ± 0.2 | 0.6 ± 0.2 | .81 |
| Glycerol, mm | 0.09 ± 0.04 | 0.09 ± 0.03 | 1.00 |
| ALP, U/L | 82.5 ± 22.9 | 81.0 ± 18.2 | .81 |
| AST, U/L | 19.6 ± 5.9 | 19.6 ± 3.0 | .35 |
| ALT, U/L | 20.6 ± 7.6 | 26.6 ± 5.8 | .014 |
| GGT, μU/L | 18.5 ± 1.0 | 24.2 ± 16.6 | .22 |
| Inflammatory markers | |||
| WBC count, ×109/L | 5.2 ± 1.0 | 6.8 ± 1.5 | .002 |
| Neutrophils, ×109/L | 2.9 ± 0.7 | 3.8 ± 0.8 | .001 |
| hsCRP, mg/L | 2.9 ± 2.0 | 4.7 ± 2.7 | .073 |
| hsIL-6, pg/mL | 1.5 ± 0.9 | 2.0 ± 0.7 | .036 |
| hsTNF, pg/mL | 1.6 ± 1.5 | 1.9 ± 2.7 | .27 |
| Haptoglobin, g/L | 1.3 ± 0.4 | 1.6 ± 0.3 | .027 |
| Orosomucoid, g/L | 0.79 ± 0.15 | 0.93 ± 0.17 | .025 |
| Uric acid, μmol/L | 282.8 ± 43.7 | 325.4 ± 47.6 | .011 |
| Energy expenditure and intake | |||
| TEE (DLW), kcal/d | 2581.0 ± 457.9 | 2637.0 ± 427.6 | .74 |
| RREE, kcal d−1 kg−1 LBM | 30.8 ± 2.2 | 32.4 ± 3.4 | .22 |
| PAEE, kcal/d | 982.0 ± 356.3 | 836.7 ± 278.8 | .26 |
| RER, VCO2/VO2 | 0.86 ± 0.05 | 0.86 ± 0.05 | .54 |
| Energy intake, kcal/d | 1973.0 ± 434.9 | 2124.0 ± 540.0 | .40 |
| Lipid intake, % DE | 33.9 ± 6.6 | 32.7 ± 5.7 | .51 |
| CHO intake, % DE | 46.3 ± 6.4 | 48.8 ± 5.9 | .31 |
| Protein intake, % DE | 17.4 ± 3.6 | 17.4 ± 2.6 | .97 |
Abbreviations: LBM, lean body mass; ApoB, apolipoprotein B; ALP, alkaline phosphatase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, γ-glutamyl transferase; WBC, white blood cells; hsCRP, high-sensitivity C-reactive protein; hsIL-6, high-sensitivity IL-6; hsTNF, high-sensitivity TNF; TEE, total energy expenditure; DLW, doubly labeled water; RREE, relative resting energy expenditure; PAEE, physical activity energy expenditure; RER, respiratory exchange ratio; CHO, carbohydrates; DE, daily energy; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol. Results are expressed as means ± SD. Statistical significance was determined with a Mann-Whitney U test.
By design, the ISO group was more insulin sensitive, having a 2-fold higher GDR during the HEC than the IRO group (Table 1). The ISO group also had lower fasting insulin, 2-hour OGTT glucose and insulin, homeostasis model assessment of insulin resistance (HOMA-IR), and glucose and insulin area under the curve (AUC) during the OGTT than the IRO group (Table 1). Altogether, these data indicate that despite similar age, BMI, and percentage fat mass, the ISO subjects clearly showed better insulin sensitivity and glucose tolerance than the IRO patients.
The IRO group also demonstrated signs of ectopic lipid deposition. Indeed, they had increased VAT area and serum alanine aminotransferase, a clinical surrogate of hepatic steatosis, and showed a trend toward higher muscle fat content (Table 1). Several circulating inflammatory markers were higher in the IRO group (Table 1).
Increased mRNA expression of genes involved in glycerolipid-FA cycling in SAT of IRO subjects
The transcript levels of genes involved in various steps of glycerolipid-FA synthesis pathway such as adipogenesis (PPARG, CIDEC), de novo lipogenesis (ACC1), FA activation (FATP1, ACSL1), and esterification (GPAT1, AGPAT2, DGAT1, DGAT2) were significantly higher in IRO than ISO patients (1.3- to 1.7-fold) (Figure 1). Interestingly, mRNA levels of genes involved in glycerolipid-FA hydrolysis such as HSL, ATGL, FABP4, and ABHD5 were also higher in IRO patients (1.3- to 1.4-fold) (Figure 1); however, the mRNA levels of SREBP1c, FASN, PLIN1, and AQP7 were not significantly different between the groups (P = .30, .36, .28, and .07, respectively) (Figure 1).
Figure 1. Increased mRNA expression of genes involved in glycerolipid-FA cycling in SAT of IRO subjects.
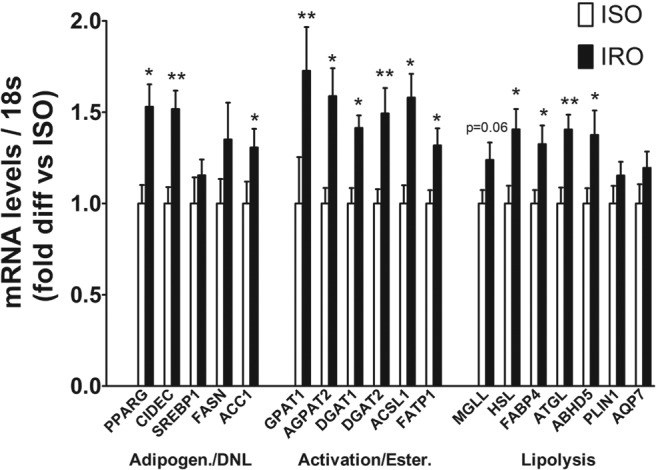
Fold difference in the mRNA levels of genes involved in various steps of glycerolipid-FA synthesis and lipolysis, in SAT of IRO relative to ISO subjects. Results are expressed as means ± SEM (n = 9–13 per group). DNL, de novo lipogenesis; Adipogen., adipogenesis; Ester., esterification. Significantly different from ISO: *, P < .05; **, P < .01.
Because VAT content is an important risk factor for insulin resistance and because IRO patients had higher VAT content than ISO individuals (Table 1), we performed a secondary analysis in which patients were classified as having high or low VAT (see Subjects and Methods). We found that even when the groups were matched for their VAT content, IRO/high VAT patients had higher expression of most glycerolipid-FA cycle genes than ISO/high VAT patients (Supplemental Figure 2). This excludes high VAT content in IRO as a potential confounder to our results and further supports the association between insulin resistance and increased glycerolipid-FA cycling in SAT.
SRM assays revealed increased protein abundance of genes involved in glycerolipid-FA cycling in SAT of IRO subjects
Based on high confidence identification in preliminary high-resolution liquid chromatography tandem-mass spectrometry analyses of the SAT samples (data not shown), mass spectrometry-based SRM assays were developed to quantify protein levels of seven of the mRNA transcripts studied by qRT-PCR (ACC1, GPAT1, FASN, PLIN1, ACSL1, HSL, and FABP4) as well as for CD36. Levels of several peptides representing the proteins CD36, PLIN1, HSL, ACSL1, FABP4, and FASN were significantly higher in IRO than in ISO patients (1.5- to 7-fold) (Figure 2, A–F). However, endogenous peptides representing ACC1 and GPAT1 were below the lower limit of quantification (data not shown). Overall, these results provide further evidence that the abundance of proteins involved in both the synthesis and lipolysis of glycerolipids is increased in IRO as compared to ISO patients.
Figure 2. Increased SAT abundance of proteins involved in glycerolipid-FA cycling and lower ACC phosphorylation in IRO relative to ISO patients.

A–F, Fold difference in the abundance of peptides representing CD36 (A), PLIN1 (B), HSL (C), ACSL1 (D), FABP4 (E), and FASN (F), as measured by SRM, in SAT of IRO relative to ISO subjects. G, Immunoblots with specific antibodies for P-ACC S79, T-ACC, P-AMPKα T172, T-AMPKα, tubulin, and β-actin as well as Ponceau Red staining of polyvinylidene difluoride membrane after transfer. H–K, Densitometric analysis of T-ACC/β-actin (H), P-ACC/T-ACC (I), P-AMPK/T-AMPK (J), and T-AMPK/β-actin (K). Results are expressed as means ± SEM (n = 6–7 per group). Abbreviated peptide sequences: VAI, VAIIDTYK; QVV, QVVLEEGTIAFK; TYL, TYLDIEPITGFTLQFAK; AFA, AFASPVENPDNYCFCTEK; LSG, LSGVFAGVR; EGQ, EGQDSEELSSLIK; LGA, LGASSWLNSFLELSGR; NIV, NIVSDCSAFVK; GFE, GFEGSFEELCR; VLQ, VLQPTVFPVVPR; GIQ, GIQVSNNGPCLGSR; VLH, VLHLTPAPAVSSTK; VLG, VLGAALAGCELAWGVAR; EVG, EVGVGFATR; STI, STITLDGGVLVHVQK; VGD, VGDPQELNGITR; TGT, TGTVSLEVR. Significantly different from ISO: *, P < .05; **, P < .01; ***, P < .001.
Increased abundance and reduced phosphorylation of ACC in SAT of IRO patients
IRO patients showed a 3-fold increase in ACC protein abundance compared to ISO subjects (Figure 2, G and H). Moreover, they had a 7-fold decrease in the level of ACC phosphorylated at S79 (Figure 2, G and I), a well-documented inhibitory site and specific downstream target of AMP-activated protein kinase (AMPK) (16). These results suggest that ACC activity and de novo lipogenesis are increased and AMPK activity is decreased in IRO compared to ISO subjects. Abundance of phospho-AMPK (P-AMPK) T172 and total-AMPK (T-AMPK) was not different between the two groups (Figure 2, G, J, and K) (P = .68 and .37, respectively). Of note, similar results were obtained when densitometric analyses of total-ACC (T-ACC) and T-AMPK were normalized by tubulin instead of β-actin (data not shown).
Effect of a 6-month dietary intervention on clinical characteristics of ISO and IRO subjects
The dietary intervention resulted in a similar decrease in BMI and an approximately 6% weight loss in both groups (Table 2). Conversely, the intervention resulted in a 30% improvement in insulin sensitivity as measured by the HEC in the IRO group, whereas this remained unchanged in the ISO group (Table 2). The dietary intervention resulted in a similar significant decrease in percentage of fat mass, VAT and abdominal SAT area, waist and hip circumferences, BP, fasting insulin, glucose, and HOMA-IR in both groups (Table 2). The intervention had no effect on measured serum lipids or inflammatory markers, however (Table 2). Altogether, these results show that the dietary intervention had beneficial metabolic and anthropometric effects on both ISO and IRO patients, but that the improvement in insulin sensitivity was predominantly seen in IRO patients.
Table 2.
Clinical Characteristics of ISO and IRO Subjects Before and After a 6-Month Hypocaloric Diet
| ISO (n = 18) |
IRO (n = 10) |
P Values |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | Interaction: Diet * Phenotype | Diet Effect in ISO | Diet Effect in IRO | ISO vs IRO Pre | ISO vs IRO Post | |
| Anthropometric parameters | |||||||||
| Weight, kg | 88.2 ± 13.8 | 82.9 ± 13.6 | 98.5 ± 17.3 | 92.8 ± 16.1 | .7821 | <.0001 | <.0001 | .093 | .093 |
| BMI, kg/m2 | 33.6 ± 3.5 | 31.6 ± 3.7 | 35.5 ± 3.7 | 33.5 ± 3.7 | .9986 | <.0001 | <.0001 | .1893 | .1893 |
| Weight loss, % BW | N/A | 6.1 ± 3.9 | N/A | 5.7 ± 3.6 | N/A | N/A | N/A | N/A | .7553 |
| Fat mass, % | 47.4 ± 3.7 | 45.9 ± 5.3 | 46.4 ± 3.8 | 45.6 ± 4.0 | .5515 | .0424 | .0424 | .7162 | .7162 |
| FFM, % | 52.7 ± 3.6 | 54.1 ± 5.3 | 53.6 ± 3.8 | 54.4 ± 4.0 | .5515 | .0424 | .0424 | .7162 | .7162 |
| VAT (L4–L5), cm2 | 182.8 ± 46.3 | 162.8 ± 42.0 | 239.6 ± 54.2 | 210.8 ± 51.7 | .3667 | <.0001 | <.0001 | .0076 | .0076 |
| SAT (L4–L5), cm2 | 517.6 ± 119.2 | 470.2 ± 119.9 | 538.9 ± 129.0 | 499.0 ± 127.0 | .6438 | <.0001 | <.0001 | .6034 | .6034 |
| Muscle fat content, cm2 | 47.3 ± 3.3 | 47.4 ± 3.8 | 49.6 ± 2.1 | 49.3 ± 1.6 | .4995 | .6417 | .6417 | .0919 | .0919 |
| Waist circumference, cm | 102.9 ± 10.8 | 98.6 ± 10.0 | 107.2 ± 8.2 | 102.5 ± 6.4 | .8688 | <.0001 | <.0001 | .273 | .273 |
| Hip circumference, cm | 116.6 ± 10.7 | 111.9 ± 9.7 | 119.5 ± 11.7 | 116.5 ± 10.4 | .3453 | .0002 | .0002 | .3628 | .3628 |
| Hemodynamic parameters | |||||||||
| Systolic BP, mm Hg | 126.7 ± 13.0 | 118.1 ± 12.9 | 131.1 ± 16.3 | 121.1 ± 14.1 | .7732 | .0005 | .0005 | .4631 | .4631 |
| Diastolic BP, mm Hg | 79.8 ± 9.4 | 73.2 ± 7.9 | 80.8 ± 7.8 | 76.0 ± 9.0 | .5768 | .0017 | .0017 | .5322 | .5322 |
| Metabolic parameters | |||||||||
| GDR, mg min−1 kg−1 FFM | 15.4 ± 2.7 | 14.4 ± 3.7 | 7.5 ± 1.9 | 9.9 ± 2.2 | .0034 | .1446 | .0069 | <.0001 | .0007 |
| HOMA-IR | 3.0 ± 1.0 | 2.6 ± 0.8 | 6.4 ± 3.4 | 4.9 ± 2.3 | .1665 | .0117 | .0117 | .0002 | .0002 |
| Fasting insulin, μIU/mL | 12.3 ± 3.7 | 10.9 ± 3.0 | 24.5 ± 11.4 | 19.5 ± 6.8 | .1064 | .0069 | .0069 | <.0001 | <.0001 |
| Fasting glucose, mm | 5.4 ± 0.6 | 5.1 ± 0.6 | 5.7 ± 0.6 | 5.5 ± 0.9 | .5754 | .0498 | .0498 | .1593 | .1593 |
| Total cholesterol, mm | 4.8 ± 0.8 | 4.8 ± 1.0 | 5.2 ± 0.8 | 5.1 ± 0.6 | .8661 | .6052 | .6052 | .2662 | .2662 |
| LDL-C, mm | 2.8 ± 0.7 | 2.8 ± 0.9 | 3.2 ± 0.7 | 3.2 ± 0.6 | .7505 | .9484 | .9484 | .1673 | .1673 |
| HDL-C, mm | 1.5 ± 0.4 | 1.5 ± 0.3 | 1.3 ± 0.2 | 1.2 ± 0.2 | .8565 | .4709 | .4709 | .0393 | .0393 |
| Triacylglycerol, mm | 1.2 ± 0.5 | 1.1 ± 0.5 | 1.8 ± 1.3 | 1.5 ± 0.6 | .1817 | .0751 | .0751 | .0677 | .0677 |
| ApoB, g/L | 0.9 ± 0.1 | 0.9 ± 0.2 | 1.0 ± 0.2 | 1.1 ± 0.2 | .9674 | .6055 | .6055 | .0572 | .0572 |
| NEFA, mm | 0.5 ± 0.2 | 0.6 ± 0.2 | 0.6 ± 0.3 | 0.6 ± 0.3 | .3352 | .6527 | .6527 | .8956 | .8956 |
| Glycerol, mm | 0.09 ± 0.04 | 0.08 ± 0.03 | 0.10 ± 0.03 | 0.09 ± 0.04 | .6380 | .4149 | .4149 | .3732 | .3732 |
| Inflammatory markers | |||||||||
| hsCRP, mg/L | 2.5 ± 1.2 | 2.0 ± 1.5 | 4.3 ± 2.5 | 4.0 ± 2.4 | .7190 | .23 | .23 | .0273 | .0273 |
| Haptoglobin, g/L | 1.3 ± 0.4 | 1.3 ± 0.4 | 1.5 ± 0.2 | 1.4 ± 0.3 | .0316 | .255 | .1386 | .0259 | .472 |
| Orosomucoid, g/L | 0.8 ± 0.2 | 0.8 ± 0.4 | 0.9 ± 0.2 | 0.8 ± 0.2 | .391 | .3327 | .3327 | .5295 | .5295 |
| Energy expenditure | |||||||||
| RREE, kcal d−1 kg−1 LBM | 30.7 ± 2.4 | 30.9 ± 2.7 | 32.7 ± 3.7 | 31.6 ± 2.7 | .1674 | .3341 | .3341 | .2052 | .2052 |
| RER (VCO2/VO2) | 0.87 ± 0.05 | 0.89 ± 0.06 | 0.86 ± 0.05 | 0.87 ± 0.03 | .582 | .2 | .2 | .6314 | .6314 |
Abbreviations: Pre, before the initiation of a 6-month hypocaloric diet; Post, after a 6-month hypocaloric diet; LBM, lean body mass; ApoB, apolipoprotein B; hsCRP, high-sensitivity C-reactive protein; RREE, relative resting energy expenditure; BW, body weight; RER, respiratory exchange ratio; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; N/A, not applicable. Results are expressed as means ± SD. Statistical significance was determined by two-way ANOVA with repeated measures followed by Wilcoxon signed rank and Mann-Whitney U tests, as appropriate.
Effect of a 6-month dietary intervention on mRNA expression of genes involved in glycerolipid-FA cycling in SAT of ISO and IRO subjects
Genes that showed differential baseline mRNA levels between the groups were assessed in paired SAT biopsies after dietary intervention. The intervention resulted in a significant decrease in SAT mRNA levels of PPARG, CIDEC, ACC1, GPAT1, AGPAT2, DGAT2, ACSL1, FATP1, and ABHD5, predominantly in IRO patients (Figure 3, A–E, G–I, and M). The intervention had no significant effect on the expression of HSL, FABP4, and ATGL in both groups (P = .85, .32, and .54, respectively) and of DGAT1 in ISO (P = .84) but showed a trend in IRO (P = .06) (Figure 3, F and J–L). Except for CIDEC, HSL, and ATGL, expression of glycerolipid-FA genes in IRO was restored to ISO levels after the intervention. Altogether, these results indicate that the diet-induced improvement in insulin sensitivity is associated with decreased mRNA expression of genes involved in glycerolipid-FA metabolism.
Figure 3. Decreased SAT expression of glycerolipid-FA metabolism genes after a 6-month hypocaloric diet intervention is predominant in IRO patients.

A–M, SAT mRNA levels of genes involved in glycerolipid-FA metabolism before and after a 6-month dietary intervention in IRO (filled box) and ISO (open circle) subjects. Results are expressed as means ± SEM (n = 6 per group, paired). Significantly different from ISO at the time point showed: a P < .05, aa P < .01, aaa P < .001; significant effect of the dietary intervention in the IRO group: b P < .05; significant effect of the dietary intervention in the ISO group: c P < .05.
Effect of a 6-month dietary intervention on protein abundance of genes involved in glycerolipid-FA cycling in SAT of ISO and IRO subjects
Figure 4 shows the average abundance of all measured peptides per protein (two to four peptides) before and after intervention, as assessed by SRM. Data for individual peptides are presented in Supplemental Figure 3. The intervention resulted in a significant decrease in protein levels FABP4 and FASN in IRO patients only (Figure 4, D and E). In addition, ACSL1, FABP4, FASN, and PLIN1 were no longer different between the ISO and IRO groups after the diet (Figure 4, C–F) (P = .86, .47, .20, and 1.0, respectively).
Figure 4. SAT abundance of glycerolipid-FA metabolism proteins is restored to similar levels in IRO and ISO patients after a 6-month dietary intervention.

A–F, SAT levels of proteins involved in glycerolipid-FA metabolism before and after 6-month hypocaloric dietary intervention in IRO (filled box) and ISO (open circle) subjects. Results are expressed as means ± SEM (n = 6 per group, paired) of the average of all peptides measured per protein and normalized to β-actin. Significantly different from ISO group at the time point showed: a P < .05, aa P < .01; significant effect of the dietary intervention in the IRO group: b P < .05.
Discussion
Although the concept of insulin-sensitive obesity is now well recognized, the underlying molecular factors that differentiate ISO from IRO patients remain largely unidentified. The results of the present study revealed a close relationship between higher SAT expression of genes involved in de novo lipogenesis and glycerolipid-FA cycling and the presence of obesity-associated insulin resistance. Accordingly, the key findings were: 1) IRO patients showed higher SAT mRNA and protein levels of genes involved in both synthesis and hydrolysis of glycerolipid-FAs than ISO subjects; 2) SAT abundance of phospho-ACC S79 was significantly lower in IRO patients, suggesting increased de novo lipogenesis and potentially decreased AMPK activity; and 3) a 6-month hypocaloric diet improved insulin sensitivity in IRO patients only and restored mRNA and protein levels of most measured glycerolipid-FA metabolism genes to ISO levels.
In keeping with previous reports (1–5), despite their similar age, BMI, and percentage fat mass, ISO subjects had a healthier cardiometabolic profile showing enhanced insulin sensitivity and glucose tolerance, favorable inflammatory profile, and decreased ectopic fat accumulation as compared to IRO patients. A novel finding was that SAT expression levels of a wide panel of molecules involved in glycerolipid-FA cycling was higher in IRO than in ISO individuals (Figures 1 and 2), even when groups were matched for high VAT content (Supplemental Figure 2). Although it remains to be experimentally verified with tracer studies, these results are indicative of enhanced SAT de novo lipogenesis, glycerolipid-FA cycling, and release of FA from SAT into the circulation in IRO patients. This provides a potential explanation for their higher ectopic lipid accumulation and altered cardiometabolic profile, and it is consistent with the notion of lipotoxicity-induced insulin resistance.
Taken individually, several of our results are consistent with previous findings. For instance, pharmacological or genetic inhibition of Gpat1 (17), Dgat1 (18, 19), Fatp1 (20), Cidec (21), or Dgat2 (22) improved lipid-induced insulin resistance in mice, whereas overexpression of Gpat1 in rats (23) and of Dgat1 in AT of FVB mice (24), but not of C57BL6 mice (25), induced insulin resistance. Moreover, CD36 protein abundance was increased in SAT of obese patients as compared to lean subjects and further increased in obese patients with type 2 diabetes (26). Taken together, these studies suggest that enhanced uptake and synthesis of glycerolipid-FAs is associated with insulin resistance and that their inhibition at various steps has beneficial effects on insulin sensitivity and metabolic health.
Interestingly, we also found that IRO patients had higher expression levels (mRNA and/or protein) of several molecules involved in neutral glycerolipid hydrolysis, such as HSL, ATGL, ABHD5, FABP4, and PLIN1. These results suggest that the overall lipid synthesis/lipolysis turnover in SAT of IRO is increased compared to ISO patients. Although not measured herein, this could result from increased SAT adipocyte size and/or number in IRO. IRO patients were reported to have larger adipocytes (27), and larger adipocytes display enhanced rates of lipid synthesis/lipolysis turnover (28). In keeping with our results of increased lipolytic gene expression in IRO, pharmacological or genetic inhibition of Abhd5 (29), Hsl (30, 31), and Atgl (32, 33) all resulted in improved insulin sensitivity in mice. In humans, there was a strong association between AT lipolysis rates and insulin resistance (30). Conversely, Jocken et al (34) reported lower SAT mRNA and protein levels of HSL and ATGL in IRO than in ISO patients. However, in contrast to our study, their patient groups were not matched for BMI, age, or sex, which could have accounted, at least in part, for the differences they observed and the divergence with our results. In addition, genes involved in glycerolipid-FA synthesis were not measured in that study. Most previous reports, as well as our results, suggest that there is a coordinate regulation of lipid synthesis and lipolysis in SAT (10). Our findings also suggest that increased glycerolipid-FA cycling in SAT could contribute to the development of insulin resistance, at least in part, by transferring FAs to ectopic tissues such as skeletal muscle.
In the present report, mRNA and/or protein expression of ACC1 and FASN, key enzymes of de novo lipogenesis, was augmented in IRO patients. These results were further supported by a 7-fold lower abundance of ACC phosphorylated at S79, a well-documented inhibitory site and specific downstream target of AMPK (16). Our results are thus suggestive of higher ACC activity and de novo lipogenesis and lower AMPK activity in SAT of IRO patients as compared to ISO subjects. Dysregulation of the fuel-sensing enzyme AMPK has been proposed as both a pathogenic factor for the development of obesity-related diseases in humans and a target for their prevention and therapy (35). AMPK activity is regulated by a complex mechanism that involves phosphorylation of the critical T172 residue of its catalytic subunit by upstream kinases such as LKB1, as well as allosteric activation by AMP (35). In the present study, despite marked differences in P-ACC S79 levels, P-AMPK T172 abundance was not significantly different between the groups. In human and rodent muscles, increases in P-ACC S79 without apparent increases in P-AMPK T172 have been reported previously and attributed to allosteric activation of AMPK (36–38). Lower AMPK activity in AT of IRO patients is in keeping with our previous findings in a different cohort of ISO and IRO patients at the Boston Medical Center (4, 5). Decreased SAT AMPK activity in IRO patients is consistent with their higher expression of GPAT1 and ACC1 because transcription of the genes encoding these enzymes is known to be down-regulated by AMPK (23, 39).
Further supporting the view that enhanced glycerolipid-FA cycling in SAT contributes to insulin resistance, we found that a 6-month dietary intervention, which led to an approximately 6% weight loss in both ISO and IRO groups but an improvement of insulin sensitivity in IRO patients only, restored most of this group's de novo lipogenesis and glycerolipid-FA metabolism mRNAs and proteins to ISO levels. Specifically, mRNA levels of PPARG, ACC1, GPAT1, AGPAT2, DGAT1, ACSL1, FATP1, and ABHD5 were decreased after the diet intervention, predominantly in the IRO patients, to values no longer different from ISO subjects. At the protein level, ACSL1, FABP4, FASN, and PLIN1, which were higher in IRO patients at baseline, were no longer different between the groups after the dietary intervention.
Our study presents some limitations. First, its cross-sectional design precludes a demonstration that altered SAT expression of glycerolipid-FA cycle genes is either a cause or a consequence of insulin resistance in obesity. However, our analyses of 18 transcripts and six protein products allowed us to observe strong associations between the expression of glycerolipid-FA metabolism genes and insulin resistance. Moreover, the longitudinal aspect of the weight-loss intervention provides strong support to these associations. An additional limit is that the study was performed only in postmenopausal obese women of the Montreal metropolitan area in Quebec, Canada, and only on abdominal SAT. New studies in men and in different age and ethnic groups are thus warranted, as well as in SAT from areas such as the gluteofemoral region, which was shown to have a distinct gene expression profile and a protective role against cardiometabolic risk (40). Although performed on a fairly small number of patients, our study has the advantage of reporting the results of well-matched patient groups that were thoroughly phenotyped with “gold standard” tests such as the HEC.
In conclusion, we showed a close relationship between insulin resistance and SAT expression of several genes involved in both the synthesis and the hydrolysis of glycerolipid-FAs. Our results also suggest that IRO patients have higher SAT de novo lipogenesis and lower AMPK activity than ISO patients. Finally, a 6-month dietary intervention was able to restore the expression of most measured genes to ISO levels. Overall, although it remains to be experimentally tested in animal models, our results suggest that enhanced de novo lipogenesis and glycerolipid-FA cycling in SAT, in association with reduced AMPK activity, contribute to obesity-associated insulin resistance.
Acknowledgments
We thank Josée Champagne, Jennifer Levasseur, Roxanne Lussier, Johane Morin, Annie Tardif, and Sylvain Tessier for technical assistance, and Drs Denis Faubert, Erik Joly, and Marie-Line Peyot for valuable advice.
This work was supported by grants from Génome Québec (to R.R.-L., R.S., and M.P.), the J-A DeSève Chair for Clinical Research (to R.R.-L.), National Institutes of Health Grant DK094749, R24 (to N.B.R. and M.P.), the Canada Institute of Health Research (to M.P. and S.R.M.M.), and Fonds de recherche du Québec-Santé (FRQ-S) (to B.C.). M.P. holds the Canada chair in diabetes and metabolism, R.R.-L. is a senior FRQ-S scholar, and M.-S.G. is a Canadian Diabetes Association fellow.
Author Contributions: M.-S.G. designed the research; acquired, analyzed, and interpreted the data; wrote the manuscript; and approved the final version to be published. J.R.P. and M.-E.L. acquired and analyzed data, critically edited and revised the manuscript for important intellectual content, and approved the final version to be published. R.S., S.R.M.M., N.B.R., B.C., M.P., and R.R.-L. designed the research, critically edited and revised the manuscript for important intellectual content, and approved the final version to be published.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by grants from Génome Québec (to R.R.-L., R.S., and M.P.), the J-A DeSève Chair for Clinical Research (to R.R.-L.), National Institutes of Health Grant DK094749, R24 (to N.B.R. and M.P.), the Canada Institute of Health Research (to M.P. and S.R.M.M.), and Fonds de recherche du Québec-Santé (FRQ-S) (to B.C.). M.P. holds the Canada chair in diabetes and metabolism, R.R.-L. is a senior FRQ-S scholar, and M.-S.G. is a Canadian Diabetes Association fellow.
Footnotes
- ABHD5
- abhydrolase domain containing 5
- ACC
- acetyl-coenzyme A carboxylase
- ACSL1
- acyl-coenzyme A synthetase long-chain family member 1
- AGPAT
- 1-acylglycerol-3-phosphate O-acyltransferase
- AMPK
- AMP-activated protein kinase
- AQP
- aquaporin
- AT
- adipose tissue
- ATGL
- adipose triglyceride lipase
- AUC
- area under the curve
- BMI
- body mass index
- BP
- blood pressure
- DGAT
- diacylglycerol O-acyltransferase
- FA
- fatty acid
- FABP4
- fatty acid-binding protein
- FASN
- fatty acid synthase
- FATP1
- FA transport protein 1
- FFM
- fat free mass
- GDR
- glucose disposal rate
- GPAT
- glycerol-3-phosphate acyltransferase
- HEC
- hyperinsulinemic-euglycemic clamp
- HOMA-IR
- homeostasis model assessment of insulin resistance
- HSL
- hormone-sensitive lipase
- IRO
- insulin-resistant obese
- ISO
- insulin-sensitive obese
- NEFA
- nonesterified fatty acid
- OGTT
- oral glucose tolerance test
- P-ACC
- phospho-ACC
- P-AMPK
- phospho-AMPK
- PLIN
- perilipin
- PPARγ
- peroxisome proliferator-activated receptor γ
- qRT-PCR
- quantitative RT-PCR
- SAT
- sc AT
- SREBP-1c
- sterol regulatory element-binding protein 1c
- SRM
- selected reaction monitoring
- T-ACC
- total-ACC
- T-AMPK
- total-AMPK
- VAT
- visceral AT.
References
- 1. Stefan N, Kantartzis K, Machann J, et al. . Identification and characterization of metabolically benign obesity in humans. Arch Intern Med. 2008;168:1609–1616. [DOI] [PubMed] [Google Scholar]
- 2. Karelis AD, Faraj M, Bastard JP, et al. . The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. 2005;90:4145–4150. [DOI] [PubMed] [Google Scholar]
- 3. Gauthier MS, Rabasa-Lhoret R, Prud'homme D, et al. . The metabolically healthy but obese phenotype is associated with lower plasma levels of persistent organic pollutants as compared to the metabolically abnormal obese phenotype. J Clin Endocrinol Metab. 2014;99:E1061–E1066. [DOI] [PubMed] [Google Scholar]
- 4. Xu XJ, Gauthier MS, Hess DT, et al. . Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53:792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gauthier MS, O'Brien EL, Bigornia S, et al. . Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meigs JB, Wilson PW, Fox CS, et al. . Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. J Clin Endocrinol Metab. 2006;91:2906–2912. [DOI] [PubMed] [Google Scholar]
- 7. Wildman RP, Muntner P, Reynolds K, et al. . The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch Intern Med. 2008;168:1617–1624. [DOI] [PubMed] [Google Scholar]
- 8. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. [DOI] [PubMed] [Google Scholar]
- 9. Unger RH, Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab. 2010;21:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prentki M, Madiraju SR. Glycerolipid metabolism and signaling in health and disease. Endocr Rev. 2008;29:647–676. [DOI] [PubMed] [Google Scholar]
- 11. Dinel AL, Kolditz C, Langin D. Metabolism of fatty acids in adipocytes. In: Clément K, Spiegelman BM, Christen Y, eds. Novel Insights into Adipose Cell Functions. Berlin, Heidelberg, Germany; Springer-Verlag; 2010;21–43. [Google Scholar]
- 12. Bézaire V, Langin D. Regulation of adipose tissue lipolysis revisited. Proc Nutr Soc. 2009;68:350–360. [DOI] [PubMed] [Google Scholar]
- 13. Rubio-Cabezas O, Puri V, Murano I, et al. . Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1:280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lavoie ME, Rabasa-Lhoret R, Doucet E, et al. . Association between physical activity energy expenditure and inflammatory markers in sedentary overweight and obese women. Int J Obes (Lond). 2010;34:1387–1395. [DOI] [PubMed] [Google Scholar]
- 15. Brochu M, Malita MF, Messier V, et al. . Resistance training does not contribute to improving the metabolic profile after a 6-month weight loss program in overweight and obese postmenopausal women. J Clin Endocrinol Metab. 2009;94:3226–3233. [DOI] [PubMed] [Google Scholar]
- 16. Hardie DG, Carling D. The AMP-activated protein kinase–fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. [DOI] [PubMed] [Google Scholar]
- 17. Neschen S, Morino K, Hammond LE, et al. . Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. [DOI] [PubMed] [Google Scholar]
- 18. Cao J, Zhou Y, Peng H, et al. . Targeting acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J Biol Chem. 2011;286:41838–41851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen HC, Smith SJ, Ladha Z, et al. . Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J Clin Invest. 2002;109:1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim JK, Gimeno RE, Higashimori T, et al. . Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest. 2004;113:756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishino N, Tamori Y, Tateya S, et al. . FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest. 2008;118:2808–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi CS, Savage DB, Kulkarni A, et al. . Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem. 2007;282:22678–22688. [DOI] [PubMed] [Google Scholar]
- 23. Gonzalez-Baró MR, Lewin TM, Coleman RA. Regulation of triglyceride metabolism. II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1195–G1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen N, Liu L, Zhang Y, Ginsberg HN, Yu YH. Whole-body insulin resistance in the absence of obesity in FVB mice with overexpression of Dgat1 in adipose tissue. Diabetes. 2005;54:3379–3386. [DOI] [PubMed] [Google Scholar]
- 25. Chen HC, Stone SJ, Zhou P, Buhman KK, Farese RV. Dissociation of obesity and impaired glucose disposal in mice overexpressing acyl coenzyme A:diacylglycerol acyltransferase 1 in white adipose tissue. Diabetes. 2002;51:3189–3195. [DOI] [PubMed] [Google Scholar]
- 26. Bonen A, Tandon NN, Glatz JF, Luiken JJ, Heigenhauser GJ. The fatty acid transporter FAT/CD36 is upregulated in subcutaneous and visceral adipose tissues in human obesity and type 2 diabetes. Int J Obes (Lond). 2006;30:877–883. [DOI] [PubMed] [Google Scholar]
- 27. Klöting N, Fasshauer M, Dietrich A, et al. . Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299:E506–E515. [DOI] [PubMed] [Google Scholar]
- 28. Salans LB, Bray GA, Cushman SW, et al. . Glucose metabolism and the response to insulin by human adipose tissue in spontaneous and experimental obesity. Effects of dietary composition and adipose cell size. J Clin Invest. 1974;53:848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown JM, Betters JL, Lord C, et al. . CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res. 2010;51:3306–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Girousse A, Tavernier G, Valle C, et al. . Partial inhibition of adipose tissue lipolysis improves glucose metabolism and insulin sensitivity without alteration of fat mass. PLoS Biol. 2013;11:e1001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park SY, Kim HJ, Wang S, et al. . Hormone-sensitive lipase knockout mice have increased hepatic insulin sensitivity and are protected from short-term diet-induced insulin resistance in skeletal muscle and heart. Am J Physiol Endocrinol Metab. 2005;289:E30–E39. [DOI] [PubMed] [Google Scholar]
- 32. Haemmerle G, Lass A, Zimmermann R, et al. . Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. [DOI] [PubMed] [Google Scholar]
- 33. Kienesberger PC, Lee D, Pulinilkunnil T, et al. . Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J Biol Chem. 2009;284:30218–30229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jocken JW, Langin D, Smit E, et al. . Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J Clin Endocrinol Metab. 2007;92:2292–2299. [DOI] [PubMed] [Google Scholar]
- 35. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wojtaszewski JF, Mourtzakis M, Hillig T, Saltin B, Pilegaard H. Dissociation of AMPK activity and ACCβ phosphorylation in human muscle during prolonged exercise. Biochem Biophys Res Commun. 2002;298:309–316. [DOI] [PubMed] [Google Scholar]
- 37. Hancock CR, Janssen E, Terjung RL. Contraction-mediated phosphorylation of AMPK is lower in skeletal muscle of adenylate kinase-deficient mice. J Appl Physiol. 2006;100:406–413. [DOI] [PubMed] [Google Scholar]
- 38. Park SH, Gammon SR, Knippers JD, Paulsen SR, Rubink DS, Winder WW. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J Appl Physiol. 2002;92:2475–2482. [DOI] [PubMed] [Google Scholar]
- 39. Li Y, Xu S, Mihaylova MM, et al. . AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pinnick KE, Nicholson G, Manolopoulos KN, et al. . Distinct developmental profile of lower-body adipose tissue defines resistance against obesity-associated metabolic complications [published online June 19, 2014]. Diabetes doi:10.2337/db14-0385. [DOI] [PubMed] [Google Scholar]